
Abstract
The quantity and quality of DNA isolated from environmental samples are crucial for getting robust high-throughput sequencing data commonly used for microbial community analysis. The differences in the nature and physicochemical properties of environmental samples impact DNA yields, and therefore, an optimisation of the protocols is always recommended. For instance, samples collected from corroded areas contain high concentrations of metals, salts, and hydrocarbons that can interfere with several steps of the DNA extraction protocols, thereby reducing yield and quality. In this study, we compared the efficiency of commercially available DNA extraction kits and laboratory-adopted methods for microbial community analysis of iron incrustations and oilfield-produced water samples. Modifications to the kits manufacturers’ protocols were included to maximise the yield and quality. For iron incrustations, the modified protocol for FastDNA Spin Kit for Soil yielded higher DNA and resulted in higher diversity, including the recovery of low-abundant and rare taxa in the samples, compared to DNeasy PowerSoil Pro Kit. The DNA extracted with modified phenol–chloroform methods yielded higher DNA but failed to pass quality control PCR for 16S sequencing with and without purification. The protocols mentioned here can be used to maximise DNA recovery from iron incrustations and oilfield-produced water samples.
Similar content being viewed by others


Introduction
Identification of microbes is crucial for finding solutions to many environmental problems. However, more than 99% of the bacteria in the environment are non-culturable and, therefore, cannot be detected through traditional culture-based methods1,2. Against this backdrop, the use of appropriate molecular methods with higher detection limits and superior sensitivity is a prerequisite for studying microbial communities in a particular environment3. Due to the incompetence of culture-based methods and the limit of detection, culture-independent high throughput sequencing (HTS) for the identification of microbial communities has recently gained much popularity4,5. HTS methods have helped to reveal the complexity of the spatial–temporal dynamics and diversity of microbial populations in different environments6,7,8. The process starts with the extraction of DNA directly from the environmental samples, followed by library preparation and HTS. The quality of DNA is crucial in generating high-quality data for HTS-based microbial community analysis.
DNA isolation from some environmental samples can be challenging due to the presence of humic acids, inhibitors, sediments, and contaminants that interfere with DNA extraction9,10. For instance, metal ions such as iron, zinc and tin can easily bind to template DNA through the formation of direct crosslink and inhibit PCR by blocking access of polymerase to the DNA11,12,13. In addition, the co-extraction of DNA from solid and liquid samples rich in chemicals and salts can reduce the quality of DNA for subsequent downstream applications14. Many studies reported optimisation of DNA extraction methods from environmental samples in the past three decades15,16,17,18,19; however, the type and diversity of environmental samples make it difficult to set a common method of DNA extraction. For instance, the basic principles for DNA extraction from solid and liquid samples are different and thus, the kits used for extractions. Furthermore, the addition of chemicals and modification of steps can result in high-concentration good-quality DNA from environmental samples20. Therefore, the choice of extraction methods and modifications have critical impacts on the DNA-based phylogenetics, diversity and abundance of any sample21,22,23. Considering the importance of DNA yield and quality for downstream applications such as PCR or HTS, selecting proper methods and optimising DNA extraction from metal-rich environments are equally important to reduce the burdens and biases from microbial community analysis.
In the energy sector, specifically in the hydrocarbon industry, microbes are associated with different types of environmental events. Among these, microbiologically influenced corrosion (MIC) is particularly complex to handle, as samples usually contain numerous inhibitors, including high concentrations of iron, salts, and hydrocarbons24,25. In MIC, microorganisms play an essential role in the deterioration of assets26; hence, assessing microbial diversity and understanding its functional capability is fundamental for preventing and mitigating undesired phenomena. Early MIC detection requires the identification of corrosive microorganisms in the system, which is currently conducted using DNA-based methods3,27,28. Currently, there are no standard protocols or established procedures for high-quality DNA extractions from corrosion-related samples; therefore, evaluating and optimising different extraction methods are paramount to obtaining meaningful data for MIC management. Successful extraction of nucleic acids will ensure the precise identification of microbial communities linked to corrosion, which will help to prevent MIC issues in the hydrocarbon industry.
This study aimed to improve the yield and quality of DNA from iron incrustations and produced water from Australian oilfields using commercial kits and manual extraction methods. We investigated whether the DNA extraction methods and modifications affect the diversity and composition of microbial communities identified through high-throughput sequencing of 16S rRNA gene.
Results
DNA yields
Both modified phenol–chloroform methods yielded a higher phenol-contaminated DNA but a lower 260/280 ratio. DNA recovery by method 1 (Barnett & Larson) was higher (322.4 ng/µl) but inferior (260/280 ratio: 1.18) than method 2 (Nishiguchi) (288.4 ng/µl, 260/280 ratio: 1.21). Both methods failed to pass the quality control in terms of 16S PCR and 1% agarose gel electrophoresis (Table S1).
