
Abstract
In the last decade, clinical studies have investigated the clinical relevance of circulating cell-free-DNA (ccfDNA) as a diagnostic and prognosis tool in various diseases including cancers. However, limited knowledge on ccfDNA biology restrains its full development in the clinical practice. To improve our understanding, we evaluated the impact of the circadian rhythm on ccfDNA release in healthy subjects over a 24-h period. 10 healthy female subjects underwent blood sampling at 8am and 20 healthy male subjects underwent serial blood sampling (8:00 AM, 9:00 AM, 12:00 PM, 4:00 PM, 8:00 PM, 12:00 AM, 4 AM (+ 1 Day) and 8 AM (+ 1 Day)). We performed digital droplet-based PCR (ddPCR) assays to target 2 DNA fragments (69 & 243 bp) located in the KRAS gene to determine the ccfDNA concentration and fragmentation profile. As control, half of the samples were re-analyzed by capillary miniaturized electrophoresis (BIAbooster system). Overall, we did not detect any influence of the circadian rhythm on ccfDNA release. Instead, we observed a decrease in the ccfDNA concentration after meal ingestion, suggesting either a post-prandial effect or a technical detection bias due to a higher plasma load in lipids and triglycerides. We also noticed a potential effect of gender, weight and creatinine levels on ccfDNA concentration.
Similar content being viewed by others

Introduction
The presence of fragmented circulating cell-free DNA (ccfDNA) within peripheral blood was first described by Mandel & Metais in 19481. The medical community has shown great interest in ccfDNA analyses across various disciplines, including infectiology, obstetrics, transplantation, surgery and oncology. In 1989, Stroun et al.2 opened the door to numerous applications when they demonstrated that, in cancer patients, a fraction of ccfDNA corresponded to cell-free tumor DNA (ctDNA). Since then, the utility of ccfDNA detection has been largely explored for cancer screening, diagnosis and prognosis3,4,5 through the analysis of somatic mutations5,6, chromosomal alterations7 and epigenetic changes8. More recently, several teams demonstrated the pertinence of ctDNA analysis to detect post-operative minimal residual disease9 and early cancer recurrence10. The gains in sensitivity and specificity, acquired through analytical methods like optimized Next Generation Sequencing (NGS), droplet-based digital PCR (ddPCR) or optimized qPCR, have allowed the detection of very small concentrations of ctDNA molecules in the bloodstream11,12.
Despite the great potential of ctDNA as a cancer biomarker, its application in clinical practice remains restricted to few indications. Amongst parameters that may limit the use of cfDNA and ctDNA in our routine practice, it is important to underline that the biological variables likely to modify the release of ccfDNA are not clearly comprehended. The origin of ccfDNA remains poorly understood and may imply several cell death processes13. Apoptosis has been proposed as a primary mechanism by which ccfDNA is released into circulation due to its distinctive fragment length of 140–180 bp, which mirrors the size of nucleosome-bound DNA fragments14. Necrosis and active release have also been described as a source of ccfDNA13. The ccfDNA fragmentation profile has been studied in different clinical situations15,16,17. Moreover, ccfDNA can be combined with diverse biological structures, including nucleosomes, extracellular vesicles (EVs; e.g., exosomes and virosomes) and neutrophile extracellular DNA trap (NETs) that could modify its integrity and physiological role18.
Moss et al. used a deconvolution methylation model in healthy subjects to show that ccfDNA mostly originates from white blood cells and erythrocyte progenitors19. Increased ccfDNA levels have been observed in various pathology such as cancers20, acute and chronic inflammatory diseases21, traumas22, and cardiovascular diseases23. The increase in ccfDNA concentration can also be observed in physiological processes such as physical activities24, pregnancy25 or obesity26. The analysis of ccfDNA concentration alone is therefore insufficient for the proper exploration of oncological processes. Analytical tests specifically targeting ctDNA by using cancer specific genetic and epigenetic alterations, have thus been developed27,28,29.
