
Abstract
Two members of the Culex pipiens subgroup, Culex pallens and Culex pipiens f. molestus, are known to occur in the Republic of Korea (ROK). These species exhibit morphologically similar features and are challenging to distinguish below the species level. Therefore, this study utilized wing geometric morphometrics (GM) on the right wing of the Culex pipiens subgroup, alongside sequencing of the cytochrome c oxidase subunit I (COI) region. Mosquitoes were collected from 11 locations between June and October (2020–2022) to minimize regional and seasonal variations. Additionally, Culex pipiens f. pipiens, which is not native to the ROK, was included in the analysis. Culex tritaeniorhynchus, Aedes albopictus, and Anopheles sinensis, the primary vectors in the ROK, were used as outgroups for comparison. All three taxa in the Culex pipiens subgroup could be identified with an 82.4%–97.0% accuracy using GM. However, a comparison of the COI regions of the Culex pipiens subgroup revealed no clear differences between the taxa. These data can be used for accurate identification, contributing to effective mosquito control, in addition to providing a foundation for evolutionary and ecological studies on wing shape differences.
Similar content being viewed by others

Introduction
Despite decades of attempts to effectively control mosquitoes (Diptera: Culicidae), mosquito-borne diseases have proliferated globally, driven by rapid urbanization, climate change, and increased trade and travel1,2. Mosquito-borne diseases are primarily result from protozoan parasites and flaviviruses, which are transmitted when female mosquitoes feed on human or animal blood to acquire nutrients for oviposition3. Within various mosquito species, the Culex pipiens subgroup, prevalent near human habitation, stands out as a significant vector of the West Nile virus (WNV)4,5,6. Importantly, although no human cases of WNV have been reported in the Republic of Korea (ROK), its detection in domestic pigeons in 2015 highlights the need for vigilance7.
The Culex pipiens subgroup comprises five species (Culex pipiens Linnaeus, Culex quinquefasciatus Say, Culex pallens Coquillett, Culex australicus Dobrotworsky & Drummond, and Culex globocoxitus Dobrotworsky)8,9,10. Following the recent update in Culicidae nomenclature, there are no longer subspecies in mosquito classification. Culex pipiens pallens has been elevated to species status and is now referred to as Culex pallens11. Culex pipiens is recognized for having two ecological forms (Culex pipiens f. pipiens and Culex pipiens f. molestus Forskal)8,9,10. Among these taxa, Cx. pallens and Cx. pipiens f. molestus are currently identified in the ROK12. Despite their close evolutionary relationship, they exhibit distinct ecological differences13. Cx. pallens primarily targets birds (ornithophilic), engages in mating while in flight (eurygamy), and requires blood for oviposition (unautogeny)14,15,16. In contrast, Cx. pipiens f. molestus shows a preference for mammals (mammalophilic), mates in enclosed spaces (stenogamy), and does not require blood for the first spawn (autogeny)13,17. These species also differ in life cycles and vector competence18,19, which highlights the importance of precise identification for effective vector control. Nevertheless, due to their high morphological similarity, accurate classification remains challenging in the ROK20.
Geometric morphometrics (GM), a technique integrating geometric analysis and biology, facilitates the quantitative analysis of an organism’s features and size of an organism through statistical methods21. Therefore, this technique serves as a valuable complement to traditional taxonomic methods. The wings of mosquitoes are less prone to damage than scales or bristles and feature a two-dimensional vein structure that enables the straight forward collection of landmarks (LMs) for GM analysis. GM is also cost-effective and time-efficient22. While molecular markers are increasingly used for accurate identification of the Culex pipiens subgroup23, alternative methods are needed to compensate for the occasional limitations of molecular approaches. Additionally, GM has been actively employed in mosquito research, proving successful in various fields24,25,26. However, there is currently a severe lack of quantitative morphological analyses of pathogen vectors in the ROK.
Therefore, this study examined Cx. pallens and Cx. pipiens f. molestus of the Culex pipiens subgroup in the ROK using GM. Furthermore, Cx. pipiens f. pipiens of the Culex pipiens subgroup, which is absent in the ROK, was included in the analysis. Furthermore, major pathogen vectors in the ROK, including Culex tritaeniorhynchus Giles (the causative agent of Japanese encephalitis), Aedes albopictus Skuse (the causative agent of Zika), and Anopheles sinensis s.s. Wiedemann (the causative agent of Malaria), served as outgroups in the analysis27,28,29. Finally, sequencing of the DNA barcoding region [cytochrome c oxidase subunit I (COI)] was conducted to compare the results of the quantitative morphological analysis using GM.
