
Abstract
Endometriosis (EMT) is an aggressive disease of the reproductive system, also called “benign cancer”. However, effective treatments for EMT are still lacking in clinical practice. Interestingly, immune infiltration is significantly involved in EMT pathogenesis. Currently, no studies have shown the involvement of cuproptosis-related genes (CRGs) in regulating immune infiltration in EMT. This study identified three CRGs such as GLS, NFE2L2, and PDHA1, associated with EMT using machine learning algorithms. These three CRGs were upregulated in the endometrium of patients with moderate/severe EMT and downregulated in patients with infertility. Single sample genomic enrichment analysis (ssGSEA) revealed that these CRGs were closely correlated with autoimmune diseases such as systemic lupus erythematosus. Furthermore, these CRGs were correlated with immune cells such as eosinophils, natural killer cells, and macrophages. Therefore, profiling patients based on these genes aid in a more accurate diagnosis of EMT progression. The mRNA and protein expression levels of GLS, NFE2L2 and PDHA1 were validated by qRT-PCR and WB studies in EMT samples. These findings provide a new idea for the pathology and treatment of endometriosis, suggesting that CRGs such as GLS, NFE2L2 and PDHA1 may play a key role in the occurrence and development of endometriosis.
Similar content being viewed by others

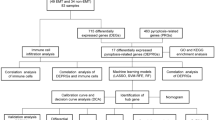

Introduction
Endometriosis (EMT) is a common estrogen-dependent disease wherein the endometrial tissue implants and grows on the peritoneal surface, ovaries, rectovaginal septum, perineum, and surgical scars1. The primary symptoms of EMT include lower abdominal pain, dysmenorrhea, menstrual abnormalities, and discomfort during intercourse. Globally, nearly 5–10% of women of reproductive age are affected by EMT, and 30–50% of women with EMT suffer from infertility2,3. However, the pathogenesis of EMT has not yet been elucidated. Several studies have identified markers associated with EMT4. However, the sample size of these studies was small, which prevents accurate diagnosis. Therefore, there is an urgent need to identify new biomarkers to understand the pathophysiological mechanisms of EMT. This would aid in designing better and more accurate diagnostic as well as therapeutic approaches for EMT.
Copper is an essential cofactor regulating several enzymes involved in various cellular functions, including mitochondrial respiration, antioxidation, and synthesis of hormones, neurotransmitters, and pigment. However, abnormal copper levels can induce oxidative stress and cytotoxicity5. Recent studies have demonstrated that copper induces cell death by altering the tricarboxylic acid cycle (TCA), which causes the accumulation of lipid-acylated proteins and a decrease in iron-sulfur cluster protein levels. This triggers a proteotoxic stress response called cuproptosis6. Moreover, studies have demonstrated that copper homeostasis is critically involved in immune cell infiltration. Tan et al. showed that copper-induced H2O2 production mediated by LOXL4 activates the type I interferon signaling pathway, which promotes PD-L1 presentation by macrophages7. A study has demonstrated a close correlation between the copper-related protein STEAP2 and the prognosis, as well as the infiltration of immune cells in gliomas8. Ji et al. indicated that cuproptosis could influence the development and prognosis of patients with renal clear cell carcinoma by regulating immune cell infiltration9.
Therefore, it is necessary to study the involvement of cuproptosis in the diagnosis and immune cell infiltration in diseases. However, no studies have determined the influence of cuproptosis-mediated immune cell infiltration on EMT pathogenesis. Therefore, in this study, we combined data on EMTs and cuproptosis-related genes (CRGs) to determine the underlying mechanism by which cuproptosis mediates immune cell infiltration in EMT. First, we obtained data on 19 CRGs from previous studies10 and the GSE7307 datasets from the "Gene Expression Omnibus (GEO)” database to perform differential and functional correlation analysis. Next, we identified three key CRGs using machine learning algorithms. Furthermore, we analyzed the correlation between key CRGs, immune cells, and pathways. Finally, we constructed a nomogram and screened for drug targets to provide insights into cuproptosis in EMT. The outcomes of this study are summarized in the following table. These results would aid in enhancing our understanding of the involvement of cuproptosis in regulating EMT.
Material and methods
Data download and pre-processing
The gene expression data set analyzed in this study came from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The search criteria used the keyword "Endometriosis" and selected the organism "Homo sapiens" from the dataset platform GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array. The number of samples in the control group and disease group of the GEO data set are both greater than 10 and are finally included in the data set 7307 and GSE7305 for subsequent analysis11,12,13,14,15,16. We use the GSE7307 data set as the training set and GSE7307 as the validation set. In this study, we first searched the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and downloaded the gene expression profiles of endometriosis (GSE7307 and GSE7305) using the "GEOQuery" package of R software (version 4.1.2). GSE7307 dataset consisting of 677 samples, however, only 18 endometriosis and 23 normal endometrium was used as a train set. Based on the same GSE7307 platform, GSE7305, from the GEO database as a validation cohort. This dataset consisting of ten ovarian endometriosis and ten normal endometrium. None of the samples were on medication or had received hormone therapy prior to surgery. In order to understand the expression of key CRGs in EMT with different severities and combined with infertility, we downloaded GSE51981 and GSE120103. GSE51981 and GSE120103 data sets are based on the GPL570 and GPL6480 Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F (Probe Name version) platforms for further verification17. Gene expression profiles were normalized using the normal Between Arrays function of the Limma package, and method = "scale". Meanwhile, 6 EMT samples were collected from EMT patients, and 6 normal endometrium samples were collected from women who underwent hysterectomy for uterine fibroids. All participants signed an informed consent form. The study was approved by the Institutional Research Ethics Committees of the Affiliated Hospital of Youjiang Medical University for Nationalities (YYFY-LL-2023-137) and conformed to the guidelines and regulations stated in the Declaration of Helsinki.