In prepared samples compared to the community standard
For four iron incrustations, the DNA concentration obtained with each protocol is shown in Table S1 and DNA quality was checked in 1% agarose gel (Fig. S1). The highest DNA concentration was obtained with MP kit (Table S1, Fig. S1). Both phenol–chloroform methods were found inefficient in recovering good quality DNA from iron incrustations, and failed to amplify in PCR; therefore, no further analyses were conducted with these methods.
For produced water samples, heating incubation for 30 min yielded higher DNA compared to others without compromising the quality (Table S2). Consequently, 30 min of thermal treatment were included in all tested protocols. DNA concentrations obtained with each protocol are presented in Table S3. The PW kit yielded significantly lower DNA concentration compared to MP and PS kits. Similarly, the highest DNA concentration for water samples was obtained with the MP kit.
The extracted DNA was purified using PC kit, followed by amplification and sequencing of V3V4 regions of 16S rRNA gene. The comparative summarised results generated from sequence data for iron incrustation and water samples using qiime2 (SILVA 138) are shown in Table S4 and S5, respectively. It was observed that all extraction protocols were able to isolate DNA from eight microbial species present in the community standard; however, the overall results suggest that MP kit had better yield and quality as well as higher efficiency in extracting and recovering community DNA from both types of samples.
From field samples
DNA concentration obtained with each protocol is presented in Table S6 and S7. As observed in the previous stage, MP kit yielded a higher (p-value < 0.001) concentration of DNA (17.5 ± 1.4 ng/µl for iron incrustation 1, 586.4 ± 145.8 ng/µl for iron incrustation 2) than the PS kit (7.1 ± 1.2 ng/µl for iron incrustation 1, 35.9 ± 4.6 ng/µl for iron incrustation 2). Similar results were obtained for the oilfield water samples wherein MP (12.6 ± 2.1 ng/µl for oilfield water 1, 30.2 ± 1.4 ng/µl for oilfield water 2) yielded significantly (p-value < 0.001) higher concentration of DNA than PS kit (2.7 ± 1.8 ng/µl for oilfield water 1, 17.4 ± 2.6 ng/µl for oilfield water 2). The PCR amplification was found negative for all DNA samples extracted with MP and PS kits. A subsequent DNA clean-up was performed with PC kit and all samples showed positive amplification after purification (Fig. S2).
Microbial diversity and composition in the field samples
Sequence statistics
Soil and water samples were represented by 1.1 million quality reads obtained from amplicon sequencing of 35 samples, ranging from 10,112 to 76,886 and an average of 28,436.5 ± 2678.2. After filtering, 1.1 million merged reads generated 1325 ASVs, 34 phyla, 162 orders, 258 genera. Each sample was sequenced at maximum depth to capture most of the diversity as revealed through a saturated rarefaction plot (Fig. S3) and high good coverage index values (Table S8).
Microbial diversity and composition in iron incrustations
Based on the observed species and Shannon as an indicator of alpha diversity measurements, MP kit captured significantly higher diversity in both iron A and B samples, compared to PS kit (Fig. 1A,B). Beta-ordination analysis showed that the selection of kit for iron incrustations’ DNA extraction had a significant influence on the identification of low-abundant rare and the richness of the most abundant bacterial communities. Principal coordinate analysis (PCoA) clearly separated microbial communities based on taxonomic dissimilarities (unweighted) and their relative abundance (weighted) as shown in Fig. 1C,D.

Diversity metrics of iron-incrustation samples. Alpha-diversity based on observed species (A) and Shannon index (B). Beta-diversity based on unweighted (C) and weighted (D) UniFrac distance metrics. The horizontal central lines in the box (A,B) showing the median values of the curated data. The box limits correspond to the 25th and 75th percentiles, and the whiskers are the 5th and 95th percentiles. ***Significant differences at α-level of 0.001.
Proteobacteria was found to be the most predominant phylum in iron incrustations extracted with both MP and PS kits. An inconsistency was observed for the second dominant phylum wherein MP favoured Actinobacteriota, and PS supported Synergistota. The second phylum Synergistota had a very high abundance with PS kit for iron B sample (Fig. 2A). At the genus level, we observed a similarity in microbial communities between samples A and B extracted with the same kit. Diverse bacterial communities were identified with MP kit despite having a significant percentage of bacteria with low abundance (< 1%) at the genus level. The iron sample A showed a higher abundance for Mycobacterium and Defluviimonas with MP, while Pseudomonas and Acetomicrobium were for PS kit (Fig. 2B).

Relative abundance of bacteria in iron samples at phylum (A) and genus (B) level. Phyla and genera represent ≥ 1% of read abundance were considered for plotting. Less than 1% and unclassified were grouped as “other”.