Methods
Sample collection and identification
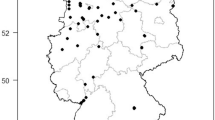
To eliminate regional and seasonal biases, specimens of the Culex pipiens subgroup were gathered from a total of 11 different sites in the ROK spanning from June to October (Fig. 1). The selected sampling sites were located around buildings in urban areas, taking into consideration the habitat preferences of the Culex pipiens subgroup BG-sentinel traps (Biogents, Regensburg, Germany) with dry ice were employed for collecting the Culex pipiens subgroup. Additionally, black light traps (BioTrap, Seoul, ROK) were deployed in a cow shed to capture Cx. tritaeniorhynchus and An. sinensis, which served as outgroups. As for Ae. albopictus, those captured alongside the Culex pipiens subgroup in Daegu were utilized. All captured mosquitoes were preserved at − 20 °C until identification.

Sample collection sites in the ROK (Culex pipiens subgroup (green), Ae. albopictus (red), Cx. tritaeniorhynchus and An. sinensis (blue)). This map was generated using R v.4.2.1 (https://www.r-project.org/) and QGIS 3.26.3 (https://www.qgis.org/ko/site).
The species within the Culex pipiens subgroup share a high morphological similarity, posing challenges for identification. Therefore, mosquito legs were detached from the bodies and utilized for identifying Cx. pallens and Cx. pipiens f. molestus, following the method described by Ryu and Choi12. Universal forward primers for Cx. pallens and Cx. pipiens f. molestus (F1457: 5′-GAG GAG ATG TGG AAT CCC AA-3′)23 were paired with species-specific reverse primers (ACEpall_R: 5′- ACA TGT CAA AAG CTC AGT TAG T -3′/ACEmole_R: 5′- TTC TCA CAG AGC CAT CAT CGA C -3′), as detailed by Ryu and Choi12 (Supplementary Fig. S1). Among the outgroup mosquitoes, An. sinensis was also typed to the species level using the molecular markers described by Bang et al.30. Universal forward primers for Anopheles species in the ROK (Universal forward primer: 5′- ATC GAT GAA GAC CGC AGC TA -3′) and eight species-specific reverse primers (An. sinensis: 5′- TAG GGT CAA GGC ATA CAG AAG G-3′/ An. koreicus: 5′- TAT CGT GGC CCT CGA CAG -3′/ An. lindesayi: 5′- ACC ATC TAC TGC CTG AAC GTG -3′/ An. kleini: 5′- TTT GTT GAT AAC TTG TAT CGT CCA TC -3′/ An. lesteri: 5′- CAG TCT CTT GCA GCC CAT TC-3′/ An. sineroides: 5′- CGC GCA CGC TCA GAT ATT -3′/ An. belenrae: 5′- TGT CCT AGG CGG TTA TCA ACA-3′/ An. pullus: 5′- CGG CGT AGT TTA TTG TGT ATA ACA TC-3′) were also used, as suggested by Bang et al.30. Cx. tritaeniorhynchus and Ae. albopictus can be clearly identified by morphological features and therefore require no molecular identification31. Table 1 provides information about the mosquitoes used in this experiment.
GM analysis
Only the right wings of the identified female mosquitoes were selectively used for the GM analysis. The right wing of each mosquito species was dissected, and wing scales were removed using a brush. Afterward, the wings were mounted on microscope slides and covered with coverslips using Canada balsam (Duksan, Seoul, ROK). These prepared wing specimens were photographed under 20 × magnification using an Olympus SZ61 Stereo Microscope (Olympus Corp., Tokyo, Japan). Wing images of Cx. pipiens f. pipiens collected in Germany, a species not distributed in the ROK, were incorporated into our experiments32,33.
Landmark coordinates were collected using TPSdig2 (2.31) (Fig. 2), and 17 LMs were established for the GM analysis of mosquito wings34. Beriotto et al.35 determined that 9, 13, and 17 LM settings produced the same total error rate in studies involving Cx. pipiens. Since this study involved three taxa of the Culex pipiens subgroup, 17 LMs were chosen for a more precise comparison. Subsequently, the TPS files for each mosquito taxon with established LMs were analyzed using various packages in R v.4.2.136.