Screening for differentially expressed CRGs
In order to find out the differences in the expression of cuproptosis-related genes (CRGs) in EMT patients, after downloading the gene chip analysis data, we normalized the gene expression and CRGs were then screened from the GSE7307 and GSE7305 datasets using the “LIMMA “ package of R software. CR-DEGS screening was performed on 28 EMT samples and 33 control endometrial tissues using P < 0.05 and |logFC|> 0.2 as the criteria for screening. The ”Ggpubr “ package of R is used to display heatmaps and box plots of the results.
Functional enrichment Analysis of DEGs
We used the “Disease Ontology (DO),” “Gene Ontology (GO),” and “Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses” to determine the biological roles of CRGs. P < 0.05 was considered statistically significant. Further, the visual enrichment maps were constructed using the "ggplot2" and "GOplot" R packages.
Machine learning algorithm to identify key CRGs
Three machine learning algorithms; the least absolute shrinkage and selection operator (LASSO), Random Forest (RF)18 and Support Vector Machines-RFE (SVM-RFE)19 were used to screen feature genes. Initial assessment of EMT differentially expressed CRGs using LASSO. Furthermore, the value of the penalty parameter (λ) filters out the tenfold cross-validation with the lowest deviation anomaly probability. After regression analysis, only genes with non-zero coefficients were retained. Use the random forest algorithm for additional screening of key genes and filter variables with a relative importance greater than 0.5. SVM-RFE was used to further screen the characteristic genes, and the top 6 genes with average ranking were retained for subsequent analysis. LASSO, RF and SVM-RFE were crossed to obtain our signature genes. Finally, the results were visualized by constructing a Venn diagram using the "venneuler" R package.
Diagnostic value of key CRGs in EMT and external validation
We determined the diagnostic value of these key CRGs in GSE7307 and validated these genes in GSE7305. For GSE51981 and GSE120103, we plotted the receiver operating characteristic (ROC) curves to determine the diagnostic value of these genes using “P < 0.05” as the threshold. We calculated the area under the ROC curve (AUC), and the AUC value between 0 and 1 was used for analysis. The greater the AUC value, the better the performance of these genes in predicting diagnosis.
Screening key regulatory pathways using Single sample genomic enrichment analysis (ssGSEA)
A set of 16 immune cell and 13 immune-related pathway20 was obtained from references. Take these 29 gene sets and the gene expression matrix as input files and run a single-sample gene set enrichment analysis (ssGSEA) on all samples using "gsva" R package21. The infiltration rates of 16 immune cell types and the activities of 13 immune system-related signaling pathways were calculated.
Immunological analysis
The relative proportions of the 22 types of infiltrating immune cells in all EMT samples were determined using “Cell-type Identification by Estimating Relative Subsets of RNA Transcripts” (CIBERSORT, http://cibersortx.stanford.edu). Next, we calculated immune scores using the “Estimation of Stromal and Immune cells in Malignant Tumor tissues using Expression data" algorithm. Finally, Spearman rank correlation analysis was performed to determine the correlation between CRGs and the number of infiltrating immune cells. The correlation was visualized using the “ggplot2” package.
Construction of nomogram prediction model and efficacy assessment for clinical diagnosis of EMT
We constructed a nomogram prediction model and validated it internally using the "rms" R package. The risk score is calculated based on the expression of individual key genes, and the total risk score is defined as the sum of the risk scores of all individual genes. The performance of the model was assessed using calibration curves, clinical decision curve analysis (DCA), and ROC curves.
Drug screening for key CRGs
The “DrugBank” database (https://go.drugbank.com/drugs) was used to predict potential therapeutic targets of key CRGs associated with EMT. The “DrugBank database is a unique bioinformatics and cheminformatics resource that contains more than 13,791 drug entries, including small molecules, biotechnology, nutraceuticals, and investigational drugs. Additionally, there are > 5236 nonredundant protein sequences associated with these drug entries.
Quantitative RealTime polymerase chain reaction (qRT-PCR)
Total RNA was isolated from EMT using Trizol reagent. The cDNA was then synthesized by reverse transcription using the RevertAid First Strand cDNA Synthesis Kit, and Real-time PCR was performed on these cDNAs using Fast SYBR Green PCR Master Mix. The qRT-PCR FRG primers used in this study were specified in Supplementary Table 1.
Western blotting
The total protein quantity from CC were lysed with RIPA lysate, electrophoresed by SDS-PAGE and transferred to PVDF membranes, then the membranes were closed with 5% skim milk powder for 2 h. Primary antibodies GLS (1:1000, abcam, UK), NFE2L2 (1:1000, abcam, UK) and PDHA1 (1:1000, abcam, UK) were incubated overnight at 4 °C and secondary antibodies were incubated at room temperature for 2 h. Bands were detected with ECL chromogenic solution, and GAPDH was used as a control for analysis.