Microbial diversity and composition in iron incrustations
Unlike soil samples, no difference between MP and PW was observed for water sample B in terms of bacterial alpha-diversity (Fig. 3A). Beta-ordinations, however, showed significant differences and the presence of new or rare species was found as the main driver linked to distinct separation with unweighted UniFrac distance metric (Fig. 3B).

Diversity and composition of microbial communities in produced water samples from oilfield. (A) Alpha-diversity based on observed species and Shannon index. (B) Beta-diversity based on unweighted and weighted UniFrac distance metrics. The horizontal central lines in the box (for A) showing the median values of the curated data. The box limits correspond to the 25th and 75th percentiles, and the whiskers are the 5th and 95th percentiles. (C) Relative abundance at phylum level. (D) Relative abundance at genus level. Phyla and genera represent ≥ 1% of read abundance were considered for plotting. Less than 1% and unclassified were grouped as “other”.
For water, both kits generated similar community composition of bacteria dominated by Proteobacteria (> 95%) and Trabulsiella (> 80%) at phylum and genus levels, respectively (Fig. 3C,D).
Discussion
DNA extraction from some environmental samples can be challenging but crucial in terms of performance, sustainability, and economy. Ample studies have been performed evaluating the efficacy of DNA extraction methods from water29, animal guts30,31, soil21,32,33, rock and coral14,34, freshwater and marine sediments35,36, and found variation in microbial communities. However, to our best knowledge, no study has yet been performed on evaluating and optimising DNA extraction from iron incrustations and produced water from oilfields. The proposed methods and modifications, therefore, could be used as a future reference for corroded samples from oilfields.
In this study, we used three of the most commonly used commercial kits for soil and water DNA extractions. We first evaluated the selectivity, specificity and accuracy of the commonly used commercial DNA extraction kits for soil and water in relation to a known microbial community (standard). Despite some variations in the relative abundance, no unwanted genera were classified from control iron powder samples that mixed with known microbial community standard. This result signifies that the used kits were accurate in the identification of microbial communities present in a sample, overruling the biases of spin filters reported earlier37,38. The accuracy of the tested kits in terms of percentages of DNA recovery for known genera from the community standard varied for the two kits. Interestingly, the DNeasy Power Water (PW) kit generated the most inferior results for water samples in terms of DNA yield and purity compared to the other two kits. Several factors could be responsible for these variations, including human gut-oriented bacteria in the microbial community standard and no bead-beating step in the power water kit. Bead beating is a critical step for the complete lysis of microbial cells during environmental DNA extraction, influencing diversity and composition more than non-bead beating methods37,39,40. This is more evident as MP kit recovered significantly higher Actinobacteriota, one of the largest Gram-positive bacterial phyla with the most diverse metabolic capabilities and ubiquitous in both terrestrial and aquatic environments41,42,43. Similar to phyla, at genus level, MP captured more ASVs for Mycobacterium, a Gram-positive environmental bacteria that can survive in extreme environments, specifically in metal-contaminated water44. Their role therefore can be crucial for the study of groundwater and soil contamination by heavy metals. Gram-positive bacteria have a thick cell wall that is difficult to lyse during DNA extraction compared to thin cell wall containing Gram-negative bacteria45. MP therefore generated a mixture of community containing both Gram-positive and Gram-negative bacteria. On the other hand, PS favoured Synergistota, Gram-negative bacteria predominantly found in anaerobic environments, including animal, gut soil, oil field, and wastewater treatment plants46,47,48. Consistent with some previous studies23,37,49, the efficacy of MP kit in yielding Gram-positive bacterial DNA from challenging environmental samples is found to be better than PS in this study. Considering the yield, quality and diversity of microbial communities, the bead-beating step is essential and recommended for all types of environmental DNA extractions.
The highest amount of DNA was recovered with MP kit outperforming PS and PW kits for soil and water, respectively. The higher yield of DNA in the present study with MP is possibly linked to unique glass beads in the lysing matrix, two different buffers, and binding matrix in comparison to other DNA extraction kits for environmental samples, as reported earlier35,36,50. Alongside DNA yield, the diversity and composition were also significantly higher with MP kit compared to PS for soil, suggesting more effective lysing of microbial cells during DNA extractions. Interestingly, more than 50% of reads for iron B samples and 25% of reads for iron A samples were unclassified with MP kit compared to significantly lower unclassified reads with PS kit, signifying identification of rare taxa by MP kit in the corroded samples. While this finding supports two other published reports from marine sediments and wastewater treatment plants35,50, it contradicts another study36 that reported no differences in observed OTUs and species in freshwater lake samples with different kits despite having higher DNA concentrations with MP kit. The type of samples might be associated with these differences as samples from wastewater treatment plants and marine sediments are quite complex and more challenging than freshwater lakes.