Although considerable efforts have been devoted to understanding the origin and the evolution of ccfDNA under various biological conditions, less attention has been paid to the daily dynamics of ccfDNA. Depending on the biological context, the estimated half-life of ccfDNA is comprised between 15 min and 1.5 h30. Understanding the potential daily variations of ccfDNA concentrations is critical to improve the reliability of ctDNA monitoring. Indeed, the circadian rhythmicity is a molecularly driven, individual clock. It generates cyclic changes through the activity of various compounds, with a periodicity of about a day31. This observation has led to chrono-pharmaceutical approaches to optimize regimen dosing and administration schedules32. Moreover, several studies have suggested that circadian rhythm could relate with the concentration of circulating nucleotides (miRNA and cfDNA) in subjects presenting with or without cancer33,34,35. The underlying mechanism is unknown. Moreover, a recent work by Diamantopoulou et al. has shown that the metastatic spread of breast cancer may accelerate during sleep36. They found that hormones implicated in the regulation of the circadian rhythm (such as melatonin, testosterone and glucocorticoids) may dictate the generation and the dynamics of circulating tumor cells, and as a consequence, that insulin may directly promote tumour cell proliferation in a time-dependent manner36.
The present study was designed to evaluate the influence of circadian rhythm on ccfDNA release in healthy subjects by focusing on its absolute quantification and fragmentation profile along the day. Such work could provide clues to determine the optimal timing for liquid biopsy sampling before its routine use in clinical practice. To reach this goal, we performed our analyses using ddPCR assays as well as BIAbooster system technology (miniaturized capillary electrophoresis) that allow for precise quantification of the total ccfDNA17. We first validated the repeatability and precision of the quantification method on commercial plasma samples from healthy subjects. We then analyzed the longitudinal ccfDNA concentration and fragmentation profile in a cohort of healthy subjects who underwent plasma sampling over a 24-h period. The impact of other variables on ccfDNA release such as weight, gender, creatinine concentration levels and ethnic origin was also evaluated.
Materials and methods
Subjects and procedures
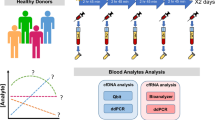
Longitudinal plasma samples collected from healthy subjects for the PSPRR cohort (previous cohort carried on in the Hospital Europeen Georges Pompidou (Plasma Soluble (Pro)Renin Receptor)) were available37. The study received the ethical approval from CPP Ile de France III (N°EUDRACT: 2010-A00365-34) ethics committee and all subjects provided with written informed consent. All processes were performed in accordance with the relevant guidelines and regulations. This healthy prospective cohort was composed of 20 healthy males and 10 healthy females for a total of 170 samples (Fig. 1). All subjects underwent blood sampling at 8 AM and only male subjects underwent longitudinal blood sampling over a 24 h period. Each male subject underwent 8 blood sampling at 8:00 AM, 9:00 AM, 12:00 PM, 4:00 PM, 8:00 PM, 12:00 AM, 4:00 AM (+ 1 Day) and 8:00 AM (+ 1 Day) using K2-EDTA tubes (Greiner Bio-One International, Cat. N°: 456043). Their meals were provided after blood sampling at 9:00 AM, 12:00 PM and 8:00 PM. Blood samples were collected in fasting conditions after 1-h rest in a semi recumbent position. To limit biases, subjects were asked to maintain a resting state throughout the study. The clinical characteristics of this healthy cohort are presented in the Supplementary Table 1. For one male, the weight and creatinine level data were not reported. Cortisol concentrations were measured using commercially available kits37. Secreted in a pulsatile fashion, it is known to display a circadian rhythm in adults were Cortisol levels are highest in the morning and subsequently decrease to a nadir in the evening38,39.

Workflow of the circadian study. In total, 20 healthy males and 10 healthy females were recruited. For each male, 8 blood samples were drawn. For each female, only one blood collection was drawn.
Repeatability
To evaluate the repeatability of the quantification method, we used 2 cohorts of commercial plasma. Cohort 1 included samples from 13 healthy individuals from BIOPREDICT International and cohort 2 included 10 healthy individuals from the Etablissement Francais du Sang (EFS). Each plasma, previously stored at − 80 °C, was aliquoted three times (1 mL) and treated independently. For each aliquot of plasma, ccfDNA was extracted as described below ensuring that aliquots originating from the same plasma sample were treated on different experiments. These ccfDNA samples were quantified by 69 bp ddPCR assay. The workflow of this analysis is summarized in the Supplementary Fig. 1.
