
Abstract
The GRAS transcription factors are multifunctional proteins involved in various biological processes, encompassing plant growth, metabolism, and responses to both abiotic and biotic stresses. Wheat is an important cereal crop cultivated worldwide. However, no systematic study of the GRAS gene family and their functions under heat, drought, and salt stress tolerance and molecular dynamics modeling in wheat has been reported. In the present study, we identified the GRAS gene in Triticum aestivum through systematically performing gene structure analysis, chromosomal location, conserved motif, phylogenetic relationship, and expression patterns. A total of 177 GRAS genes were identified within the wheat genome. Based on phylogenetic analysis, these genes were categorically placed into 14 distinct subfamilies. Detailed analysis of the genetic architecture revealed that the majority of TaGRAS genes had no intronic regions. The expansion of the wheat GRAS gene family was proven to be influenced by both segmental and tandem duplication events. The study of collinearity events between TaGRAS and analogous orthologs from other plant species provided valuable insights into the evolution of the GRAS gene family in wheat. It is noteworthy that the promoter regions of TaGRAS genes consistently displayed an array of cis-acting elements that are associated with stress responses and hormone regulation. Additionally, we discovered 14 miRNAs that target key genes involved in three stress-responsive pathways in our study. Moreover, an assessment of RNA-seq data and qRT-PCR results revealed a significant increase in the expression of TaGRAS genes during abiotic stress. These findings highlight the crucial role of TaGRAS genes in mediating responses to different environmental stresses. Our research delved into the molecular dynamics and structural aspects of GRAS domain-DNA interactions, marking the first instance of such information being generated. Overall, the current findings contribute to our understanding of the organization of the GRAS genes in the wheat genome. Furthermore, we identified TaGRAS27 as a candidate gene for functional research, and to improve abiotic stress tolerance in the wheat by molecular breeding.
Similar content being viewed by others

Introduction
Wheat (Triticum aestivum L.) is an important cereal crop, contributing 20% of the daily caloric intake, is an rich source of protein and carbohydrates in the human diet. Grain yield is the most important economic parameter and is influenced by several biotic and abiotic factors. The demand for wheat is forecast to increase by 50% by 2050 (https://www.openaccessgovernment.org/demand-for-wheat/83189/). To cope with abiotic challenges such as salinity, drought, and heat, plants employ a variety of stress-responsive pathways and activated defense mechanisms1. Globally, these environmental constraints lead to average production losses of more than 50% in main crops2. To meet the food demands of the growing human population, breeding stress-tolerant varieties resistant to various environmental challenges, as well as improved quality and yield3 are future goals. Since traditional breeding has had little success in abiotic stress due to its multigenic nature and narrow genetic pool, using transgenic technology to develop stress-tolerant cultivars is a viable option. For the development of stress-tolerant plants, understanding the molecular mechanisms and exploring the stress-responsive genes that regulate plant responses to abiotic stresses4, 5. Transcription factors (TFs) are essential signaling elements in protecting plants from abiotic stress. Therefore, identifying relevant TFs is still essential for studying signaling pathways and abiotic stress. Translational genomics is a successful strategy to identify wheat genes.
After the first three functionally recognized members, gibberellic acid insensitive (GAI), a repressor of GAI (RGA), and a scarecrow (SCR), GRAS, a key TF gene family in plants, was verified6,7,8. The carboxyl terminus of GRAS proteins contains the highly conserved sequences LHR I, VHIID, LHR II, PFYRE, and SAW, which range in length from 400 to 770 amino acids9,10,11. These conserved motifs play an important role in protein function12, 13. For example, in Arabidopsis, mutations of the PFYRE and SAW motifs in the RGA and SLR1 regions cause major phenotypic alterations8, 12. GRAS proteins have more diverse amino-termini in length and sequence, implying functional specialisation11. By far, the GRAS gene family has been explored in several plant species11. Initially, based on shared features and sequence similarities, eight subfamilies of the GRAS gene family were identified: SCL3, SCR, SHR, LS, LISCL, PAT1, DELLA, and HAM. Each subfamily may have a unique function in a plant's physiological activities11. For example, the Arabidopsis SCR gene, which governs the radial form of the root, was the first GRAS member discovered6. Through the SCR/SHR complex, Arabidopsis SHR, a member of the GRAS gene family, is also necessary for root development13,14,15. AtSCL3 (SCARECROW-LIKE 3) has been demonstrated to positively control the gibberellin (GA) pathway and operate as a DELLA repressor, regulating GA homeostasis in root development. In comprehensive studies