The 17 LMs set up on the right wing of a female adult mosquito for GM analysis.
Procrustes analysis was conducted with the ‘geomorph’ package (v. 4.0.5) to transform the difference in position and direction of each data coordinate37. The results were superimposed to generate Procrustes coordinates. The adequacy of the established LMs was confirmed using the ‘LamBDA’ package (v. 0.1.1)38. Centroid size (CS), defined as the square root of the sum of the squared distances of all landmarks on the object from their centroid, was calculated from the Procrustes coordinates to estimate wing size39. The allometric effect, which represents the relationship between shape and size, was also evaluated using the ‘geomorph’ package (1000 permutations)40. Mean CS values for each species were compared using analysis of variance (ANOVA), and pairwise comparisons were conducted using t-tests with the ‘agricolae’ (v.1.3–7) package (Bonferroni adjusted p-value)41. To analyze differences in wing shape between groups, linear discriminant analysis (LDA) was performed using the ‘tidyverse’ (v.2.0.0) and ‘MASS’ (v.7.3–60) packages42,43. The convex hull algorithm, encompassing all points for each group in the scatterplot, was also applied. To compare the wing shape of each taxon, mean shapes for each LM coordinate were calculated and compared. The Mahalanobis distance, a statistical indicator of the difference between each group, was calculated using the ‘CVA’ function of the ‘Morpho’ package (v. 2.11) with 10,000 permutations44. Finally, a jackknife leave-one-out cross-validation was performed to determine the accuracy of the classification based on wing shape.
COI barcoding
Genomic DNA was extracted from the whole body, except the right wing, of the species-identified mosquitoes using the Clear-S™ Quick DNA Extraction Kit (InVirusTech, Gwangju, ROK), following the manufacturer’s protocol.
Following genomic DNA extraction, universal primers (LCO1490: 5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′/HCO2198: 5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′) were employed to amplify the COI region45. The polymerase chain reaction (PCR) amplification mixture (total: 25 μL) consisted of 1 μL of extracted genomic DNA, 1 × PCR buffer, 0.2 mM dNTPs, 1.5 mM MgCl2, 0.4 μL of each primer, and 0.5 units of Taq DNA polymerase (TaKaRa, Shiga, Japan). The PCR cycling conditions were as follows: initial denaturation at 94 °C for 5 min, followed by 30 cycles of 94 °C for 30 s, 56 °C for 30 s, 72 °C for 1 min, and a final extension at 72 °C for 5 min.
PCR amplification products were confirmed by electrophoresis, and samples with successful amplification were characterized via Sanger sequencing in both directions (Macrogen, Daejeon, Korea). The sequences were analyzed using BLAST, organized using the ‘msa’ (v. 1.28.0) package in R and Bioedit, and aligned using Clustal X46,47,48. The COI sequences obtained in this study were deposited in the NCBI GenBank database [accession numbers: OR835698 ~ OR835753]. The COI sequences of Cx. pipiens f. pipiens individuals were compared using sequences deposited in NCBI GenBank database (accession numbers: MT731275 ~ 6). For an accurate comparison, only the COI sequences of Cx. pipiens f. pipiens obtained by Sauer et al.32 used in this study were included in the phylogenetic analysis. The COI sequences (550 bp) were analyzed using the ‘ape’ (v. 5.7–1), ‘phangorn’ (v. 2.11.1), and ‘phytools’ (v. 1.5–1) packages49,50,51. A maximum likelihood (ML) tree was generated for phylogenetic analysis. After determining the nucleotide substitution model, GTR + I was identified as the best fit model. To assess the robustness of the tree, a bootstrap analysis with 1,000 replications was conducted. The ‘ggplot2’ (v. 3.4.2) and ‘ggtree’ packages (v. 3.4.4) were utilized to visualize the results of the phylogenetic analysis52,53.
Results
Wing GM and COI barcoding
The right wing samples for GM analysis consisted of 47 Cx. pallens, 67 Cx. pipiens f. molestus, 17 Cx. pipiens f. pipiens, 45 Cx. tritaeniorhynchus, 40 Ae. albopictus, and 35 An. sinensis samples (Table 1). To assess the sampling adequacy of LMs, we examined the LM sampling evaluation curve using the ‘LamBDA’ package, revealing no oversampling (fit = 0.90: 12 LMs; fit = 0.99: 16 LMs) (Supplementary Fig. S2).