Plasma ccfDNA preparation
The collection tubes (K2-EDTA) were processed within 4 h to prevent the destruction of white-blood cells at 1200g for 15 min at 4 °C. For each sample, 1 mL of plasma was aliquoted and stored at − 80 °C. Before extraction plasma were centrifugated a second time at 8000g for 10 min at 4 °C. The DNA was extracted using the Maxwell RSC ccfDNA Plasma Kit (Promega, Cat No./ID: AS1480) following the manufacturer’s instruction and was eluted in 62µL of elution buffer. The DNA concentration was measured from 2µL by Qubit 2.0 fluorometer (ThermoFisher Scientific) using the dsDNA HS Assay (ThermoFisher Scientific, Cat No. Q32851). Extracted DNA samples were stored frozen at − 20 °C before testing.
ccfDNA analysis
Droplet-based digital PCR assays
We designed ddPCR assays to target the wild-type sequence of KRAS gene exon 2, codon 13. These assays allowed amplifying two sequences with lengths of 69 and 243 bp, which reflected the quantification of DNA fragments > 69 bp and > 243 bp, respectively. In both cases, a probe bearing a VIC fluorophore (λex 538 nm/λem 554 nm) detected the wild-type sequence of the KRAS gene (Supplementary Table 2).
ddPCR assays and DNA integrity index (DII) calculation
A total of 11 μL of ddPCR Supermix for Probes (2X) (No dUTP) (Ref: 1863024, BIO-RAD laboratories) was mixed with 1.1 µL of assay Mix solution (20X) containing 8 µM of forward (69 or 243 bp) and reverse primers, and 4 µM VIC Taqman® labeled-probes. The extracted plasma DNA was added to a final reaction volume of 22 μL.
The PCR assay mixes were prepared in a pre-PCR room to limit the risk of contamination. The probe bearing VIC-fluorophore was designed to be specific to the KRAS exon 2 WT allele. Emulsifications of DNA samples were generated according to manufacturer protocol (Ref: 1864002, QX200™ Droplet Generator, BIO-RAD laboratories). The emulsions were thermal cycled following different PCR programs described in the Supplementary Table 3. After completion, the emulsions were either stored at 4 °C or processed immediately to measure the end-point fluorescence signal from each droplet.
The concentration of ccfDNA in the plasma samples was determined as previously described8. Briefly, considering that an haploid genome equivalent (approx. 3 pg) is contained in a positive droplet, DNA concentrations were calculated by taking into account the volume of extracted plasma as well as the elution volume used for DNA extraction and the tested volume of DNA.
Since the number of positive droplets reveals the quantity of amplifiable target DNA, the fraction of amplifiable DNA in each sample could be determined. The number of DNA copies per well was determined using the QuantaSoft™ Analysis Pro Software (BIO-RAD laboratories). DII was defined as a ratio between the number of amplified copies of “long” (243 pb) and “short” (69 pb) DNA fragments. DII is calculated as below (Eq. 1):
BIAbooster analysis
The fragmentation profiles and concentration analyses of ccfDNA from 10 males (80 DNA samples) were also performed using BIAbooster System (ADELIS) as previously described40. The system is based on the principle of DNA fragment migration by capillary electrophoresis coupled to LED induced fluorescence (LEDIF) detectors. It allows performing size and concentration analyses of double stranded DNA with a sensitivity of 10 fg/μL in an operating time of 20 min. Based on the quantity of the different fragments obtained, a ratio highlighting the size distribution of the tested sample is calculated by BIAbooster technology as previously described40 (Eq. 2):
Statistical analyses
In order to evaluate potential differences between DNA concentrations and integrity index at each collection time, paired non-parametric Friedman tests and Wilcoxon tests were performed. Mann Whitney test were performed for un-paired data. Post-hoc Dunn’s multiple comparison test was performed after significant Friedman test. Prism 9 software was used for statistical analysis.
Results
Evaluation of the repeatability of the method
The preliminary evaluation of the repeatability by ddPCR—69 bp assay was carried out. Figure 2 shows a representation of the variation of ccfDNA concentrations within replicates. The coefficient of variation was 12.6% in cohort 1 and 12.96% in cohort 2. No significant differences were observed between the plasma triplicates (Friedman test, p-value = 0.37 for cohort 1 and 0.97 for cohort 2). Both results suggest a repeatability of the pre-analytical and analytical methods. The ccfDNA concentrations in these cohorts were comprised between 1.2 and 9.2 ng/mL.