of their regulatory mechanisms, DELLA proteins were revealed to be repressors of GA signaling8. The members of the DELLA subfamily GAI, RGA, RGL1 (RGA-LIKE1), RGL2, and RGL3 can be distinguished from other GRAS proteins by the conserved DELLA motif in their N-terminal region. The PAT1 subfamily member linked to AtSCL13 is largely a positive regulator of phytochrome B (phyB) signaling, in contrast to other members of this subfamily, such as PAT1 and SCL21, which are involved in phytochrome A (phyA) signalling. Rice MONOCULM 1 (OsMOC1) expression in axillary buds is critical for regulating rice tillering16. Tomato (Ls) and Arabidopsis (LAS/SCL18) possess OsMOC1-like genes that are also important for the control of axillary meristem outgrowth17. BrLAS, a GRAS transcription factor from Brassica rapa L., is involved in drought stress tolerance in transgenic Arabidopsis18. Several GRAS members, including NSP1 and NSP2 (or their protein complex), are required for nodulation in Medicago truncatula Gaertn. as putative regulators of Nod-factor-inducible gene expression19, 20. Furthermore, NSP1 and NSP2 are necessary for strigolactone production in M. truncatula and Oryza sativa L.5. A recent study has shown that the GRAS transcription factors SCL6/SCL6-IV, SCL22/SCL6-III, and SCL27/SCL6-II are regulated by miRNA171 in Arabidopsis21,22,23. There are GRAS members with stress-related effects in Arabidopsis, cabbage (SCL13)24, rice (CIGR1 and CIGR2)25, tobacco (GRAS1)26, and poplar (SCL7)27, foxtail millet28, cucumber29. Researchers conducted a study to investigate the impact of abiotic stress on the DELLA gene, gibberellic acids (GA), and grain development in Sorghum bicolor (L.) Moench30. Similarly, studies on cucumber have shown that certain subfamilies such as SHR, SCR, and DELLA have a strong impact on the control of this species29. On the other hand , when studying cassava, scientists analysed how a specific gene called MeGRAS responds to various abiotic stresses, such as drought, salinity, cold stress, and exposure to hydrogen peroxide (H2O2)31. The findings of these studies strongly suggest that this gene plays diverse roles in cassava biology31.
Our study involved an extensive genome-wide study of 177 members of the wheat GRAS gene family. Identification of these GRAS genes was achieved through a multifaceted approach including their classification characterisation, expression profiling, and molecular dynamic simulations. To confirm the expression pattern obtained from in-silico analysis, we performed a quantitative real-time polymerase chain reaction. The findings strongly suggest that TaGRAS27 is a promising candidate for enhancing drought, heat, and salt tolerance through genome-editing techniques. The results of this study are important for the systematic elucidating of the functional roles played by the TaGRAS family genes in wheat.
Materials and methods
Identification and annotation of TaGRAS genes in T. aestivum
To identify the GRAS genes in wheat, the Phytozome database32 and the Rice Annotation Project (RAP) (https://rapdb.dna.afrc.go.jp/) was used to identify the GRAS protein sequences of related plant species, viz Arabidopsis thaliana (L.) Heynh., Brassica napus L., and Hordium vulgare L. The bread wheat proteome sequences (fp://fp.ensemblgenomes.org/pub/-plants/release-51/fasta/triticumaestivum/pep/) were used as the database for BLASTp33, while sequences from other species, such as A. thaliana, were used as query sequences O. sativa, B. napus and H. vulgare are used. These techniques were used to identify potential TaGRAS candidates. After eliminating duplicate results, the remaining sequences were scanned using HMMscan (https://www.ebi.ac.uk/Tools/hmmer/search/hmmscan), the SMART database34 (http://smart.embl-heidelberg. de/), NCBI CDD35 and pfam36 among others to confirm the GRAS domain (PF03514) (http://pfam.Sanger.Ac.United Kingdom/). Calculations of the number of amino acids, molecular weights (MW), and isoelectric points (pI) of each TaGRAS protein were also performed using ExPASy tools (http://www.expasy.ch/tools/pitool.html).
Phylogenetic analysis and classification of the TaGRAS gene family
Clustal W software was used to analyse all GRAS protein sequences from rice, Arabidopsis, brassica, and wheat37. Following that, an unrooted phylogenetic tree was built in MEGA-X with 1000 bootstrap repetitions using Maximum likelihood (ML) using default parameters e.g., used (JTT model) with uniform rates and 4 number of threads38. TaGRAS members in wheat were classified into subfamilies based on their Arabidopsis and rice homologues.
Using Blast2GO mapping (https://www.blast2go.com/) for molecular functions, biological processes and cellular components, it was possible to identify the functional annotation of the target genes. Metabolic pathways were annotated using maps from the KEGG database39.
Motif prediction and Gene structure
The MEME program was used for the conserved motif analysis of GRAS40, whose parameters were set as the standard. Annotation and visualization of identified motifs were performed using the TBtools41. The online tools Gene Structure Display Server (GSDS 2.0; http://gsds.gao-lab.org/)42 based on the CDS and matching to genetic sequences were also used to study the exon–intron patterns.