In the present study, phenol–chloroform extraction yielded higher concentration but poor quality DNA in terms of 260/280 ratio and showed negative results in 16S PCR and 1% agarose gel. This result upholds the complex nature of iron incrustation samples and produced water from oilfield, wherein quality is more important than quantity. Here, we collected samples from oilfields and corrosion sites rich in iron, manganese, and crude oil. Therefore, most of the samples were found negative in 16S PCR after extraction due to a poor 260/280 ratio and the presence of inhibitors. A higher 260/280 ratio, positive 16S PCR and greater recovery of microbial communities were ensured by an additional purification step with PowerClean following the extraction of samples. This further purification is reported to increase the yield of DNA and diversity of microbial communities from some challenging environmental samples such as oil-contaminated soil51, acidic soil52,53, soil from volcanic desert54, saline soil and water55,56, heavy metal contaminated soil57, and iron incrustations from mines49,58. Like previous reports, we found that the combination of DNA extraction and purification methods is very effective in removing PCR inhibitors from samples collected in challenging environmental conditions.
The overall data showed that the MP kit is better in characterising the low abundant and rare taxa in iron incrustations. PS kit on the other hand, was found very selective in capturing highly abundant or rich taxa in iron incrustations with low resolution for the unclassified communities. Nevertheless, the diversity in terms of richness was found to be significantly higher with the MP kit than the PS kit for iron incrustations. Even with water samples, the MP kit was ahead of PS kit in terms of unshared ASVs and species diversity. Both kits have a certain level of selectivity in DNA-based profiling of bacterial communities wherein the results can be influenced by the abundance of taxa as found in the weighted UniFrac distance metric. For instance, the MP kit showed lower sensitivity in detecting Gram-negative (-ve) bacteria for water samples. The overall findings also suggest that purification of DNA following extraction can increase DNA purity through the removal of inhibitors and, thereby, increasing the efficiency of 16S PCR and recovery of microbial communities in amplicon sequencing.
Materials and methods
Study design
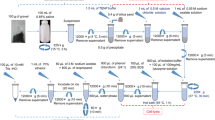
A three-phase study was designed to evaluate the efficiency of commonly used DNA extraction methods and kits for soil and water samples from the perspective of iron incrustation samples and produced water from oilfield. An outline of the study design has been depicted in Fig. 4.

A flow diagram showing the study design and methods used for the data analysis.
Preparation of microbial standard
We first tested the influence of chemicals and metals on DNA yields and quality by preparing a mock sample with known microbial communities (ZymoBIOMICS) containing eight different species in various abundance. The aim was to test the accuracy of different methods in terms of microbial strain recovery from the standard from iron and water samples and to detect any kit-based contaminations (kit-ome) (Table S4 and S5).
Preparation of iron samples
An iron incrustation sample received from an Australian oil production field was used to obtain the iron powder for the laboratory preparation of an iron incrustation sample. The iron powder was aliquoted in Eppendorf tubes (500 mg/tube) and autoclaved to degrade the DNA present in the sample. Following DNA extraction with FastDNA Spin kit for Soil (MP Biomedicals, USA), the DNA degradation by autoclaving was confirmed by the absence of DNA on 1% agarose gel and negative PCR amplification of bacterial 16S, 27F and 1492R59. The outcomes and efficiency of autoclaved samples were comparable to non-autoclaved ones that showed bands on the gel for DNA but not for 16S PCR (Table S9; Fig. S3).
Amplification of iron samples
In the next step, 75 µl of microbial community standard (ZymoBIOMICS) was added to each of the four autoclaved iron powder tubes. The MP-extracted DNA samples were amplified with PCR using bacterial 16S primers covering the complete gene and V3-V4 hypervariable region. Only the positive control (DNA from Shewanella sp.) showed a band on the gel after the visualisation of PCR products (Fig. S5A). Considering positive DNA on gel but negative amplification in PCR, DNA purification from one sample per treatment (T2 and T3) was performed with the DNeasy PowerClean Clean-Up (PC) kit (Qiagen) to clean the sample from possible PCR inhibitors. The purified DNA sample (T3) from the non-autoclaved group showed a positive band on agarose gel, while the autoclaved sample (T2) was found negative (Fig. S5B).
Preparation of water samples
Mock samples for screening of optimum incubation temperature were prepared by filtration of 200 ml of produced water from oilfield on each filter. The standard sample was produced by filtration of 25 ml of nuclease-free water mixed with 75 µl of ZymoBiomics Microbial Community standard.