Repeatability of the pre-analytical and analytical process. The repeatability of the quantification method was evaluated using 2 independent cohorts of healthy plasma obtained from Biopredic international (A, n = 13) and Etablissement Francais du Sang (EFS, B, n = 10). In short, three aliquots were realized for each plasma. For each aliquot ccfDNA was extracted and its concentration determined by ddPCR (69 bp fragment assay). Aliquots originating from the same plasma sample were treated on different experiments.
Implication of the circadian rhythm on circulating DNA concentration
CcfDNA quantification by ddPCR
The 69 bp assay targeting the KRAS exon 2 codon 13 sequence was used to precisely quantify ccfDNA. Using this quantification, significant differences between ccfDNA concentrations were observed between blood collection times (Fig. 3A, Friedman non-parametric test, p-value = 0.0002). Post-hoc Dunn’s multiple comparison test was subsequently performed (Supplementary Table 4). The ccfDNA concentration was significantly decreased in samples drawn 3 to 4 h after meal ingestion (Time 12:00 PM, 4:00 PM & 12:00 AM). More specifically, ccfDNA concentration decreased between 9:00 AM and 12:00 PM and between 8:00 PM and 12:00 AM. It increased between 4:00 PM and 8:00 PM and between 12:00 AM and Day 1 + 4:00 AM. On the contrary, we observed no differences in ccfDNA concentrations between the samples drawn at distance from meal ingestion (Friedman non-parametric test, p-value = 0.39; Fig. 3B and Supplementary Table 5 for Post-hoc Dunn’s multiple comparison test). Normalization of ccfDNA against the value observed at 8am (time of arrival) did not modify the observed results (Supplementary Fig. 2A,B). As observed in this figure, there were some extreme values (2 values higher than 100ng/mL) of ccfDNA concentrations when compared to the commercial healthy cohorts (ie. cohorts 1 and 2) that can probably be attributed to pre-analytical sample treatment. However, these values did not modify the reported observations (data not shown).

Impact of the circadian rhythm on ccfDNA release measured by ddPCR. (A) ccfDNA quantity (ng/mL) at each blood collection point over 24 h. (B) ccfDNA quantity (ng/mL) at each blood collection after exclusion of the post-prandial periods.
The variation of cortisol concentration during the 24-h inclusion period, indicative of circadian rhythm, is shown in Supplementary Fig. 3. The ccfDNA and cortisol concentration were not correlated (Spearman r coefficient = 0.22). Between the healthy individual’s entrance in hospital (blood sampling at 8:00 AM) and the last sampling (the next day at the same time), ccfDNA concentration and proportion remained stable (respectively 3.87 and 3.12 ng/mL of plasma for 8:00 AM & Day 1 + 8:00 AM) (Supplementary Fig. 4).
CcfDNA quantification by BIAbooster system
The ccfDNA concentration was also determined at each time-point and compared using the BIAbooster as previously described. As observed with ddPCR determination, we showed significant differences in ccfDNA concentrations between time-points of blood collection over 24 h period, related with meal ingestion (Kruskal–Wallis test, p-value = 1e−04) (Fig. 4a). Apart from the samples collected after meal ingestion (12:00 PM, 4:00 PM & 12:00 AM), we noticed no differences between other blood collection points (Kruskal–Wallis test, p-value = 0,068; Fig. 4b).

Impact of the circadian rhythm on ccfDNA release measured by BIAbooster system. (a) ccfDNA proportion at each timepoint over a 24-h period. (b) ccfDNA proportion for blood collection after exclusion of the post-prandial.
Evaluation of the DNA integrity index over 24 h
Using the ddPCR − 243 bp assay, we quantified the corresponding DNA fragments. The DII index was determined for each DNA sampling-point following Eq. (2). Overall, no significant differences in ccfDNA integrity were observed between the different times-points (Friedman test, p-value = 0.34; Fig. 5A).

Influence of the circadian rhythm on ccfDNA integrity. (A) ccfDNA integrity index (DII) for each blood collection point over 24 h period. (B) ccfDNA ratio (Eq. 2).
The integrity ratio calculated using the BIAbooster System showed similar results (Fig. 5B; Friedman test, p-value = 0.096).
Influence of clinical parameters on ccfDNA concentration in healthy subjects
Gender (n = 20 men & n = 10 women): Within the whole group, the ccfDNA concentration measured by ddPCR (69bp fragment) was higher in men (Mann Withney, p-value < 0.0001, Fig. 6A). This difference remains significant when excluding extreme ccfDNA concentration values (see above).