Chromosomal locations, Orthologous events, and interaction network analysis
Using the T. aestivum genome annotation dataset, the chromosomal locations of all TaGRAS members were verified43. To find the orthologous genes, wheat TaGRAS gene sequences were blasted by BLASTn33 using parameters e-value of 1e−10 to detect duplication of genes. Using MCScanX's default settings, the pattern of duplicated TaGRAS was categorized as segmental and tandem duplications44. A region of the chromosome that is smaller than 200 kb and has two or more genes is known as a tandem duplication45. However, synonymous (Ks) and non-synonymous (Ka) substitutions of each duplicated TaGRAS gene as well as synteny between interspecies were determined by TBtools41. The STRING v1054 databases (https://string-db.org/) were used to identify functional protein–protein interactions46. The GRAS protein sequences were input into the STRING46 program, and H. vulgare was used as the reference species to search in the database. With blast software set to an e-value of 1e−10, the H. vulgare genome was searched against all known interaction partners. The best-hit gene for each gene was chosen using Cytoscape58 to create a PPI network47. Finally, Cytoscape cyto-Hubba plugin was used to determine the top hub genes from the interaction network (Cytoscape Consortium 2016).
TaGRAS promoter analysis
Cis-elements are important in controlling plant growth and environmental adaptation. The sequences 1500 bp upstream of the start codon were extracted, used as a hypothetical promoter and then submitted to the online tool PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) to predict the cis-elements and identify the putative cis-elements in the promoter regions of TaGRAS48.
Prediction of miR genes targeting TaGRAS
The Ensembl plant database was used to find the transcript sequences of TaGRAS gene. Now the transcript sequences of TaGRAS and the matured miRNA sequences49 were examined using the default parameters of the psRNATarget service50, 51.
RNA-seq derived gene expression profiling
To identify the in-silico gene expression patterns across a range of tissues under control and stressed conditions, RNAseq data was utilised. We acquire transcripts per million (TPM) values for each TaGRAS from the expVIP database (http://www.wheat-expression.com/), and created heatmaps to visually represent these expression profiles using Clustvis 2.052 (https://biit.cs.ut.ee/clustvis/). Furthermore, we validate the expression of 20 GRAS genes identified by in-silico analysis under abiotic stress conditions using qRT-PCR as described in section "RNA isolation and real-time quantitative PCR analysis.".
Molecular modelling and molecular dynamics (MD) simulations
The three-dimensional structure was generated using the Rosettafold53 as it employs deep learning techniques for accurate prediction of protein structure without relying on homology. MD simulations were carried out for the modeled GRAS protein structures using the Desmond MD simulation package (release 2018) of Schrodinger (Desmond Research) to investigate the conformational changes of the protein with the solvent system. The OPLS_2005 force filed54 was used for the proteins. Utilizing the system builder module of Desmond, the proteins were solvated in the cubical water box (TIP3P water model) keeping a 10 Å distance between the box edge and protein atom. Systems were neutralized by the addition of counter ions and 0.15 M ionic concentration was maintained by the addition of Na+ and Cl ions. The minimization of the solvated built systems was performed with 10,000 steepest descent steps followed by gradual heating from 0 to 300 K, under Berendsen NVT ensemble55. Prior to the production run, the systems underwent heat relaxation for 5 ns using the Nose–Hoover Chain thermostat56 and Martyna-Tobias-Klein barostat method for maintaining temperature and pressure scale at 300 K and 1 atm. Lastly, a 20 ns production run was carried out for each system using a cut-off distance of 12 Å for non-bonded interactions. The coordinates of the simulations were recorded every 10 ps. The figures were rendered through UCSF chimera57.
Plant material and growth conditions
In the current study, seeds of six contrasting wheat genotypes (C306, WL711, RAJ3765, HS240, HD2009, and KRL213) were obtained from the Germplasm Unit of the ICAR-Indian Institute of Wheat and Barley Research in Karnal, India. Under controlled conditions, the seeds were germinated in Petri dishes at 22 °C after being sterilized with 1% sodium hypochlorite for 10 min and washed three times with distilled water. After five days of germination, seedlings were transferred to full-strength Hoagland's solution phyta-jars and incubated for 14 days in a BOD incubator with two sets of three biological replicates of each genotype. To study the effects of drought stress, two different wheat cultivars C306 and WL711-which are drought tolerant and susceptible, respectively, were used. At the seedlingstage, an osmotic stress using 20% (v/v) polyethylene glycol (PEG) 6000 was given after growing in Hoagland's solution for 14 days, and samples were collected at time intervals of 0 h, 3 h, 24 h, and 48 h. Unstressed seedlings served as the control58. For the expression study, leaf samples from control and stressed seedlings were also collected at the aforementioned intervals. Two contrast wheat genotypes RAJ3765 (heat tolerant) and HS240 (heat sensitive) were selected for heat stress. These plants were kept at a basal temperature of 37 °C for 3 h, and then kept at room temperature for 3 h, and finally 42 °C for 3 h (at Acquired). Leaf samples from the baseline and acquired stress levels were taken at the time interval mentioned above. For salt stress, two contrasting wheat genotypes HD2009 (salt sensitive) and KRL213 (salt tolerant) were used. Both genotypes at two leaves seedling stage were stressed with 150 mM NaCl. After the treatment, the leaf samples were taken at 0, 3, 24, and 48 h. For total RNA isolation, all acquired samples were immediately wrapped in foil and frozen in liquid nitrogen at − 80 °C.