Statistical analysis
R 4.0.5 statistical software was used to complete the analysis. We used the t-test and Wilcoxon rank-sum test for quantifying variables. Furthermore, Spearman correlation analysis was used to investigate the correlation between CRGs and immune cell infiltration. P < 0.05 was considered statistically significant.
Results
Screening for DEGs in patients with EMT
After downloading the gene chip analytic data, we normalized the gene expression and the data are shown in Supplementary Fig. S1. We combined 19 CRGs from previously published studies and DEGs screened from patients with EMT. A total of 10 DEGs were screened from GSE7307, of which six were upregulated, and four were downregulated (Fig. 1A, B).

The dysregulated genes were shown in the A heat map via analyzing GSE7307 datasets. 10 CRGs were identified between EMT and normal samples. B CRGs expression in EMT and normal samples, blue: normal samples, red: tumor samples (*p < 0.05, **p < 0.01, ***p < 0.001).
Functional enrichment analysis of DEGs
We performed the DO, GO, and KEGG pathway enrichment analyses on DEGs. The DO enrichment analysis demonstrated that these key CRGs were mainly enriched in the nutrient deficiency disease pathway associated with carbohydrate metabolism disorder (Fig. 2A). The GO enrichment analysis revealed that these key CRGs were enriched in various processes such as acetyl-CoA biosynthesis from pyruvate, acetyl-CoA metabolism, and thioester, acyl-a CoA, a sulfur compound, nucleoside bisphosphate, ribonucleoside, and acyl-CoA biosynthesis processes (Fig. 2B). The KEGG enrichment analysis showed pathways, including TCA, Glycolysis/Gluconeogenesis, pyruvate, and carbon metabolism, as well as central carbon metabolism in cancer, resistance to platinum drugs, and the glucagon and HIF-1 signaling pathways were enriched by these CRGs. However, no significant difference was observed between these CRGs (Fig. 2C).

A DO enrichment analysis of 10 CRGs in EMT. B GO enrichment analysis of 10 CRGs in EMT. C KEGG pathway analyses of 10 CRGs in EMT.
Screening of EMT markers using machine learning algorithms
A total of ten DEGs were identified using machine learning as signature genes. Five signature genes, including NFE2L2, NLRP3, DLD, PDHA1, and GLS, were identified using the “LASSO regression analysis” (Fig. 3A, B). Next, six signature genes, including GLS, PDHB, DLD, NFE2L2, PDHA1, and SLC31A1, were identified using the “SVM-RFE” algorithm (Fig. 3C, D). Finally, five signature genes, including PDHA1, GLS, NFE2L2, DLAT, and MTF1, were identified using the “RF” algorithm (Fig. 3E, F). Further, genes identified using these three machine learning algorithms were intersected, and three signature genes, including GLS, NFE2L2, and PDHA1, were identified. The three genes were intersected to obtain three signature genes, GLS, NFE2L2, and PDHA1, and plotted on a Wayne diagram (Fig. 3G, H).

Selection procedure of diagnostic markers for EMT diagnoses. (A, B) Tuning feature selection in the LASSO model. (C, D) Biomarker signature gene expression validation through the SVM-RFE arithmetic. (E) RandomForest error rate versus the number of classification trees. F The top 10 relatively important genes. (G, H) Venn graph presenting 3 diagnostic biomarkers shared by the LASSO, SVM-RFE and RandomForest arithmetic methods.
Expression and validation of the diagnostic significance of the signature genes
First, we determined the differential expression and diagnostic value of GLS, NFE2L2, and PDHA1 in GSE7307. Further, these genes were externally validated in GSE7305. The results revealed a significant increase in GLS and NFE2L2 expression and a significant decrease in PDHA1 expression in both datasets (Fig. 4A, B). The AUC values of PDHA1 in the two datasets were 0.879 and 0.920, respectively (Fig. 5A, B). The AUC values of all three diagnostic genes were > 0.800, thereby indicating these CRGs had high diagnostic values.

The expression pattern of the 3 critical genes in EMT. A GLS and NFE2L2 were highly expressed in EMT, and PDHA1 was lowly expressed in EMT. B The expression pattern of the 3 critical genes was further demonstrated in GSE7305 datasets.

A The diagnostic value of the 3 critical genes was studied using ROC assays in GSE7307. B Three genes (GLS, NFE2L2, and PDHA1) were further demonstrated to be diagnostic genes in GSE7305.