Influence of others clinical and biological parameters on ccfDNA release. (A) Gender. Comparison of plasma DNA concentrations between male and female. (B) Ethnicity. Comparison of plasma DNA concentrations between caucasian and non caucasian subjects. (C) Weight. The median weight of the cohort (female and male) was 67.5 kg. We defined two groups (< 67.5 kg and ≥ 67.5 kg). (D) Creatinine concentration. The median creatinine concentration of the cohort (female and male) 75 mol/L. We defined two groups (≤ 75 mol/L and > 75 mol/L).
Ethnic status (n = 10 non-Caucasian men & n = 10 Caucasian men): In male subjects, we showed non-significant difference in ccfDNA concentrations according to the ethnical status (Mann Withney test, p-value = 0.075) Fig. 6B.
Weight (n = 15 with a weight ≤ 67.5 kg, n = 14 with a weight > 67.5 kg): The median weight of the whole cohort (women and men) was 67.5 kg. We therefore defined two groups, one with a weight ≤ 67.5 kg and one with a weight > 67.5 kg. The higher weight group was associated with higher ccfDNA concentrations (Mann Withney test, p-value = 0.026, Fig. 6C).
Creatinine (n = 15 with a creatinine concentration ≤ 75 µmol /L, n = 14 with a creatinine concentration > 75 µmol/L): Assuming the absence of renal failure in this population, the creatine level could be considered as a biomarker reflecting the muscular mass41. The median creatinine concentration level in this cohort (male and female) was 75 mol/L. We defined two groups, one with a creatinine concentration ≤ 75 mol/L and one with a creatinine concentration > 75 mol/L. The high creatinine concentration group showed higher levels of ccfDNA concentration (Mann Withney test, p-value = 0.01), suggesting a potential effect of muscular mass on ccfDNA release (Fig. 6D). Yet, creatinine level is often higher in males which may create bias in the interpretation of our results.
Discussion
The ctDNA detection is a powerful marker that can be used for various applications including the detection of minimal residual disease and the diagnosis of cancer progression42,43,44. Several specific clinical applications are under evaluation in randomized control trials45,46,47. The fraction of ctDNA is small within cfDNA which can complicate its detection. To properly interpret those studies, it is crucial to pay close attention to the pre-analytical and analytical procedures, along with considering confounding factors associated with physiological conditions. The present trial is one of the rare studies that analyzed the link between the circadian rhythm and the detection of cfDNA.
In the first part of this study, we validated the absence of technical variability which ensured the pertinence of our biological data. We also confirmed that our technic benefited from a reproducible pre-analytical process starting from plasma preparation up to DNA extraction48,49.
Then we observed that the ccfDNA concentration and fragmentation profile remained stable during the day at each blood collection time (Figs. 3A, 4b, 5), apart from the samples drawn after meal ingestion. The circadian rhythm showed no interaction with ccfDNA release.
We observed a decrease in ccfDNA concentration after meal ingestion which could be related to a real postprandial effect on ccfDNA released. Yet, it could also be explained by technical bias. Indeed, difficulties to extract ccfDNA fragments have already been described after a meal absorption when the plasma is enriched in lipids and triglycerides50. In 2019, Meddeb et al. observed a decreased concentration of ccfDNA between 9:00 AM and 12:00 PM after meal ingestion51. In 2020, Lois Gardner et al. demonstrated that biomolecule corona of lipid nanoparticles may contain ccfDNA, suggesting an interaction between lipids and ccfDNA52.
We also noticed a significant difference in ccfDNA concentrations between women and men. However, the average weight between these groups was also different, which appears as a major confounding factor. Due to the small size of the present cohort, it was not possible to discriminate between gender or weight contribution on ccfDNA release. Such difference has not been observed for ctDNA in the ALGECOLS cohort53. The difference that we observed between the low and high weight groups (Fig. 6C) is in line with the literature26,54,55. Relationship between the adipocyte inflammatory process and the ccfDNA concentration in overweighted individuals has already been described26.
We also found a relation between creatinine and ccfDNA concentration level41. Yet all the female participant in our cohort were in the low creatinine group which may create bias. Further analyses, including the measurement of the muscular mass could be performed to clarify this observation.