RNA isolation and real-time quantitative PCR analysis
TRIzol reagent (Thermo Scientific, USA) was used to isolate the total RNA as per manufacturer's instructions. The extracted RNA was subjected to DNase I (NEB, USA) treatment. RNA integrity was checked by 1.2% native agarose gel electrophoresis and concentration was measured using the Nano-Drop ND-1000 spectrophotometer (Thermo Scientific, USA). Using Superscript-III reverse transcriptase (Thermo Scientific, USA), 1 µg of total RNA was converted into the first strand of cDNA. PCR reactions were carried out on BIO-RAD CFX96 Touch Real-Time PCR System (Bio-Rad, USA) in 10 µl reaction volumes, comprising 5 µl of 2 × SYBR Green Master Mix (Thermo Scientific, USA), 0.5 µl of each primer (0.5 µM) and 1 µl of the cDNA diluted to a ratio of 1:2. The primers were designed using the PrimerBlast tool59, with a final product size of approximately 150 base pairs (Supplementary Table S1). The primer efficiency was calculated by using the formula: E = 10 (-1/slope)60. The thermal cycling conditions for qRT-PCR was: 95 °C for 5 min, then 40 cycles of 94 °C for 15 s, 55 °C for 30 s and 72 °C for 30 s, and a final melting curve step from 65 °C to 95 °C with a ramp speed of 0.5 °C per 5 s. Actin, a housekeeping gene, was used as an internal control61 (F: 5′-GGAGAAGCTCGCTTACGTG-3′ & R: 5′-GGGCACCTGAACCTTTCTGAA-3′). The standard deviation was measured by averaging the threshold cycle values (Ct) of the experimental triplicates. The comparative 2∆∆−Ct method was used to determine the relative expression level of genes62.
Ethical approval
The use of plant parts in the study complies with institutional guidelines.
Results
Identification and annotation of TaGRAS genes in wheat
In total, we identified 177 wheat genes (Supplementary Table S2) belonging to the GRAS gene family, which were designated as TaGRAS1 to TaGRAS180. However, it should be noted that the sequence lengths of three genes, namely TaGRAS71, TaGRAS72, and TaGRAS103, were relatively short, posing challenges to the alignment procedure. Therefore, to facilitate the study, these genes were excluded from further analysis. As a result, our current investigation focused on the remaining 177 GRAS genes. For the comparative genomic evaluation, GRAS protein sequences were retrieved from the reference genomes of four different species (Supplementary Table S3) and 422 GRAS proteins were identified. These proteins were used to assess the expression of GRAS transcription factors that reflect the major evolutionary divergence of wheat species. The protein molecular weight (MW) of the identified GRAS family members ranged from 16.76596 kDa to 163.7962 kDa, and the isoelectric point (pI) ranged from 4.8 to 9.0. However, the theoretical pI values of 23 proteins were higher than 7, and most proteins contained acidic amino acids (Supplementary Table S4).