Optimisation of incubation for water samples
Heating is an optional step for DNA extraction from environmental samples. Thermal treatment of the samples during the isolation procedure helps to denature proteins and increase the speed of chemical reactions60. Nonetheless, heating may also lead to degraded DNA61. In this research, we evaluated three heating incubation times to select the optimal time that increases the yield of DNA without compromising its quality. DNA extraction from filter membranes was performed using 10, 30, and 60 min of heating incubation at 70 °C. DNA concentration and quality were used to select the optimal time.
DNA extractions methods
DNA extraction was conducted using the following protocols: (i) Modified FastDNA Spin Kit for Soil: 3 × 3 cycles of homogenisation in FastPrep Ribolyser, extended centrifugation for 15 min at step 5, five minutes incubation at step 9, optional incubation (step 16) for 5 min at 55 °C. (ii) Modified DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany): 3 × 3 cycles of homogenisation in FastPrep Ribolyser in place of vortexing at step 2. (iii) Modified DNeasy Power Water Kit (Qiagen, Hilden, Germany), only for water samples. (iv) Modified phenol–chloroform method reported by Barnett & Larson, 201262: Resuspension of dried pellet with 100 µl TE buffer. (v) Modified phenol–chloroform method reported by Nishiguchi et al., 2002 with substantial modifications63 (Supplementary data M1.1). Following concentration measurements in Nanodrop spectrophotometer 2000 cc (Thermo Fisher Scientific, MA, USA), the DNA quality was checked in 1.5% agarose gel. The acronym MP, PS and PW were used to mention FastDNA Spin Kit for Soil, DNeasy PowerSoil Pro Kit, and DNeasy Power Water Kit, respectively. MP kit yielded the highest DNA for iron incrustations (Table S1, Fig. S1), and the modified PW kit generated poor data for prepared samples from the membrane filter compared to MP and PS (Table S3) and, therefore, not considered for field samples.
Evaluation of DNA extraction methods using field samples
Two iron incrustation samples received from Australian oilfields were used in this phase to evaluate the extraction protocols with a more diverse community. These samples were collected from ballast tanks exhibiting internal corrosion defects. A total of 10 aliquots, each of 0.53 ± 0.007 mg were prepared from each sample. DNA was extracted in quintuplicate with the modified MP kit and the modified PS protocols described earlier. For water samples, two produced water samples received from Australian oilfields were used in this phase to evaluate the extraction protocols. Sample A was obtained from the high-pressure separator within an oil production facility, whereas, Sample B was collected from a wellhead of an oilfield. A total of eight membrane filters were prepared by filtering 200 ml of water. Each filter was equally divided, and DNA was extracted in quintuplicate with the modified MP and PS protocols. The concentration and quality of DNA were checked in agarose gel and qubit. For water, sample A extracted with PS failed to pass the required QC (16S PCR amplification) and was eventually removed for further analysis.
PCR amplification, amplicon library preparation and sequencing
The amplicon for 16S V3V4 regions was generated in two-step PCR methods following “Illumina 16S metagenomic sequencing library preparation”64,65. The first PCR was run for 30 cycles, followed by beads purification of positive amplicons, and indexing via second PCR (12 cycles). Paired-end sequencing (v3 kit, 600 cycles) was performed with Illumina MiSeq platforms (Illumina Inc., CA, USA). The PCR conditions are available in supplementary data (Supplementary data M1.2).
Data processing and statistical analysis
The raw sequence data was imported into qiime2 (v2021.4) for paired-end processing66. Quality trimming (denoising) of demultiplexed sequences was performed in q2-dada2 plug-in with the following parameters: -p-trim-left-l 0; -p-trunc-len-f 280; -p-trim-left-r 0; -p-trunc-len-r 22067. The DADA2 output as a feature frequency ASV (Amplicon Sequence Variants) table that represents biological features of amplicon sequence was classified taxonomically against SILVA 138 release using consensus blast68. The chimeric features and singletons were removed and the feature ASV table was filtered based on the lowest non-zero frequency 10. We removed chloroplast and mitochondrial sequences from the final data. Each sample was set to an even depth of 10,112 for downstream analysis of alpha–beta diversity and microbial composition. The rarefied ASV table, taxonomy table and metadata were used for alpha–beta diversity analysis with the R statistical software (v4.22)69. Observed species and Chao1 were chosen for the changes in diversity, while Shannon and Simpson's indices were used for the calculation of evenness among groups. Weighted (relative abundance) and unweighted (presence-absence) UniFrac distance metrics were used for the calculation of beta diversity. Alpha–beta diversity measurements were performed using phyloseq70, microbiomeSeq71, microbiome72 and vegan73 R packages in support of plotting packages ggplot2 . The relative abundance of bacteria at various taxa levels was calculated with the phyloseq R package. Clostridium sensu stricto 1 has been renamed as Clostridium. Kruskal–Wallis ranks test was used to compare alpha-diversity among groups. Centroid analysis of beta dispersion among the groups was performed as permutational multivariate analysis (PERMANOVA) with the vegan R package. Significantly abundant bacterial genera were identified using Linear Discriminant Analysis Effect Sizes (LefSe) at LDA cut-off value of 2.0 and more74. At every stage of data analysis, p-value of less than 0.05 was considered as statistically significant.