Expression of key CRGs in EMT with different severity and combined infertility
To validate the confidence and applicability of the key CRGs, external datasets such as GSE51981 and GSE120103 were selected to validate the correlation between key CRGs, EMT severity, and combined infertility. Since PDA1 was not expressed in GSE120103, PDHA1 was not analyzed in EMT samples. The patients with micro/light EMT were categorized into one group, and patients with moderate/severe EMT were categorized into another group. The results revealed high GLS and NFE2L2 expression in patients with moderate/severe EMT (Fig. 6A). ROC curves were performed on these upregulated key CRGs to analyze the severity of EMT (P < 0.05) and the AUC values > 0.7 (Fig. 6B). Next, we used GSE120103, consisting of patients with EMT who may or may not be suffering from infertility, for subsequent validation. The results revealed an increase in GLS and NFE2L2 expression in the endometrium of patients with EMT; however, a decrease in GLS and NFE2L2 expression was observed in the endometrium of patients with infertility (Fig. 7A). On the contrary, we performed ROC analysis to determine the significance of key CRGs in diagnosing patients with EMT. The AUC values validated the sensitivity and specificity of CRGs (Fig. 7B).

The expression pattern of the 3 critical genes in different severity groups of endometrioses. A Expression of CRGs in different severity groups of endometrioses. B ROC analysis of CRGs predicting the severity of endometriosis in GSE51981.

Expression of CRGs in the endometria of the infertile and fertile females with endometriosis. A Upregulated CRGs were decreased in the infertile group. B The ROC analysis of CRGs predicting infertility of endometriosis in GSE120103.
Analyzing Regulatory pathway enriched by key CRGs
The ssGSEA results showed that the complement and coagulation pathways, Legionellosis, Malaria, Pertussis, Systemic lupus erythematosus (SLE), etc., were enriched in the high GLS and high NFE2L2 expression groups (Fig. 8A, B). In addition, the cell cycle, the Fanconi anemia and protein export pathways, DNA replication, and Homologous recombination are enriched in the PDHA1 expression group (Fig. 8C).

GSEA revealed the enriched pathways of the hub genes. A The main signaling pathways that are significantly enriched by the high expression of GLS, NFE2L2 and PDHA1. B The main signaling pathways that are significantly enriched by the low expression of key CRGs.
Correlation between three key CRGs and proportion of immune cell infiltration in EMT
We utilized the “CIBERSORT” method to determine the proportion of 22 immune cell subpopulations infiltrating in patients with EMT. GLS, NFE2L2, and PDHA1 expression were evaluated as a function of the immune microenvironment. A significant positive correlation was observed between GLS and immune cells like M2 macrophages, Plasma cells, naïve CD4 T cells, and activated Mast cells. GLS was negatively correlated with activated NK cells (Fig. 9A). NFE2L2 was significantly correlated with immune cells such as Plasma cells, memory B cells, resting memory CD4 T cells, Eosinophils, M1 and M2 macrophages, resting mast cells, and CD8 T cells (Fig. 9B). Furthermore, a significant positive correlation was observed between NFE2L2 and immune cells such as activated NK cells, CD8T cells, M1 and M2 Macrophages, resting mast cells, naïve B cells, resting memory CD4 T cells, eosinophils, memory B cells, and Plasma cells (Fig. 9C).

A Correlation between GLS and infiltrating immune cells in EMT. B Correlation between NFE2L2 and infiltrating immune cells in EMT. C Correlation between PDHA1 and infiltrating immune cells in EMT.
Construction and validation of the Nomogram prediction model
We constructed a nomogram prediction model using GLS, NFE2L2, and PDHA1 for predicting the risk of EMT development (Fig. 10A). Nomogram was externally validated using GSE7305. The rate of incidences was derived from each risk factor score and summed to obtain a total score. Next, we plotted the calibration curve (Fig. 10B). The results revealed that the three curves were consistent and converged, thereby indicating a good agreement between the actual and predicted outcomes of the model. In addition, DCA showed the clinical benefit of the nomogram prediction model (Fig. 10C). Finally, we plotted the ROC curve (the AUC value = 0.942) to determine the predictive ability of the columnar graph model (Fig. 10D). The AUC value of the nomogram prediction model was 0.942, thereby indicating that the model had a good predictive ability.

Construction and validation of the EMT diagnostic column line graph model. A Column line graphs are used to predict the occurrence of EMT. B Calibration curves to assess the predictive power of the column line graph model. C DCA curves to assess the clinical value of the column line graph model. D ROC curves to assess the clinical value of the column line graph model.
Drug prediction
We identified five drugs targeting three key CRGs using data from the “DrugBank database (Fig. 11), and all of these drugs have been approved. First, L-glutamine (DB00130) was identified as a drug targeting CRGs. It is an important energy source in cells and is involved in metabolic processes. It is used to treat patients with sickle cell disease gastric and duodenal ulcers, improving brain function. Next, we identified Glutamate (DB00142), a common amino acid and an excitatory neurotransmitter. It is a precursor molecule required for GABA synthesis in GABAergic neurons. It is mainly used as total parenteral nutrition and as an adjunct for treating psycho-neurological disorders. Next, NADH (DB00157) targets PDHA1. Finally, dimethyl fumarate (DMF, DB08908) binds to NFE2L2. It is used for treating patients with relapsing multiple sclerosis.

Drug screening for targeting key CRGs.
Validation of GLS, NFE2L2 and PDHA1 Expression in EMT by using qRT-PCR and Western blot
The above analysis revealed that GLS, NFE2L2 and PDHA1 have the potential as prognostic biomarkers for EMT. We verified the expression of GLS, NFE2L2 and PDHA1 in EMT and normal endometrium using qRT-PCR and Western blot. qRT-PCR and Western blot showed that GLS and NFE2L2 were expressed at higher levels in EMT, and PDHA1 were expressed at lower levels in EMT (P < 0.05, Fig. 12A–E).