Analysis of phylogenetic tree of GRAS transcription factor
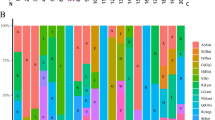
The phylogenetic tree was constructed based on the maximum likelihood method to analyse the evolutionary correlation of GRAS genes (Fig. 1). TaGRAS genes were classified into fourteen groups. Group-I (LISCL subfamily) comprised 61 members from the wheat, whereas 9 proteins were from Arabidopsis and 19, 15, and 19 were from rice, barley and oilseed rape, respectively. Group-II (HAM subfamily) consisted of 13, 7, 4, 6 and 13 GRAS proteins in wheat, rice, Arabidopsis, barley, and oilseed rape, respectively. Like the Arabidopsis GRAS proteins have distinct and necessary biological functions in the HAM subfamily that are required for maintaining the shoot apical meristem63. Whereas, 12 TaGRAS proteins from T. aestivum, belong to Group-III (SHR subfamily). However, in Group-IV, PAT1, consists of 23, 5, 5, 5, and 17 individuals from the different crops of wheat, rice, A. thaliana, barley, and oilseed rape, respectively. Like A. thaliana, three GRAS proteins (TaGRAS106, TaGRAS107, and TaGRAS108) may also play significant roles in the wheat phytochrome signal transduction pathways. Group-V includes three proteins TaGRAS65, TaGRAS66, and TaGRAS67, Group-VI of TaGRAS40, TaGRAS41 and TaGRAS42 and Group-VII of TaGRAS15, TaGRAS16, and TaGRAS17of wheat proteins (Fig. 1). Group-VIII includes the SCR protein, having two GRAS proteins from Arabidopsis (AthGRAS21 and AtGRAS29) and thirteen GRAS proteins from wheat. Group-IX and -X (RAM 1 and RAD subfamily) are basically involved in mycorrhizal signalling. Three wheat genes (TaGRAS43, TaGRAS44 and TaGRAS45) and one each in rice (OsGRAS50) and barley gene (HvGRAS8) were clustered in RAM1 subfamily. However, seven genes from wheat, two each from rice and barley were grouped in the RAD subfamily. Thirteen wheat proteins comprised of all DELLA proteins formed Group-XI. According to prior reports on Arabidopsis, the majority of these proteins including RGL2 (AtGRAS9), RGL3 (AtGRAS10), GAI (AtGRAS3), and RGA, are negative regulators of GA signalling (AtGRAS7)8, 64,65,66,67. The rest of the Groups-XII, XIII and XIV had 3, 6 and 14 genes, respectively.

Characterised sequences of five species (wheat, Arabidopsis, rice, barley and oilseed rape) were used to draw this phylogenetic tree (Maximum Likelihood) with the wheat GRAS proteins using MEGA X34. Combined phylogenetic analysis of GRAS proteins from T. aestivum (Ta), A. thaliana (At), O. sativa (Os), H. vulgare (Hv) and B. napus (Bn). The GRAS proteins are clustered into 14 subgroups, marked by different colours.
To assess the activities of the TaGRAS genes, we examined the enrichment of GO (Gene Ontology) and KEGG (Kyoto Encyclopaedia of Genes and Genomes) terms. The 177 TaGRAS family members were classified into three ontology categories based on GO enrichment analysis: biological process, cellular component, and molecular function (Supplementary Table S5). From these GO terms, 30 functional terms were then created (Supplementary Fig. 1). With 146 genes involved in biological regulation, cellular activities, and metabolic processes, we expected that the TaGRAS gene family in wheat is involved in many plant regulatory processes. In addition, 26 genes revealed putative transcription factor activity and binding roles.
Analysis of gene structures and conserved motifs
To investigate the properties of the GRAS genes, all of the TaGRAS genes were utilized to assess the distribution of exons, introns, and UTR (Supplementary Fig. 2). The analysis revealed that most TaGRAS genes were intron-less 72.8% (129 out of 177) particularly, all genes in Os19, DLT, HAM, NSP2, SCL4/7, and RAD were intron-less. However, DELLA and SHR subfamilies were intron-less except two genes each (TaGRAS30, TaGRAS32, and TaGRAS85, TaGRAS87, respectively). The LAS subfamily also has no intron genes except one gene (TaGRAS21), whereas, members of LISCL, PAT1, SCR, and RAM1 subfamilies showed numerous introns (Supplementary Fig. 2). Additionally, the intron of the SCL3 subfamily member TaGRAS3 was exceptionally lengthy, covering upto 1000 bp. In general, gene structure varied across members of the same subfamily, indicating gene family diversification. The conserved motifs for each GRAS protein family were discovered (Supplementary Fig. 3) and concluded that the majority of GRAS proteins in the same group had similar motifs. MEME was used to determine the LOGO of these protein motifs (Supplementary Table S6; Supplementary Fig. 4). The GRAS domain, a common structure, was found in the TaGRAS. To understand each group, fourteen motifs were identified and utilised. Members of the same group were highly similar as far as motif composition but differed from other groups. The C-terminal portion of the GRAS proteins appeared to be more conserved than the N-terminal, as evidenced by the likelihood motifs9.
Chromosome distribution of the GRAS transcription factor family
The physical location map of the TaGRAS was drawn based on the physical location information of the wheat genome. 177 TaGRAS genes were widely and irregularly distributed on the 21 chromosomes of T. aestivum (Fig. 2). Chromosomes 4A (n = 17, 9.6%), 4B (n = 16, 8.9%), and 4D (n = 16, 8.9%) each contained the majority of the GRAS transcription factors. By contrast, chromosome 2A, 2B contains 10 genes (5.6%) and 2D has 11 genes (6.11%). whereas many GRAS transcription factors were gathered at chromosomes 5A, 5B and 5D (13, 8, 7) respectively. However, 9, 8, and 8 GRAS transcription factors were also located on chromosomes 3A, 3B, and 3D, respectively. Chromosome 1A has 6 genes, chromosome 1B contains 8 genes and chromosome 1D has 9 genes (Fig. 2). The localization of the GRAS transcription factor was lower on chromosome 6 and 7.