Data availability
The 16S rRNA sequences were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive under BioProject number PRJNA1004675.
References
Chen, L. et al. Profiling total viable bacteria in a hemodialysis water treatment system. J. Microbiol. Biotechnol. 27, 995–1004 (2017).
Ding, L., Su, X. & Yokota, A. Research progress of VBNC bacteria—A review. Acta Microbiol. Sin. 51, 858–862 (2011).
Eckert, R. B. & Skovhus, T. L. Advances in the application of molecular microbiological methods in the oil and gas industry and links to microbiologically influenced corrosion. Intl. Biodeter. Biodegr. 126, 169–176 (2018).
Vasudeva, K., Kaur, P. & Munshi, A. Metagenomics to Bioremediation 685–708 (Elsevier, Berlin, 2023).
Wydro, U. Soil microbiome study based on DNA extraction: A review. Water 14, 3999 (2022).
Saraiva, J. P. et al. Mining synergistic microbial interactions: A roadmap on how to integrate multi-omics data. Microorganisms 9, 840 (2021).
Song, M. et al. Response mechanism of meiofaunal communities to multi-type of artificial reef habitats from the perspective of high-throughput sequencing technology. Sci. Total Environ. 863, 160927 (2023).
Michan, C., Blasco, J. & Alhama, J. High-throughput molecular analyses of microbiomes as a tool to monitor the wellbeing of aquatic environments. Microb. Biotechnol. 14, 870–885 (2021).
Howarth, A., Drummond, B., Wasef, S. & Matheson, C. D. An assessment of DNA extraction methods from blood-stained soil in forensic science. Forensic Sci. Int. 341, 111502 (2022).
Wilson, I. G. Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol. 63, 3741–3751 (1997).
Hall, N. E. & Axelrod, D. E. Interference of cellular ferric ions with DNA extraction and the application to methods of DNA determination. Anal. Biochem. 79, 425–430 (1977).
Kuffel, A., Gray, A. & Daeid, N. N. Impact of metal ions on PCR inhibition and RT-PCR efficiency. Int. J. Legal Med. 135, 63–72 (2021).
Patterson, C., Gray, S., Wendt, F. R. & Roy, R. Inhibition of DNA amplification caused by metal in extracted bloodstains and in direct amplification. Forensic Sci. Int. Genetics 55, 102598 (2021).
Herrera, A. & Cockell, C. S. Exploring microbial diversity in volcanic environments: A review of methods in DNA extraction. J. Microbiol. Methods 70, 1–12 (2007).
Amin, H. et al. Optimization of bacterial DNA and endotoxin extraction from settled airborne dust. Sci. Total Environ. 857, 159455 (2023).
Guerra, V., Beule, L., Lehtsaar, E., Liao, H.-L. & Karlovsky, P. Improved protocol for DNA extraction from subsoils using phosphate lysis buffer. Microorganisms 8, 532 (2020).
Hwang, C., Ling, F., Andersen, G. L., LeChevallier, M. W. & Liu, W.-T. Evaluation of methods for the extraction of DNA from drinking water distribution system biofilms. Microb. and Environ. 27, 9–18 (2012).
Miller, D., Bryant, J., Madsen, E. & Ghiorse, W. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 65, 4715–4724 (1999).
Medina Caro, D. et al. An improved method for intracellular DNA (iDNA) recovery from terrestrial environments. MicrobiologyOpen 12, e1369 (2023).
Alibrandi, A., di Primio, R., Bartholomäus, A. & Kallmeyer, J. A modified isooctane-based DNA extraction method from crude oil. mLife 2, 328–338 (2023).
Zielińska, S. et al. The choice of the DNA extraction method may influence the outcome of the soil microbial community structure analysis. MicrobiologyOpen 6, e00453 (2017).
Munoz-Colmenero, M. et al. Evaluation of DNA extraction methods and bioinformatic pipelines for marine nano-and pico-eukaryotic plankton analysis. Frntr. Mar. Sci. 7, 584253 (2021).
Kennedy, N. A. et al. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS One 9, e88982 (2014).
Javaherdashti, R. & Javaherdashti, R. Microbiologically influenced corrosion (MIC) (Springer, 2017).
Telegdi, J., Shaban, A. & Trif, L. Microbiologically influenced corrosion (MIC). Trends in oil and gas corrosion research and technologies, 191–214 (2017).