The correlation of key CRGs expression in EMT. A The levels of GLS, NFE2L2 and PDHA1 mRNA expression in EMT tissues. B The levels of GLS, NFE2L2 and PDHA1 protein expression in EMT.
Discussion
Currently, invasive techniques, such as laparoscopic histopathological examination, are used for EMT diagnosis. The most common treatment modalities include hormonal suppression or surgical excision/ablation of visible lesions. However, the therapeutic outcomes are unsatisfactory and often accompanied by adverse effects. Studies have identified several key molecules, such as adhesion molecules, matrix metalloproteinases, lncRNA HOTAIR, oxidative stress, and regulatory T cells, that are involved in EMT development22,23. However, the exact mechanism is still unclear. Therefore, there is an urgent need to identify new biomarkers associated with EMT pathogenesis. This would aid in developing non-invasive diagnostic and screening tools for patients with EMT.
Copper is an essential cofactor of enzymes. It is critically involved in various physiological processes and activities. Turgut et al. showed that copper was associated with the pathogenesis of EMT and oxidative stress24. Cuproptosis is a newly discovered form of cell death involving TCA regulation. Lipid-acylated proteins induce cytotoxic stress, which leads to cell death 6. However, no studies have shown the involvement of cuproptosis in EMT. Therefore, in this study, we combined data on EMT and CRGs from GEO. We identified 10 DEGs, including NFE2L2, NLRP3, SLC31A1, DLAT, DLD, PDHB, PDHA1, MTF1, CDKN2A, and GLS. The DO, GO, and KEGG pathway enrichment analysis showed that key CRGs were primarily enriched in the acetyl-CoA and pyruvate metabolism, TCA, and Glycolysis/Gluconeogenesis. Bradbeer et al. showed a correlation between TCA enzyme succinate dehydrogenase and EMT25. Interestingly, alteration in TCA occurs in cuproptosis, which induces cell death. Therefore, these results indicate that these CRGs could be a promising target for modulating cuproptosis in EMT, which could be a new therapeutic strategy for treating EMT.
Next, we used three machine learning algorithms, such as “LASSO regression,” “SVM-RFE,” and “RF,” to screen for EMT and cuproptosis-related biomarkers. We identified three CRGs associated with immune cell infiltration in EMT. Of these three CRGs, an increase in GLS and NFE2L2 expression and a decrease in PDHA1 expression were observed. Furthermore, the ROC curve analysis validated the diagnostic value of these key CRGs in distinguishing the endometrium of patients with EMT and healthy controls. Finally, we validated our results using the external validation dataset GSE7305, and the results obtained were consistent.
Glutaminase (GLS) is located on chromosome 2. It is a key enzyme mediating glutamine-to-glutamate conversion. GLS is converted to alpha-ketoglutarate, which enters TCA to produce energy. GLS is an oncogene, and an increase in GLS expression was observed in pancreatic ductal adenocarcinoma. Under oxidative stress, SUCLA2 increases glutamine catabolism and the production of nicotinamide adenine dinucleotide phosphate as well as glutathione by activating GLS, thereby ameliorating oxidative stress and promoting tumor growth26. Our results revealed a significant correlation between CRGs like GLS and EMT progression. Nuclear transcription factor E2-related factor 2 (NFE2 like bZIP transcription factor 2, NFE2L2, also known as Nrf2) is primarily localized in the cytoplasm. It is an antioxidant transcription factor that regulates inflammatory responses and oxidative stress in cells27. NFE2L2 variants are closely linked to the risk of human disease. Interestingly, oxidative damage promotes EMT progression. Previous studies have shown that NFE2L2 increases EMT risk by inhibiting processes such as oxidative stress against the growth and development of EMT28,29. The pyruvate dehydrogenase complex E1 alpha subunit (PDHA1) is the E1 subunit of the pyruvate dehydrogenase complex. It is primarily localized in the mitochondria and contains three serine residues, which could be activated by four inhibitory PDHK1-4 and two reactivated phosphatases (PDP1-2). These PDP1-2 are reversibly phosphorylated and regulate glycolysis and TCA30,31. Spakova et al. demonstrated that silencing HIF-1α/miR210 expression increases PDHA1 and decreases MITF-M expression. This promotes mitochondrial respiratory activity, which aids in eliminating reactive oxygen species in melanoma cells32. Our results indicate a significant role of GLS, NFE2L2 and PDHA1 in EMT pathogenesis. ROC curve analysis revealed that these key CRGs are significantly involved in the onset and progression of EMT, thereby indicating the potential diagnostic value of these CRGs in clinical settings. Moreover, risk modeling results suggested that GLS, NFE2L2 and PDHA1 could be risk factors for EMT. Based on previous studies and our results, CRGs may play an important role in EMT development, and the underlying mechanisms should be further investigated.