The mean CS differed significantly between mosquito species (F = 125.7, p < 0.0001). On average, An. sinensis exhibited the largest CS, whereas Ae. albopictus had the smallest CS (Fig. 3). Pairwise comparisons of Cx. pallens vs. Cx. pipiens f. molestus, Cx. pallens vs. Cx. pipiens f. pipiens, and Cx. pipiens f. molestus vs. Cx. pipiens f. pipiens showed no statistically significant differences in CS (p = 1) (Supplementary Table S1). The allometric effect was found to be statistically significant (R2 = 3.3%, p < 0.0001), and it was analyzed together with the shape data, as it could provide useful information for the identification process54,55.

Boxplot showing the results of centroid size (CS) comparisons for each taxon (SIN = An. sinensis, PAL = Cx. pallens, MOL = Cx. pipiens f. molestus, PIP = Cx. pipiens f. pipiens, TRI = Cx. tritaeniorhynchus, and ALB = Ae. albopictus). The box represents the first and third quartiles, and the black line in the middle represents the median. The asterisks represent outliers.
Among the outgroup species, An. sinensis and Ae. albopictus were clearly separated, as shown in the LDA scatterplot results for all species analyzed together (Fig. 4a). Cx. tritaeniorhynchus, also set as an outgroup, was generally well distinguished from mosquitoes in the Culex pipiens subgroup, with minor overlap in some specimens. Species within the genus Culex pipiens subgroup clustered together, displaying slight overlap for some species. Differences in mean wing shapes were observed in LM1-2 and LM16-17 for An. sinensis and Ae. albopictus, and mainly in LM1-2 and LM12-17 for An. sinensis and Culex mosquitoes (Fig. 4b). Significant differences in mean wing shape between Ae. albopictus and Culex mosquitoes were noted, particularly at LM2 and LM16-17. Subsequent LDA using only Culex mosquitoes revealed a clear separation of each taxon (Fig. 5a). A comparison of mean wing shapes within the genus Culex (Fig. 5b) indicated no major differences in wing shape for each taxon, with some distinctions primarily in LM16-17. The Mahalanobis distance calculated using canonical variate analysis showed that the closest taxa were Cx. pallens and Cx. pipiens f. pipiens (4.3152, p < 0.0001), followed by Cx. pallens and Cx. tritaenioryhnchus (4.4569, p < 0.0001). The most dissimilar species were An. sinensis and Cx. pipiens f. pipiens (17.2230, p < 0.0001) (Supplementary Table S2). Cross-validation (leaving-one-out method) for classification accuracy showed an overall average accuracy of 96.8% (Table 2). The outgroups An. sinensis and Ae. albopictus achieved 100% (An. sinensis, 35/35; Ae. albopictus, 40/40) accuracy, whereas Cx. tritaeniorhynchus achieved 97.7% (44/45) accuracy. Within the Cx. pipiens subgroup, Cx. pallens achieved 95.8% (45/47) accuracy, Cx. pipiens f. molestus achieved 97.0% (65/67), and Cx. pipiens f. pipiens achieved 82.4% (14/17), with the lowest accuracy, resulting in many individuals being misidentified as Cx. pallens (3/17).

(a) Scatterplot of LDA results based on wing shape variation from the six taxa (LD1: 62%, LD2: 29%). (b) Comparison of mean wing shape for each taxon.

(a) Scatterplot of LDA results based on wing shape variation among species in the genus Culex (LD1: 80%, LD2: 12%). (b) Comparison of mean wing shape for each taxon.
As shown in the ML tree, An. sinensis, Ae. albopictus, and Cx. tritaeniorhynchus, designated as outgroups, formed a monophyletic clade with strong bootstrap support. However, species within the Cx. pipiens subgroup were unresolved (Fig. 6).

Maximum likelihood (ML) phylogenetic tree (GTR-I). The bootstrap value is shown next to each node (1,000 replicates). Unresolved taxa are shown in red text. Drosophila melanogaster (GenBank accession number: MT807020) was used as the outgroup.