Kotu, S. P., Mannan, M. S. & Jayaraman, A. Emerging molecular techniques for studying microbial community composition and function in microbiologically influenced corrosion. Intl. Biodeter. Biodegr. 144, 104722 (2019).
Salgar-Chaparro, S. J., Lepkova, K., Pojtanabuntoeng, T., Darwin, A. & Machuca, L. L. Microbiologically influenced corrosion as a function of environmental conditions: A laboratory study using oilfield multispecies biofilms. Corros. Sci. 169, 108595 (2020).
Zhang, Y. et al. Microbiologically influenced corrosion of steel in coastal surface seawater contaminated by crude oil. NPJ Mater. Degrad. 6, 35 (2022).
Brandt, J. & Albertsen, M. Investigation of detection limits and the influence of DNA extraction and primer choice on the observed microbial communities in drinking water samples using 16S rRNA gene amplicon sequencing. Front. Microbiol. 9, 2140 (2018).
Burbach, K., Seifert, J., Pieper, D. H. & Camarinha-Silva, A. Evaluation of DNA extraction kits and phylogenetic diversity of the porcine gastrointestinal tract based on Illumina sequencing of two hypervariable regions. Microbiologyopen 5, 70–82 (2016).
Kashinskaya, E. N., Andree, K. B., Simonov, E. P. & Solovyev, M. M. DNA extraction protocols may influence biodiversity detected in the intestinal microbiome: a case study from wild Prussian carp. Carassius gibelio. FEMS Microbiol. Ecol. 93, fiw240 (2017).
Echeverría-Beirute, F., Varela-Benavides, I., Jiménez-Madrigal, J., Carvajal-Chacon, M. & Guzmán-Hernández, T. eDNA extraction protocol for metagenomic studies in tropical soils. BioTechniques 71, 580–586 (2021).
Tanase, A.-M. et al. Comparison of total DNA extraction methods for microbial community form polluted soil. Agric. Agric. Sci. Procedia 6, 616–622 (2015).
Pratte, Z. A. & Kellogg, C. A. Comparison of preservation and extraction methods on five taxonomically disparate coral microbiomes. Frntr. Marine Sci 8, 938 (2021).
Ramírez, G. A., Graham, D. & D’Hondt, S. Influence of commercial DNA extraction kit choice on prokaryotic community metrics in marine sediment. Limnol. Oceanogr. Methods 16, 525–536 (2018).
Shi, Z. et al. The effects of DNA extraction kits and primers on prokaryotic and eukaryotic microbial community in freshwater sediments. Microorganisms 10, 1213 (2022).
Guo, F. & Zhang, T. Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl. Microbiol. Biotechnol. 97, 4607–4616 (2013).
Xue, M., Wu, L., He, Y., Liang, H. & Wen, C. Biases during DNA extraction affect characterization of the microbiota associated with larvae of the Pacific white shrimp, Litopenaeus vannamei. PeerJ 6, e5257 (2018).
Anderson, S. R. & Thompson, L. R. Optimizing an enclosed bead beating extraction method for microbial and fish environmental DNA. Environ. DNA 4, 291–303 (2022).
Yuan, J., Li, M. & Lin, S. An improved DNA extraction method for efficient and quantitative recovery of phytoplankton diversity in natural assemblages. PLoS One 10, e0133060 (2015).
Anandan, R., Dharumadurai, D. & Manogaran, G. P. Actinobacteria-basics and biotechnological applications (IntechOpen, 2016).
Stach, J. E., Maldonado, L. A., Ward, A. C., Goodfellow, M. & Bull, A. T. New primers for the class Actinobacteria: Application to marine and terrestrial environments. Environ. Microbiol. 5, 828–841 (2003).
Ward, A. C. & Bora, N. Diversity and biogeography of marine actinobacteria. Curr. Opin. Microbiol. 9, 279–286 (2006).
Primm, T. P., Lucero, C. A. & Falkinham, J. O. III. Health impacts of environmental mycobacteria. Clin. Microbiol. Rev. 17, 98–106 (2004).
Mahalanabis, M., Al-Muayad, H., Kulinski, M. D., Altman, D. & Klapperich, C. M. Cell lysis and DNA extraction of gram-positive and gram-negative bacteria from whole blood in a disposable microfluidic chip. Lab Chip 9, 2811–2817 (2009).
Fan, Q. et al. Microbial community evolution, interaction, and functional genes prediction during anaerobic digestion in the presence of refractory organics. J. Environ. Chem. Eng. 10, 107789 (2022).
Hardy, J. et al. Similar methanogenic shift but divergent syntrophic partners in anaerobic digesters exposed to direct versus successive ammonium additions. Microbiol. Spectr. 9, e00805-00821 (2021).