We also determined the expression of key CRGs in patients with varying severity of EMT and those suffering from infertility. The results revealed an increase in GLS and NFE2L2 expression in patients with moderate/severe EMT. Furthermore, an increase in the expression of key CRGs could lead to infertility in patients with EMT. Qiao et al. showed that isoniazid activates the Keap1/Nrf2 signaling pathway by inducing oxidative stress and apoptosis, thereby impairing the reproductive system and reducing fertility in mammals33. Ivanov et al. showed that melatonin could protect the developing embryo from oxidative stress by modulating NFE2L2, SOD1, and GPX1 expression34. These results indicate a close correlation between NFE2L2 and infertility, consistent with our results.
Next, we performed ssGSEA to verify the involvement of key CRGs in EMT. Our results revealed significant enrichment of the complement and coagulation pathways, Legionellosis, Malaria, Pertussis, and SLE in the high GLS and high NFE2L2 expression groups. On the contrary, the cell cycle, the Fanconi anemia and protein export pathways, DNA replication, and homologous recombination were enriched in the high PDHA1 expression group. SLE is an autoimmune disease characterized by the presence of specific autoantibodies, including antinuclear antibodies. Studies have shown a correlation between EMT and autoimmune diseases such as SLE and Sjögren's syndrome35,36.
However, the underlying mechanism of the pathogenesis of EMT is still unclear. Implantation of reflux menstrual blood, endocrine, genetic factors, angiogenesis, and stem cell differentiation are important factors associated with EMT pathogenesis. Of these factors, abnormal immune responses could be the underlying mechanism of EMT pathogenesis. In fact, the immune component of EMT has received widespread attention from researchers. Some studies have demonstrated that an increase in the secretion of proinflammatory cytokines due to immune dysfunction, impaired immune surveillance, and altered immune cell profiles contribute to EMT progression. Prolonged persistent immune dysregulation could lead to a state of chronic inflammation, thereby creating a conducive environment for promoting adhesion and angiogenesis. This could lead to a vicious cycle of EMT development and progression37. The “CIBERSORT” method is widely used to study EMT in humans. It is based on the principles of linear support vector regression to deconvolute the expression matrix of immune cell subtypes.
Therefore, we used CIBERSORT to assess infiltrating immune cells for determining the role of immune cell infiltration in EMT. Our results revealed that these three CRGs were associated with various immune cell types, including eosinophils, activated NK cells, and macrophages. Eidukaite et al. showed that epithelial cells in endometriotic foci secrete high levels of eosinophil-specific chemokines compared to in situ endometrial cells. These eosinophils and other myeloid cells enter the peritoneum, which triggers inflammatory and allergic responses38. Ścieżyńska et al. showed that endometriotic cells' survival and growth in the peritoneal cavity might also be due to their recognition and clearance by local immune cells such as macrophages and NK cells39. Ramírez-Pavez et al. demonstrated that macrophages accumulate in the peritoneal cavity of patients with EMT; however, their ability to clear migrating endometrial debris is reduced40. Furthermore, combining the results of differential immune cell infiltration and the correlation between cuproptosis and immune cell infiltration revealed that immune cells such as eosinophils, activated NK cells, and M2 Macrophages could be critically involved in the modulation of CRG-regulated immune function in EMT.
EMT is an aggressive disease. Koninckx et al. have suggested that oxidative stress and peritoneal microbiota due to retrograde menstruation could cause EMT. Therefore, pharmacological treatment could prevent new lesions and be prescribed post-surgery. Additionally, Sirohi et al. showed that inhibiting copper-induced toxicity increases the survival of endometriotic cells41. Therefore, copper chelators could be used as therapeutic agents for treating patients with EMT42. We identified five drugs from the DrugBank database targeting these three CRGs. Qinpi methicin (DB13155) has anti-inflammatory and antioxidant activities. Hence, qinpi methicin could be used for treating diseases caused by inflammation and oxidative stress, such as EMT. As-Sanie et al. demonstrated high forebrain insula glutamine (DB00130)-glutamate (DB00142) binding in EMT patients with chronic pelvic pain. High islet activity could cause chronic pelvic pain in patients with EMT; therefore, reducing islet glutamate levels could alleviate chronic pelvic pain43. DMF is an immunomodulatory and antioxidant molecule commonly used to treat patients with relapsing multiple sclerosis and psoriasis. It binds with NFE2L2. Chen et al. demonstrated that DMF, an Nrf2 agonist, could significantly influence immune responses and oxidative stress. Additionally, Yan et al. suggested that DMF could attenuate inflammation, oxidative stress, and iron death via the NRF2/ARE/NF-kB signaling pathway. Moreover, DMF improves cognitive dysfunction in rats with chronic hypoperfusion44. Also, EMT is closely associated with oxidative stress45. Therefore, it is necessary to investigate if DMF could be used for treating patients with EMT. NADH is a nutritional supplement. It has antioxidant properties, reduces anxiety, promotes neurotransmitter synthesis, and prevents dementia. Govatati et al. showed that altering the mitochondrial membrane complex I could be a risk factor for EMT. Mitochondrial membrane complex I catalyze the transfer of electrons from NADH to ubiquinone and is closely correlated with EMT, which could be a new therapeutic approach46.