Discussion
Cx. pipiens f. molestus and Cx. pallens differ in the D/V ratio of male genitalia and ommatidial number of females56. However employing such methods for identification is challenging for inexperienced researchers, time-consuming, and expensive, particularly when dealing with large numbers of individuals. Molecular biological approaches serve as valuable tools to overcome the challenges of morphological identification23,57,58 and remain the most powerful methods for accurate species identification. In response to these challenges, the analysis of mosquito wings using GM has gained acceptance and has proven to be a reliable and efficient method, complementing traditional morphological classification methods24,32,54,55. This study compared three taxa within the Culex pipiens subgroup (Cx. pipiens f. molestus, Cx. pipiens f. pipiens, and Cx. pallens) with Cx. tritaeniorhynchus, Ae. albopictus, and An. sinensis as outgroups. The results demonstrated that species identification using GM can be successfully applied not only at the level of evolutionarily distant genera but also in very closely related relationships. However, cross-validation results indicated that reliable classification of Cx. pipiens f. pipiens could not be achieved with high accuracy (14/17:82.4%). For example, Cx. pipiens f. pipiens and Cx. torrentium, known to co-occur sympatrically in Europe, cannot be easily differentiated via traditional morphological approaches, whereas GM was able to successfully discriminate them59. There are also examples of high heterogeneity in spatially separated populations of the same species60. However, in the case of Cx. pipiens f. pipiens used in this study32,33, it was not clearly identified using GM, despite its absence in the ROK and geographical separation. Therefore, further studies with larger geographic sampling are necessary. Nevertheless, only Cx. pipiens f. molestus and Cx. pallens of the Culex pipiens subgroup are currently known to occur in the ROK. GM-based methods are thus expected to be successful in identifying various pathogen vector mosquitoes, including the Culex pipiens subgroup in the ROK. Analyzing wing size using CS as an indicator revealed no statistically significant differences in wing size among the three taxa of the Culex pipiens subgroup. Furthermore, wing size in insects, including mosquitoes, is strongly influenced by the environment61, suggesting that using wing size for discrimination is not suitable for classifying close relatives like the Culex pipiens subgroup. However, LDA based on wing shape variation demonstrated that wing vein structure is homologous, species-specific, and less influenced by the environment than wing size. This makes it an extremely useful morphological trait for species identification, phylogenetic analysis, and tracking evolutionary relationships62.
In this study, the analysis of the Culex pipiens subgroup was based on data from Cx. pallens and Cx. pipiens f. molestus from the ROK and Cx. pipiens f. pipiens from Germany32,33. While Cx. quinquefasciatus, another member of the Culex pipiens subgroup, was not included in this experiment, its wide distribution in tropical regions and tendency to co-occur with other subgroup members suggests the need for further experiments to determine the feasibility of accurate species identification using GM compared to other members of the Culex pipiens subgroup8,16,63. As suggested by Dujardin et al.64, the wing images used in this study will be made available through an open-access repository, allowing researchers to use them in their own studies (https://doi.org/10.5061/dryad.gtht76ht6).
The sequences of the COI region of Cx. pallens and Cx. pipiens f. molestus from the ROK was compared with that of Cx. pipiens f. pipiens, but no clear differences were observed. The COI region is commonly used for accurate species delimitation in insects and identifying genetic divergence in closely related species, and it has been extensively successful65. In the case of the Cx. pipiens subgroup, the influence of Wolbachia infection on mitochondrial diversity and the occurrence of sympatric populations capable of hybridization may contribute to limited genetic variation in the COI region. Thus, future comparisons between populations should adopt a multi-loci approach, considering both mitochondrial and nuclear genes12,66,67.
In the ROK, despite the Culex pipiens subgroup being a diverse pathogen vector, its accurate identification been challenging due to difficulties in morphological classification5,20,68. The successful use of GM in classifying the Culex pipiens subgroup, along with Cx. tritaeniorhynchus, Ae. albopictus, and An. sinensis, suggests the potential applicability of GM-based classification to various mosquito species in the ROK. This method could benefit researchers unfamiliar with mosquito taxonomy and those facing challenges with molecular biological approaches, providing a means for accurate species identification. Furthermore, GM analysis is not confined to interspecific variation and has been applied in various studies, including sexual dimorphism and parasite detection69,70. Therefore, the results of this study can serve as foundational data for future ecological and evolutionary research on the Culex pipiens subgroup. The wing images utilized in this study are also expected to be valuable resources for researchers in various fields.