Wang, Y. et al. Anaerobic co-digestion of food waste and sewage sludge in anaerobic sequencing batch reactors with application of co-hydrothermal pretreatment of sewage sludge and biogas residue. Bioresour. Technol. 364, 128006 (2022).
Li, A.-D. et al. Effects of sample preservation and DNA extraction on enumeration of antibiotic resistance genes in wastewater. FEMS Microbiol. Ecol. 94, fix189 (2018).
Florczyk, M., Cydzik-Kwiatkowska, A., Ziembinska-Buczynska, A. & Ciesielski, S. Comparison of three DNA extraction kits for assessment of bacterial diversity in activated sludge, biofilm, and anaerobic digestate. Appl. Sci. 12, 9797 (2022).
Ong, S. Y. & Sudesh, K. Effects of polyhydroxyalkanoate degradation on soil microbial community. Polym. Degradation Stab. 131, 9–19 (2016).
Zheng, M. M. et al. Changes of acid and alkaline phosphatase activities in long-term chemical fertilization are driven by the similar soil properties and associated microbial community composition in acidic soil. Eur. J. Soil Biol. 104, 103312 (2021).
Zheng, M. M., Wang, C., Li, W. X., Song, W. F. & Shen, R. F. Soil nutrients drive function and composition of phoC-harboring bacterial community in acidic soils of Southern China. Front. Microbiol. 10, 2654 (2019).
Yoshitake, S. et al. Successional changes in the soil microbial community along a vegetation development sequence in a subalpine volcanic desert on Mount Fuji, Japan. Plant Soil 364, 261–272 (2013).
Endreny, T., Burke, D. J., Burchhardt, K. M., Fabian, M. W. & Kretzer, A. M. Bioretention column study of bacteria community response to salt-enriched artificial stormwater. J. Environ. Qual. 41, 1951–1959 (2012).
Wang, X. et al. Long-term phytoremediation of coastal saline soil reveals plant species-specific patterns of microbial community recruitment. Msystems 5, e00741-e1719 (2020).
Kushwaha, P., Neilson, J. W., Maier, R. M. & Babst-Kostecka, A. Soil microbial community and abiotic soil properties influence Zn and Cd hyperaccumulation differently in Arabidopsis halleri. Sci. Total Environ. 803, 150006 (2022).
Hurt, R. A. Jr. et al. Improved yield of high molecular weight DNA coincides with increased microbial diversity access from iron oxide cemented sub-surface clay environments. PLoS One 9, e102826 (2014).
Frank, J. A. et al. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74, 2461–2470 (2008).
Dashti, A. A., Jadaon, M. M., Abdulsamad, A. M. & Dashti, H. M. Heat treatment of bacteria: A simple method of DNA extraction for molecular techniques. Kuwait Med. J. 41, 117–122 (2009).
Elsanhoty, R. M., Ramadan, M. F. & Jany, K. D. DNA extraction methods for detecting genetically modified foods: A comparative study. Food Chem. 126, 1883–1889 (2011).
Barnett, R. & Larson, G. A phenol–chloroform protocol for extracting DNA from ancient samples. Ancient DNA: Methods and Protocols, 13–19 (2012).
Nishiguchi, M. K. et al. DNA isolation and procedures: In techniques in molecular systamatics and evolution 249–287 (Springer, 2002).
Hellequin, E. et al. Specific recruitment of soil bacteria and fungi decomposers following a biostimulant application increased crop residues mineralization. PLoS One 13, e0209089 (2018).
Illumina. 16s metagenomic sequencing library preparation (Illumina, San Diego, 2013).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
R Core Team. R: A language and environment for statistical computing. (2013).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013).
Ssekagiri, A., Sloan, W. & Ijaz, U. Z. in ISCB Africa ASBCB Conference. (ISCB Kumasi).
Lahti, L. & Shetty, S. Introduction to the microbiome R package. Preprint at https://github.io/tutorials/microbiome (2018).
Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60–R60 (2011).
Acknowledgements
We thank the members of the Microbiologically Influenced Corrosion (MIC)—Joint Industry Project (JIP) “Preventing and Managing Microbiologically Influenced Corrosion in the Oil and Gas Industry” for the financial support to conduct this research.
Author information
Authors and Affiliations
Contributions
S.S. and J.F. designed the experiments, J.F. executed the experiments, carried out the microbial analysis, and prepared the manuscript with the contribution of S.S. Both authors approved the final version of the manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Foysal, M.J., Salgar-Chaparro, S.J. Improving the efficiency of DNA extraction from iron incrustations and oilfield-produced water. Sci Rep 14, 2954 (2024). https://doi.org/10.1038/s41598-024-53134-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53134-9
- Springer Nature Limited