Distribution of TaGRAS genes on 21 wheat (T. aestivum) chromosomes constructed using Mapchart38.
microRNA targeting GRAS transcription factors in wheat
Previously, it was found that three Arabidopsis GRAS genes were targeted by miRNA17168. We identified 10 TaGRAS genes targeted by miRNA171 in wheat (TaGRAS82, TaGRAS81, TaGRAS80, TaGRAS79, TaGRAS77, TaGRAS78, TaGRAS74, TaGRAS76, TaGRAS75, and TaGRAS73), which were located on various chromosomes e.g., 1A, 1B, 1D, 4A, 4B, 4D, 5A, 6A, 6B, 6D. 7A, and 7D. In our analysis, miRNA171 was complementary to the internal region of these twelve GRAS transcription factors. These proteins belong to the HAM subfamily, which is necessary for maintaining the shoot apical meristem. miRNA160 targets PAT1 subfamily (TaGRAS95, TaGRAS96 and TaGRAS97), basically participate in phytochrome signaling. miRNA164 targets four genes that participating in nodulation signalling pathways (Table1). However, miR399 targeted different GRAS subfamilies i.e., SHR, LISCL, DLT and DELLA. Whereas, miR167 and miR397-5p target the SCR subfamily and is involved in radial patterning in both embryonic roots and shoots.
Identification of orthologous and paralogous GRAS genes
Comparative analysis was utilized to evaluate the orthologous GRAS gene triplication in wheat and Aegilops tauschii. We discovered 85 orthologous gene pairs among all the GRAS proteins from wheat and A. tauschii. Contrarily, there were 124 GRAS orthologous gene pairs discovered between wheat and rice, 117 between wheat and Triticum dicoccoides, and just 8 between wheat and Arabidopsis (Supplementary Table S7). We discovered that each rice GRAS gene contained several wheat orthologs among the orthologous gene pairs, indicating that many GRAS transcription factors in wheat experienced duplication along with genome triplication. Since wheat and rice have a close lineage, there are more GRAS orthologous genes between the two than between wheat and Arabidopsis (Fig. 3). Additionally, in wheat, Arabidopsis, and rice, only 60, 7, and 22 paralogous GRAS gene pairs, respectively were found. Analysis of collinearity relationships can provide orthologous relationships between different species. GRAS gene pairings between the genomes of T. aestivum and A. thaliana were synthesized. The findings revealed that 16 T. aestivum GRAS genes shared syntenic relationships with AtGRAS genes (Fig. 3A), indicating that these genes may have aided in the development of the TaGRAS gene family. Similarly, Fig. 3B showed syntenic relationships with OsGRAS (Fig. 3B).

(a) Interspecies synteny of T. aestivum and A. thaliana (b) T. aestivum and O. sativa on the basis of orthologous genes. Gray lines in the background indicate the collinear blocks within T. estivum (Ta) and A. thaliana (At), T. aestivum (Ta) and O. sativa (Os) while the red lines highlight the syntenic GRAS gene pairs by using TBtools41.
However, Ks values, Ka values, Ka_Ks ratios, and divergence times of paralogous and orthologous GRAS family genes were calculated to assess the effectiveness of evolutionary constraints (Table 2). Ka_Ks ratios were generally greater than 1 for segmentally duplicated TaGRAS gene pairs. Moreover, segmental gene divergence occurred between 18.76 and 34.97 Mya. These results suggested that the TaGRAS gene family may have been the target of strong purifying selection during evolution.
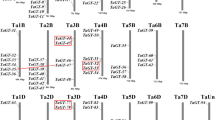
We analyzed nine segmental events on various chromosomes and one tandem duplication event on the same chromosome. The results showed that segmental duplication events were essential for the expansion of TaGRAS genes in the wheat genome and that certain TaGRAS genes may have been created through gene duplication. The tandem and segmental duplicated genes belong to LISCL, PAT1, DLT, and RAM1. We also investigated how frequently tandem duplications occur. This region contained 60 TaGRAS gene pairs, all of which were closely linked. The identities of these, however, were > 80%, indicating that they were incorporated into instances of tandem duplication. Since gene duplication has a significant influence on the creation of novel abilities and gene families, we examined the TaGRAS gene duplication events in the wheat genome. The paralogous gene pairs were used to draw the circos (Fig. 4).