The present work shows several limitations. Firstly, the cohort is small and only composed of 20 healthy male subjects. Another drawback is that the age of healthy individuals (18–35 years old) does not reflect the age of patients developing cancer (> 60 years old). Since aging, inflammatory processes and cell senescence may increase the ccfDNA concentration, a dedicated study should be performed on a larger cohort of healthy volunteers aged over 60 years old and include women. Further investigations on the potential postprandial effect on circulating DNA variation are also required.
In conclusion, circadian rhythm in healthy individuals did not clearly contribute to the variation in ccfDNA concentrations. However, these finding suggest that blood should be drawn ideally before meal ingestion to optimize the ccfDNA concentration. Further analyses are required to confirm those observations.
Data availability
The data that support the findings of this study are available from the corresponding authors VT or LB, upon reasonable request.
References
Mandel, P. & Metais, P. Nuclear acids in human blood plasma. C R Seances Soc. Biol. Fil. 142, 241–243 (1948).
Stroun, M. et al. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 46, 318–322 (1989).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018).
Mathai, R. A. et al. Potential utility of liquid biopsy as a diagnostic and prognostic tool for the assessment of solid tumors: Implications in the precision oncology. J. Clin. Med. 8, 373 (2019).
Wang, Z. et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): A phase 2, single-arm, multicentre clinical trial. Lancet Respir. Med. 6, 681–690 (2018).
Bachet, J. B. et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 29, 1211–1219 (2018).
McCoach, C. E. et al. Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non-small cell lung cancer. Clin. Cancer Res. 24, 2758–2770 (2018).
Garrigou, S. et al. A study of hypermethylated circulating tumor DNA as a universal colorectal cancer biomarker. Clin. Chem. 62, 1129–1139 (2016).
Tie, J. et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 8, 219 (2016).
Cheng, F.T.-F. et al. Liquid biopsy detects relapse five months earlier than regular clinical follow-up and guides targeted treatment in breast cancer. Case Rep. Oncol. Med. 2019, 1–4 (2019).
Postel, M., Roosen, A., Laurent-Puig, P., Taly, V. & Wang-Renault, S.-F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 18, 7–17 (2018).
Pécuchet, N. et al. Analysis of base-position error rate of next-generation sequencing to detect tumor mutations in circulating DNA. Clin. Chem. 62, 1492–1503 (2016).
Aucamp, J., Bronkhorst, A. J., Badenhorst, C. P. S. & Pretorius, P. J. The diverse origins of circulating cell-free DNA in the human body: A critical re-evaluation of the literature. Biol. Rev. 93, 1649–1683 (2018).
Underhill, H. R. et al. Fragment length of circulating tumor DNA. PLoS Genet. 12, e1006162 (2016).
Thierry, A. R. et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 38, 6159–6175 (2010).
Mouliere, F. et al. high fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 6, e23418 (2011).
Poulet, G. et al. Characterization of plasma cell-free DNA integrity using droplet-based digital PCR: Towards the development of circulating tumor DNA-dedicated assays. Front. Oncol. 11, 639675 (2021).
Thierry, A. R., El Messaoudi, S., Gahan, P. B., Anker, P. & Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 35, 347–376 (2016).
Moss, J. et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 9, 1–12 (2018).
Strijker, M. et al. Circulating tumor DNA quantity is related to tumor volume and both predict survival in metastatic pancreatic ductal adenocarcinoma. Int. J. Cancer 146, 1445–1456 (2020).
Duvvuri, B. & Lood, C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. Front. Immunol. 10, 502 (2019).
Qi, Y. et al. Perioperative elevation in cell-free DNA levels in patients undergoing cardiac surgery: Possible contribution of neutrophil extracellular traps to perioperative renal dysfunction. Anesthesiol. Res. Pract. 2016, e2794364 (2016).
Salzano, A. et al. Circulating cell-free DNA levels are associated with adverse outcomes in heart failure: Testing liquid biopsy in heart failure. Eur. J. Prev. Cardiol. 28, e28–e31 (2021).
Stawski, R. et al. Repeated bouts of exhaustive exercise increase circulating cell free nuclear and mitochondrial DNA without development of tolerance in healthy men. PLoS ONE 12, e0178216 (2017).
Lo, Y. M. et al. Presence of fetal DNA in maternal plasma and serum. Lancet 350, 485–487 (1997).
Nishimoto, S. et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci. Adv. 2, e1501332 (2016).