The mRNA and protein expression levels of GLS, NFE2L2 and PDHA1 were validated by qRT-PCR and WB studies in EMT samples. However, our study has a few limitations. First, the sample size of our study was relatively small since data were only obtained from GEO. Therefore, additional studies using a larger sample size should be performed to validate our results. Second, although we have successfully identified three CRGs as potential biomarkers for immunophenotyping of EMT, no in vivo or in vitro studies were conducted to validate these results. Thus, future studies should focus on conducting in vivo or in vitro studies to validate these biomarkers.
Conclusions
GLS, NFE2L2, and PDHA1 could be novel diagnostic markers for EMT, representing a significant breakthrough. Additionally, the correlation between these CRGs and immune cell infiltration could aid in developing effective immunotherapy for patients with EMT.
Data availability
Microarray data for human endometriosis and control specimens were obtained from the GEO database datasets GSE7307, GSE7305, GSE51981 and GSE120103.
References
Kapoor, R., Stratopoulou, C. A. & Dolmans, M. Pathogenesis of endometriosis: New insights into prospective therapies. Int. J. Mol. Sci. 22, 11700 (2021).
Taylor, H. S., Kotlyar, A. M. & Flores, V. A. Endometriosis is a chronic systemic disease: Clinical challenges and novel innovations. Lancet. 397, 839–852 (2021).
Hodgson, R. M., Lee, H. L., Wang, R., Mol, B. W. & Johnson, N. Interventions for endometriosis-related infertility: A systematic review and network meta-analysis. Fertil. Steril. 113, 374–382 (2020).
Anastasiu, C. V. et al. Biomarkers for the noninvasive diagnosis of endometriosis: State of the art and future perspectives. Int. J. Mol. Sci. 21, 1750 (2020).
Ge, E. J. et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer. 22, 102–113 (2022).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated Tca cycle proteins. Science. 375, 1254–1261 (2022).
Tan, H. Y. et al. Lysyl oxidase-like 4 fosters an immunosuppressive microenvironment during hepatocarcinogenesis. Hepatology. 73, 2326–2341 (2021).
Wang, X. et al. The copper-associated protein Steap2 correlated with glioma prognosis and immune infiltration. Front. Cell. Neurosci. 16, 944682 (2022).
Ji, Z., Ren, W., Wang, H., Gao, W. & Yuan, B. Molecular subtyping based on cuproptosis-related genes and characterization of tumor microenvironment infiltration in kidney renal clear cell carcinoma. Front. Oncol. 12, 919083 (2022).
Jiang, X. et al. A novel cuproptosis-related gene signature of prognosis and immune microenvironment in head and neck squamous cell carcinoma cancer. J. Cancer. Res. Clin. Oncol. 149, 203–218 (2023).
Jiang, L. et al. Exploring diagnostic M6a regulators in endometriosis. Aging 12, 25916–25938 (2020).
Jiang, L., Zhang, M., Wang, S., Han, Y. & Fang, X. Common and specific gene signatures among three different endometriosis subtypes. Peerj. 8, e8730 (2020).
Chen, S., Chai, X. & Wu, X. Bioinformatical analysis of the key differentially expressed genes and associations with immune cell infiltration in development of endometriosis. BMC Genomic Data. 23, 20 (2022).
Zhao, S. et al. Igf2Bp2 promotes the progression of ovarian endometriosis by regulating M6a-modified Meis2 and Gata6. Int. J. Biochem. Cell Biol. 152, 106296 (2022).
Hever, A. et al. Human endometriosis is associated with plasma cells and overexpression of B lymphocyte stimulator. Proc. Natl. Acad. Sci. 104, 12451–12456 (2007).
Hu, W., Xie, Q., Xu, Y., Tang, X. & Zhao, H. Integrated bioinformatics analysis reveals function and regulatory network of Mir-200B-3P in endometriosis. Biomed. Res. Int. 2020, 1–7 (2020).
He, Y. et al. Identification and analysis of potential immune-related biomarkers in endometriosis. J. Immunol. Res. 2023, 1–21 (2023).
Albaradei, S. et al. Machine learning and deep learning methods that use omics data for metastasis prediction. Comp. Struct. Biotechnol. J. 19, 5008–5018 (2021).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. Svm-Rfe: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19 (2018).
Wei, P. et al. Identification and validation of a signature based on macrophage cell marker genes to predict recurrent miscarriage by integrated analysis of single-cell and bulk Rna-sequencing. Front. Immunol. 13, 1053819 (2022).
Hanzelmann, S., Castelo, R. & Guinney, J. Gsva: Gene set variation analysis for microarray and Rna-Seq data. BMC Bioinform. 14, 7 (2013).
Zhang, L. et al. Exosomal lncrna hotair promotes the progression and angiogenesis of endometriosis via the Mir-761/Hdac1 axis and activation of Stat3-mediated inflammation. Int. J. Nanomed. 17, 1155–1170 (2022).
Koninckx, P. R. et al. Pathogenesis Based Diagnosis and Treatment of Endometriosis. Front. Endocrinol. 12, e070750 (2021).
Turgut, A. et al. Copper, ceruloplasmin and oxidative stress in patients with advanced-stage endometriosis. Eur. Rev. Med. Pharmacol. Sci. 17, 1472–1478 (2013).