Data availability
Sequencing data used in this study are deposited in the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/) under the accession numbers (OR835698 ~ OR835753). The wing images used in this study are available at https://doi.org/10.5061/dryad.gtht76ht6.
References
Gubler, D. J. The global emergence/resurgence of arboviral diseases as public health problems. Arch. Med. Res. 33, 330–342 (2002).
Kilpatrick, A. M. & Randolph, S. E. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet 380, 1946–1955 (2012).
Chamberlain, R. & Sudia, W. D. Mechanism of transmission of viruses by mosquitoes. Annu. Rev. Entomol. 6, 371–390 (1961).
Fonseca, D. M. et al. Emerging vectors in the Culex pipiens complex. Science 303, 1535–1538 (2004).
Turell, M. J. Members of the Culex Pipiens complex as vectors of viruses. J. Am. Mosq. Control Assoc. 28, 123–126 (2012).
Vogels, C. B. F., Goertz, G. P., Pijlman, G. P. & Koenraadt, C. J. M. Vector competence of European mosquitoes for West Nile virus. Emerg. Microbes Infect. 6, e96 (2017).
Kim, C. Y. et al. First detection of West Nile virus in domestic pigeon in Korea. J. Vet. Sci. 17, 587–589 (2016).
Harbach, R. E. Culex Pipiens: Species versus species complex—Taxonomic history and perspective. J. Am. Mosq. Control Assoc. 28, 10–23 (2012).
Russell, R. C. A review of the status and significance of the species within the Culex Pipiens group in Australia. J. Am. Mosq. Control Assoc. 28, 24–27 (2012).
Aardema, M. L., vonHoldt, B. M., Fritz, M. L. & Davis, S. R. Global evaluation of taxonomic relationships and admixture within the Culex pipiens complex of mosquitoes. Parasites Vectors 13, 8 (2020).
Harbach, R. E. & Wilkerson, R. C. The insupportable validity of mosquito subspecies (Diptera: Culicidae) and their exclusion from culicid classification. Zootaxa 5303, 1–184 (2023).
Ryu, J. & Choi, K. S. Species diversity of the Culex pipiens complex in the Republic of Korea. Entomol. Res. 52, 376–381 (2022).
Mogi, M. The forms of the Culex Pipiens complex in East Asia, with ecological thoughts on their origin and interrelation. J. Am. Mosq. Contr. 28, 28–52 (2012).
Harbach, R. E., Dahl, C. & White, G. B. Culex (Culex) Pipiens-Linnaeus (Diptera, Culicidae)—Concepts, type designations, and description. Proc. Entomol. Soc. Wash. 87, 1–24 (1985).
Sawabe, K. et al. Host-feeding habits of Culex pipiens and Aedes albopictus (Diptera: Culicidae) Collected at the urban and suburban residential areas of Japan. J. Med. Entomol. 47, 442–450 (2010).
Farajollahi, A., Fonseca, D. M., Kramer, L. D. & Kilpatrick, A. M. “Bird biting” mosquitoes and human disease: A review of the role of Culex pipiens complex mosquitoes in epidemiology. Infect. Genet. Evol. 11, 1577–1585 (2011).
Haba, Y. & McBride, L. Origin and status of mosquito ecotypes. Curr. Biol. 32, R237–R246 (2022).
Turell, M. J. et al. Laboratory transmission of Japanese encephalitis and West Nile viruses by molestus form of Culex pipiens (Diptera: Culicidae) collected in Uzbekistan in 2004. J. Med. Entomol. 43, 296–300 (2006).
Jiang, S. F. et al. Infection and dissemination of West Nile virus in China by the potential vector, Culex pipiens pallens. J. Vector Ecol. 39, 78–82 (2014).
Lim, A. Y., Cheong, H. K., Chung, Y., Sim, K. & Kim, J. H. Mosquito abundance in relation to extremely high temperatures in urban and rural areas of Incheon Metropolitan City, South Korea from 2015 to 2020: An observational study. Parasites Vectors 14, 559 (2021).
Bookstein, F. L. Biometrics, biomathematics and the morphometric synthesis. Bull. Math. Biol. 58, 313–365 (1996).