McCabe, M. J. et al. Development and validation of a targeted gene sequencing panel for application to disparate cancers. Sci. Rep. 9, 17052 (2019).
Cimmino, F., Lasorsa, V. A., Vetrella, S., Iolascon, A. & Capasso, M. A targeted gene panel for circulating tumor DNA sequencing in neuroblastoma. Front. Oncol. 10, 596191 (2020).
Barault, L. et al. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut 67, 1995–2005 (2018).
Khier, S. & Lohan, L. Kinetics of circulating cell-free DNA for biomedical applications: critical appraisal of the literature. Future Sci. OA 4, FS0295 (2018).
Patke, A., Young, M. W. & Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 21, 67–84 (2020).
Koyanagi, S. Chrono-pharmaceutical approaches to optimize dosing regimens based on the circadian clock machinery. Biol. Pharm. Bull. 44(11), 1577–1584 (2021).
Rekker, K. et al. Circulating miR-200-family micro-RNAs have altered plasma levels in patients with endometriosis and vary with blood collection time. Fertil. Steril. 104(4), 938–946 (2015).
Heegaard NH, Carlsen AL, Lilje B, Ng KL, Rønne ME, Jørgensen HL, Sennels H, Fahrenkrug J. Diurnal Variations of Human Circulating Cell-Free Micro-RNA.PLoS One. 2016 Aug 5;11(8):e0160577.
Tóth, K. et al. Circadian rhythm of methylated septin 9, cell-free DNA amount and tumor markers in colorectal cancer patients. Pathol. Oncol. Res. 23(3), 699–706 (2017).
Diamantopoulou, Z. et al. The metastatic spread of breast cancer accelerates during sleep. Nature 607(7917), 156–162 (2022).
Nguyen, G. et al. Plasma soluble (pro)renin receptor is independent of plasma renin, prorenin, and aldosterone concentrations but is affected by ethnicity. Hypertension 63, 297–302 (2014).
Ivars, K. et al. Development of salivary cortisol circadian rhythm and reference intervals in full-term infants. PLoS ONE 10, e0129502 (2015).
Rostami, A. et al. Senescence, necrosis, and apoptosis govern circulating cell-free DNA release kinetics. Cell Rep. 31, 107830 (2020).
Andriamanampisoa, C.-L. et al. BIABooster: online DNA concentration and size profiling with a limit of detection of 10 fg/μL and application to high-sensitivity characterization of circulating cell-free DNA. Anal. Chem. 90, 3766–3774 (2018).
Baxmann, A. C. et al. Influence of muscle mass and physical activity on serum and urinary creatinine and serum cystatin C. Clin. J. Am. Soc. Nephrol. 3, 348–354 (2008).
Schøler, L. V. et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin. Cancer Res. 23, 5437–5445 (2017).
Pantel, K. & Alix-Panabières, C. Liquid biopsy and minimal residual disease: Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 16, 409–424 (2019).
Groot, V. P. et al. Circulating tumor DNA as a clinical test in resected pancreatic cancer. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-19-0197 (2019).
Heidrich, I., Ačkar, L., Mossahebi Mohammadi, P. & Pantel, K. Liquid biopsies: Potential and challenges. Int. J. Cancer 148, 528–545 (2021).
Ulz, P., Gerger, A., Belic, J. & Heitzer, E. Potentials, challenges and limitations of a molecular characterization of circulating tumor DNA for the management of cancer patients. J. Lab. Med. 40, 323–334 (2016).
Taïeb, J. et al. Decision for adjuvant treatment in stage II colon cancer based on circulating tumor DNA: The CIRCULATE-PRODIGE 70 trial. Dig. Liver Dis. 52, 730–733 (2020).
Meddeb, R., Pisareva, E. & Thierry, A. R. Guidelines for the preanalytical conditions for analyzing circulating cell-free DNA. Clin. Chem. 65, 623–633 (2019).
Lampignano, R., Neumann, M. H. D., Weber, S. & Heitzer, E. Multicenter evaluation of circulating cell-free DNA extraction and downstream analyses for the development of standardized (pre)analytical work flows. Clin. Chem. 65, 306837 (2019).
Dallongeville, J. et al. The plasma and lipoprotein triglyceride postprandial response to a carbohydrate tolerance test differs in lean and massively obese normolipidemic women. J. Nutr. 132, 2161–2166 (2002).