The synteny analysis of TaGRAS family in T. aestivum. Different colours represent GRAS subfamilies on A, B and D sub-genome red lines indicate duplicated TaGRAS subfamily gene pairs on A sub-genome, green lines indicated on B sub-genome and blue represented D sub-genome. The chromosome number is indicated at the bottom of each chromosome by using TBtools41.
Analysis of promoter sequences in GRAS genes in wheat
One hundred and twelve cis-elements were identified in the ~ 1.5 kb region upstream of the transcription initiation site of the TaGRAS gene (Supplementary Fig. 5). Several key cis-elements including the ABRE (ABA-responsive element), MYB (drought–responsive) and LTRE (low temperatures) were found. It was found that cis-element types exist in the genes LISCl, PAT1, DLT, NSP2, and HAM subfamilies. Finally, the cis-elements investigation showed that separate subfamily genes may be controlled in diverse ways and that the majority of TaGRAS genes can respond to a range of environmental stresses.
Interaction network of TaGRAS proteins
In our study, we utilized homology analysis of GRAS proteins in wheat and Arabidopsis via the STRING database to create a protein interaction network for TaGRAS proteins. We identified four key hub genes, TaGRAS63, TaGRAS84, TaGRAS80, and TaGRAS89, based on their high degrees of interactions, specifically TaGRAS18, TaGRAS21, and TaGRAS5, respectively (Fig. 5). These hub genes play significant roles in various biological processes. TaGRAS63 encodes an SCR protein influencing mesophyll and bundle sheath cell development, as well as radial patterning in embryonic roots and shoots. TaGRAS80 is involved in the nodulation signalling pathway, impacting nodulation component production, root development, and interactions with other phytohormones. TaGRAS89, resembling a scarecrow protein with DNA binding transcription factor activity, resides in the nucleus, controlling transcription and gene expression. It interacts with transcriptional activators genes regulating root growth, stamen formation, cell expansion, and flowering time. Lastly, TaGRAS84 interacts with genes such as jasmonate O-methyltransferase (JMT), a key player in plant defence by converting jasmonate into methyl-jasmonate.

Interaction network among TaGRAS families in wheat. Specific protein interactions between GRAS transcription factors in wheat were determined using String42.
Expression Patterns of GRAS genes
To examine the expression profiles of wheat GRAS members in different tissues, we used the TPM values of all TaGRAS genes obtained from abiotic stress experiments and different developmental stages. The expression levels of different GRAS genes varied greatly within the same tissue (Supplementary Fig. 6). About one-tenth of the 177 GRAS genes were not expressed. In this study, five different tissues were obtained at three different developmental stages. Different TaGRAS subfamilies exhibit varying levels of induction in various tissues (Supplementary Fig. 6A). In the Z39 stage, GRAS genes such as TaGRAS27, TaGRAS28, TaGRAS106, TaGRAS110, TaGRAS111, and TaGRAS112 showed more than fivefold up-regulation. However, at the Z75 stage, the expression of genes TaGRAS27, TaGRAS28, TaGRAS98, TaGRAS100, TaGRAS104, TaGRAS106, TaGRAS107, TaGRAS108, TaGRAS176, and TaGRAS179 was up-regulated in leaf tissues. Notably, at the Z39 stage, some GRAS genes showed up-regulation. At the Z10 stage, the expression of 16 TaGRAS family genes increased more than threefold root tissues. This suggests that different TaGRAS genes may be involved in the development of various tissues at different stages. Many GRAS genes, including TaGRAS98, TaGRAS99, TaGRAS100, TaGRAS106, TaGRAS107, TaGRAS108, TaGRAS111, TaGRAS112, TaGRAS117, TaGRAS160, TaGRAS164, TaGRAS177, and TaGRAS179 showed induction at different stages of the wheat grain aleurone layer. Only a few members of the TaGRAS subfamily showed no induction at any of the several embryonic phases.
We also assessed in-silico expression analysis of GRAS genes in wheat tissues under abiotic stresses (Supplementary Fig. 6B). Across all examined tissues, GRAS genes from the DELLA, PAT1, and LISCL subfamilies exhibited high expression levels (log2-based values > 5), suggesting their potential role in tissue development based on their tissue-specific expression patterns. Notably, several genes similar to AtPAT1 showed strong expressions in leaves, aligning with AtPAT1's role as a positive regulator in the phyA signal pathway. Several TaGRAS genes showed constitutive expression levels during the majority of the phases of wheat growth. For instance, a > 5-folds increased level was observed for six genes (TaGRAS27, TaGRAS28, TaGRAS98, TaGRAS99, TaGRAS110, and TaGRAS112) under drought stress, as that of TaGRAS159, TaGRAS160, TaGRAS171, and TaGRAS179 for heat stress. However, TaGRAS179, TaGRAS160, TaGRAS159, and TaGRAS99 showed a considerable level of expression for both stresses. Other GRAS genes were active at low expression levels in a variety of tissues and at different developmental stages in wheat. These findings showed that TaGRAS genes exhibit a wide range of expression pattern, and that even genes belonging to the same subfamily have distinct expression patterns.