Lorenz, C. et al. Geometric morphometrics in mosquitoes: What has been measured?. Infect. Genet. Evol. 54, 205–215 (2017).
Smith, J. L. & Fonseca, D. M. Rapid assays for identification of members of the Culex (Culex) pipiens complex, their hybrids, and other sibling species (Diptera: Culicidae). Am. J. Trop. Med. Hyg. 70, 339–345 (2004).
Lorenz, C., Marques, T. C., Sallum, M. A. M. & Suesdek, L. Morphometrical diagnosis of the malaria vectors Anopheles cruzii, An. homunculus and An. bellator. Parasites Vectors 5, 257 (2012).
Laurito, M., Almiron, W. R. & Luduena-Almeida, F. F. Discrimination of four Culex (Culex) species from the Neotropics based on geometric morphometrics. Zoomorphology 134, 447–455 (2015).
Christe, R. D., Wilke, A. B. B., Vidal, P. O. & Marrelli, M. T. Wing sexual dimorphism in Aedes fluviatilis (Diptera: Culicidae). Infect. Genet. Evol. 45, 434–436 (2016).
Ree, H. I. Studies on Anopheles sinensis, the vector species of vivax malaria in Korea. Korean J. Parasitol. 43, 75–92 (2005).
Solomon, T. Control of Japanese encephalitis–within our grasp?. N. Engl. J. Med. 355, 869–871 (2006).
Weetman, D. et al. Aedes mosquitoes and Aedes-borne arboviruses in Africa: Current and future threats. Int. J. Environ. Res. Public Health 15, 220 (2018).
Bang, W. J. et al. Multiplex PCR assay for the identification of eight Anopheles species belonging to the Hyrcanus, Barbirostris and Lindesayi groups. Malar. J. 20, 287 (2021).
Ree, H. I. Taxonomic review and revised keys of the Korean mosquitoes (Diptera: Culicidae). Entomol. Res. 33, 39–52 (2003).
Sauer, F. G. et al. Geometric morphometric wing analysis represents a robust tool to identify female mosquitoes (Diptera: Culicidae) in Germany. Sci. Rep. 10, 17613 (2020).
Sauer, F. G. et al. Geometric morphometric wing analysis represents a robust tool to identify female mosquitoes (Diptera: Culicidae) in Germany. Dryad https://doi.org/10.5061/dryad.zs7h44j5s (2020).
Rohlf, F. J. The tps series of software. Hystrix 26, 9–12 (2015).
Beriotto, A. C., Garzon, M. J. & Schweigmann, N. Is there a minimum number of landmarks that optimizes the geometric morphometric analysis of mosquito (Diptera, Culicidae) wings?. J. Med. Entomol. 58, 576–587 (2021).
Ihaka, R. & Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 5, 299–314 (1996).
Adams, D. C. & Otarola-Castillo, E. geomorph: An r package for the collection and analysis of geometric morphometric shape data. Methods Ecol. Evol. 4, 393–399 (2013).
Watanabe, A. How many landmarks are enough to characterize shape and size variation?. Plos One 13(6), e0198341 (2018).
Bookstein, F. L. Morphometric Tools for Landmark Data (Cambridge University Press, 1991).
Klingenberg, C. P. Size, shape, and form: Concepts of allometry in geometric morphometrics. Dev. Genes Evol. 226, 113–137 (2016).
de Mendiburu, F. & de Mendiburu, M. F. Package ‘agricolae’. R package, version 1 (2019).
Venables, W. N. & Ripley, B. D. Modern Applied Statistics with S-PLUS (Springer Science & Business Media, 2013).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686 (2019).
Schlager, S. Morpho and Rvcg-shape analysis in R: R-packages for geometric morphometrics, shape analysis and surface manipulations. In Statistical Shape and Deformation Analysis (ed. Schlager, S.) 217–256 (Elsevier, 2017).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Hall, T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids Symp. Ser. 41, 95–98 (1999).
Larkin, M. A. et al. Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Bodenhofer, U., Bonatesta, E., Horejš-Kainrath, C. & Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 31, 3997–3999 (2015).
Schliep, K. P. phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 (2010).
Revell, L. J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012).
Paradis, E. & Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Wickham, H. Ggplot2: Elegant Graphics for Data Analysis (Springer, 2009).