Bradbeer, J. N., Lindsay, P. C. & Reeve, J. Fluctuation of mineral apposition rate at individual bone-remodeling sites in human iliac cancellous bone: Independent correlations with osteoid width and osteoblastic alkaline phosphatase activity. J. Bone. Miner. Res. 9, 1679–1686 (1994).
Tong, Y. et al. Sucla2-coupled regulation of Gls succinylation and activity counteracts oxidative stress in tumor cells. Mol. Cell. 81, 2303–2316 (2021).
Lee, D. H. et al. Sqstm1/P62 activates Nfe2L2/Nrf2 via Ulk1-mediated autophagic Keap1 degradation and protects mouse liver from lipotoxicity. Autophagy. 16, 1949–1973 (2020).
Siracusa, R. et al. The methyl ester of 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid reduces endometrial lesions development by modulating the Nfkb and Nrf2 pathways. Int. J. Mol. Sci. 22, 3991 (2021).
Osiński, M. et al. The assessment of Gwas-identified polymorphisms associated with infertility risk in polish women with endometriosis. Ginekol. Pol. 89, 304–310 (2018).
Echeverri Ruiz, N. P. et al. Dynamic regulation of mitochondrial pyruvate metabolism is necessary for orthotopic pancreatic tumor growth. Cancer Metab. 9, 39 (2021).
Xu, X., Tang, Y., Zhu, J. & Luo, J. Endoplasmic reticulum stress-related four-biomarker risk classifier for survival evaluation in esophageal cancer. J. Oncol. 2022, 1–14 (2022).
Špaková, I., Rabajdová, M., Mičková, H., Graier, W. F. & Mareková, M. Effect of hypoxia factors gene silencing on Ros production and metabolic status of a375 malignant melanoma cells. Sci. Rep. 11, 10325 (2021).
Qiao, P. et al. Oral isoniazid causes oxidative stress, oocyte deterioration and infertility in mice. Toxicology. 455, 152749 (2021).
Ivanov, D. et al. Melatonin, its beneficial effects on embryogenesis from mitigating oxidative stress to regulating gene expression. Int. J. Mol. Sci. 22, 5885 (2021).
Geng, R. et al. Gene expression analysis in endometriosis: immunopathology insights, transcription factors and therapeutic targets. Front. Immunol. 13, 1037504 (2022).
Shigesi, N. et al. The association between endometriosis and autoimmune diseases: A systematic review and meta-analysis. Hum. Reprod. Update. 25, 486–503 (2019).
Jiang, I., Yong, P. J., Allaire, C. & Bedaiwy, M. A. Intricate connections between the microbiota and endometriosis. Int. J. Mol. Sci. 22, 5644 (2021).
Eidukaite, A. & Tamosiunas, V. Activity of eosinophils and immunoglobulin E concentration in the peritoneal fluid of women with endometriosis. Clin. Chem. Lab. Med. 42, 590–594 (2004).
Ścieżyńska, A. A., Komorowski, M. M., Soszyńska, M. M. & Malejczyk, J. J. Nk cells as potential targets for immunotherapy in endometriosis. J. Clin. Med. 8, 1468 (2019).
Ramírez-Pavez, T. N. et al. The role of peritoneal macrophages in endometriosis. Int. J. Mol. Sci. 22, 10792 (2021).
Sirohi, D., Al, R. R. & Knibbs, L. D. Environmental exposures to endocrine disrupting chemicals (Edcs) and their role in endometriosis: A systematic literature review. Rev. Environ. Health. 36, 101–115 (2021).
Delsouc, M. B. et al. Antiproliferative and antiangiogenic effects of ammonium tetrathiomolybdate in a model of endometriosis. Life Sci. 287, 120099 (2021).
As-Sanie, S. et al. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J. Pain 17, 1–13 (2016).
Yan, N., Xu, Z., Qu, C. & Zhang, J. Dimethyl fumarate improves cognitive deficits in chronic cerebral hypoperfusion rats by alleviating inflammation, oxidative stress, and ferroptosis via Nrf2/are/Nf-Κb signal pathway. Int. Immunopharmacol. 98, 107844 (2021).
Samimi, M., Pourhanifeh, M. H., Mehdizadehkashi, A., Eftekhar, T. & Asemi, Z. The role of inflammation, oxidative stress, angiogenesis, and apoptosis in the pathophysiology of endometriosis: Basic science and new insights based on gene expression. J. Cell. Physiol. 234, 19384–19392 (2019).
Govatati, S., Deenadayal, M., Shivaji, S. & Bhanoori, M. Mitochondrial Nadh: Ubiquinone oxidoreductase alterations are associated with endometriosis. Mitochondrion. 13, 782–790 (2013).
Author information
Authors and Affiliations
Contributions
Prof. Dong and Prof. Wang was responsible for study design and writing supervision, while J.W. primarily participated in the experiment. J.W. and Y.L. primarily participated in sample collection. J.W. participated in statistical analysis and drafting the initial manuscript. Prof. Dong contributed to bioinformatics and machine learning analyses. All authors made significant contributions to this article and approved its submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, J., Lu, Y., Sun, G. et al. Machine learning algorithms for a novel cuproptosis-related gene signature of diagnostic and immune infiltration in endometriosis. Sci Rep 13, 21603 (2023). https://doi.org/10.1038/s41598-023-48990-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48990-w
- Springer Nature Limited