Meddeb, R. et al. Quantifying circulating cell-free DNA in humans. Sci. Rep. 9, 1–16 (2019).
Gardner, L. et al. The biomolecule corona of lipid nanoparticles contains circulating cell-free DNA. Nanoscale Horiz. 5, 1476–1486 (2020).
Benhaim, L. et al. Circulating tumor DNA is a prognostic marker of tumor recurrence in stage II and III colorectal cancer: multicentric, prospective cohort study (ALGECOLS). Eur. J. Cancer 159, 24–33 (2021).
Livergood, M. C., LeChien, K. A. & Trudell, A. S. Obesity and cell-free DNA ‘no calls’: Is there an optimal gestational age at time of sampling?. Am. J. Obstet. Gynecol. 216(413), e1-413.e9 (2017).
Hou, Y. et al. Factors affecting cell-free DNA fetal fraction: statistical analysis of 13,661 maternal plasmas for non-invasive prenatal screening. Hum. Genom. 13, 62 (2019).
Acknowledgements
The authors thank the Clinical investigation Center of the hospital Européen Georges-Pompidou, AP-HP for their collaboration in this work.
Funding
This work was supported by a joint grant from the Assistance Publique des Hôpitaux de Paris (Contrat de Recherche Clinique grant no. CRC06043), the INSERM-DGOS (Contrat de Recherche Translationnelle), the Agence Nationale pour la Recherche Physique et Chimie du Vivant, the Ministère de l’Enseignement Supérieur et de la Recherche, the Université de Paris, the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM), the Centre de Recherche des Cordeliers and the ligue nationale contre le cancer (LNCC, Program “Equipe labelisée LIGUE”; no. EL2016.LNCC). Geoffroy Poulet was supported by a CIFRE (N°2018/0368) funding from ANRT (Association Nationale Recherche Technologie) and Eurofins-Biomnis.
Author information
Authors and Affiliations
Contributions
The individual contributions for this work are: G.P.: Experimental work and analysis; Data acquision; Writing—Original Draft; Writing—Review & Editing. J.S.H.: Data acquision; Formal analysis; Investigation; Methodology; Resources; Data Curation; Writing—Original Draft; Writing—Review & Editing; Funding acquisition. A.B.: Formal analysis; Investigation; Methodology; Resources; Data Curation; Writing—Review & Editing. D.B.: Data acquision; Investigation; Methodology; Resources; Data Curation; Writing—Review & Editing. W.X.: Data interpretation; Writing—Review & Editing. F.G.: Data acquision; Experimental work and analysis; Writing—Review & Editing. A.B.: Data acquision; Experimental work and analysis; Writing—Review & Editing. G.B.: Experimental work and analysis; Writing—Original Draft; Writing—Review & Editing. V.G.: Supervision; Writing—Review & Editing. L.P.: Supervision; Writing—Review & Editing. M.A.: Investigation; Resources; Supervision; Funding acquisition; Writing—Review & Editing. P.L.P.: Conceptualization ; Methodology; Investigation ; Resources ; Supervision. L.B.: Formal analysis; Data Curation; Writing—Original Draft; Writing—Review & Editing. V.T.: Conceptualization; Methodology; Resources; Data Curation ; Writing—Original Draft; Writing—Review & Editing; Visualization; Supervision; Funding acquisition. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
VT: Honoraria from Raindance Technologies and Boerhinger Ingehleim; cofounder emulseo; board emulseo. PLP: Honoraria and board: Amgen, Merck-Serono, Boehringer Ingelheim, Sanofi, Roche, Lilly. AB: Honoraria and board: Amgen. JSH: speaker, advisory board or consultancy fees from Amgen, Astra Zeneca, Bayer, Bristol-Myers Squibb, Novartis, Novo Nordisk, Vifor Pharma, all unrelated to the present work. GP: Eurofins-Biomnis employee. PL: Eurofins-Biomnis employee. VG: Eurofins-Biomnis employee. AB (Dr Audrey Boutonnet): ADELIS employee. FG: ADELIS employee. The other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poulet, G., Hulot, JS., Blanchard, A. et al. Circadian rhythm and circulating cell-free DNA release on healthy subjects. Sci Rep 13, 21675 (2023). https://doi.org/10.1038/s41598-023-47851-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-47851-w
- Springer Nature Limited