Wilke, A. B. B. et al. Morphometric wing characters as a tool for mosquito identification. Plos One 11, e0161643 (2016).
de Souza, A., Multini, L. C., Marrelli, M. T. & Wilke, A. B. B. Wing geometric morphometrics for identification of mosquito species (Diptera: Culicidae) of neglected epidemiological importance. Acta Trop. 211, 105593 (2020).
Noguchi, Y. & Asahina, S. Ommatidial number as a diagnostic character for Japanese autogenous Culex Molestus. J. Med. Entomol. 3, 146–148 (1966).
Kasai, S. et al. PCR-based identification of Culex pipiens complex collected in Japan. Jpn. J. Infect. Dis. 61, 184–191 (2008).
Zittra, C. et al. Ecological characterization and molecular differentiation of Culex pipiens complex taxa and Culex torrentium in eastern Austria. Parasites Vectors 9, 197 (2016).
Börstler, J. et al. The use of morphometric wing characters to discriminate female Culex pipiens and Culex torrentium. J. Vector Ecol. 39, 204–212 (2014).
Carvajal, T. M. et al. Spatial analysis of wing geometry in dengue vector mosquito, Aedes aegypti (L.) (Diptera: Culicidae), populations in Metropolitan Manila, Philippines. J. Vector Borne Dis. 53, 127–135 (2016).
Gómez, G. F., Márquez, E. J., Gutiérrez, L. A., Conn, J. E. & Correa, M. M. Geometric morphometric analysis of Colombian Anopheles albimanus (Diptera: Culicidae) reveals significant effect of environmental factors on wing traits and presence of a metapopulation. Acta Trop. 135, 75–85 (2014).
Perrard, A., Baylac, M., Carpenter, J. M. & Villemant, C. Evolution of wing shape in hornets: Why is the wing venation efficient for species identification?. J. Evol. Biol. 27, 2665–2675 (2014).
Vidal, P. O., Peruzin, M. C. & Suesdek, L. Wing diagnostic characters for Culex quinquefasciatus and Culex nigripalpus (Diptera, Culicidae). Rev. Bras. Entomol. 55, 134–137 (2011).
Dujardin, J.-P.A.L., Kaba, D. & Henry, A. B. The exchangeability of shape. BMC Res. Notes 3, 266 (2010).
Hebert, P. D., Cywinska, A., Ball, S. L. & de Waard, J. R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 270, 313–321 (2003).
Atyame, C. M., Delsuc, F., Pasteur, N., Weill, M. & Duron, O. Diversification of endosymbiont in the mosquito. Mol. Biol. Evol. 28, 2761–2772 (2011).
Ohashi, K., Tsuda, Y., Kasai, S., Kawada, H. & Takagi, M. Hybridization between sympatric populations of Culex pipiens pallens and Culex pipiens f. molestus (Diptera: Culicidae) in Nagasaki, Japan. Med. Entomol. Zool. 65, 67–72 (2014).
Jung, S. H., Kim, D., Jung, K. S. & Lee, D. K. Color preference for host-seeking activity of Aedes albopictus and Culex pipiens (Diptera: Culicidae). J. Med. Entomol. 58, 2446–2452 (2021).
Yeap, H. L. et al. Assessing quality of life-shortening-infected mosquitoes in the field based on capture rates and morphometric assessments. Parasite Vector https://doi.org/10.1186/1756-3305-7-58 (2014).
Virginio, F., Vidal, P. O. & Suesdek, L. Wing sexual dimorphism of pathogen-vector culicids. Parasite Vector https://doi.org/10.1186/s13071-015-0769-6 (2015).
Acknowledgements
The authors thank the Basic Science Research Program of the National Research Foundation of Korea (NRF), Korean Ministry of Education (NRF-2020R1I1A2066186).
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1I1A2066186).
Author information
Authors and Affiliations
Contributions
J.J. and K.S.C. designed the study. D.Y.L., Y.J., J.R. and E.K. collected and identified samples. J.J. analyzed the data and drafted the manuscript. K.S.C. helped draft the manuscript with analysis of the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeon, J., Lee, D.Y., Jo, Y. et al. Wing geometric morphometrics and COI barcoding of Culex pipiens subgroup in the Republic of Korea. Sci Rep 14, 878 (2024). https://doi.org/10.1038/s41598-024-51159-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51159-8
- Springer Nature Limited