TaGRAS genes are involved in the response to abiotic stresses
Overall, many TaGRAS genes were significantly induced/repressed by the various forms of stresses as shown in Fig. 6. The expression levels of these genes changed over time or in different tissues depending on the specific treatments. The relative expression levels of six genes (TaGRAS8, TaGRAS27, TaGRAS53, TaGRAS54, TaGRAS98, and TaGRAS122) significantly showed their expression > tenfold under drought stress. However, the expression of nine genes (TaGRAS106, TaGRAS108, TaGRAS117, TaGRAS118, TaGRAS123, TaGRAS124, TaGRAS159, TaGRAS160, and TaGRAS176) were not obviously changed, followed by downregulation of TaGRAS29, TaGRAS63, TaGRAS84, TaGRAS99, and TaGRAS111 genes expression (Fig. 6A) in leaf tissues of wheat variety C306. Whereas in root tissues, 19 gene expression levels was more than two-fold. Likewise, the expression level of TaGRAS8, TaGRAS54 and TaGRAS159 was ~ 10-folds in leaf tissues followed by expression > threefold in five genes at different time intervals. The expression patterns in root tissue of WL711 showed that eight genes were expressed 15-folds while seven genes showed moderate expression at different time intervals followed by down-regulated of remaining genes (Supplementary Fig. 7).


qRT-PCR based differential expression analysis of 20 TaGRAS genes under abiotic stress. (A) drought stress at 20% (v/v) PEG treatment in C306 and WL711 for 24 h leaf tissues, (B) Salt stress treatment at 150 mM NaCl in Kharchia65 and HD2687 at 0 h, 24 h and 48 h leaf tissues, (C) Heat stress treatment in Raj3765 and HD2009 at 37 °C and 42 0C.
Under salt stress conditions, the expression level in leaf tissue of HD2009 at 0 h, three genes (TaGRAS8, TaGRAS98, and TaGRAS29) were expressed more than tenfold and three genes (TaGRAS27, TaGRAS99, and TaGRAS106) were expressed more than two folds (Fig. 6B). While KRL213 wheat six genes (TaGRAS8, TaGRAS27, TaGRAS29, TaGRAS98, TaGRAS99 and TaGRAS53) were expressed more than 10-folds. Further, the expression level in root tissues of HD2009 was shown to be higher only in TaGRAS27 and TaGRAS99 and five genes (TaGRAS8, TaGRAS29, TaGRAS98, TaGRAS106, and TaGRAS118) were moderate. Whereas, expression level in root tissue of KRL213 was found > 20-fold in TaGRAS8, TaGRAS27, and TaGRAS98 (Supplementary Fig. 8). After 3 h of stress, the expression in leaf tissue of HD2009 genotype was more than tenfold in four genes (TaGRAS8, TaGRAS29, TaGRAS53 and TaGRAS98). However, in KRL213, expression of TaGRAS27 was more than forty-five folds in leaf tissue followed by TaGRAS29 of ten folds. To check the GRAS gene expression in root tissue in HD2009 genotype, it was found that only three genes (TaGRAS8, TaGRAS27, and TaGRAS29) were expressed more than two folds. Whereas, expression of KRL213 root tissue was ~ 25-folds in TaGRAS29. At 24 h expression level was lower in HD2009 than KRL213 in leaf tissues. Whereas, the expression in root tissues two genes expressed more than 30-fold and five genes were expressed more than two fold in the HD2009 genotype. While in the root tissue of KRL213, only one gene (TaGRAS27) is expressed more 30-fold and three genes were expressed more than six folds. Expressions at 48 h in leaf tissue of HD2009 were found to be highly expressed in five genes (Fig. 6B) while it was low at KRL213. After that, the expression was calculated in the root tissues of HD2009 and only two genes (TaGRAS8 and TaGRAS27) showed moderate expression (Supplementary Fig. 8) as compared to that of KRL213 where three genes (TaGRAS8, TaGRAS27 and TaGRAS29) were expressed with ~ 15 folds after 48 h of stress. Apart from this, the expression of other genes was very low in both leaf and root tissues.
Similarly, GRAS gene expression under heat stress with two contrasting genotypes RAJ3765 (resistant variety) and HS277 (susceptible variety) was studied (Fig. 6C). To check the expression of GRAS genes in RAJ3765 under acquired conditions, six genes (TaGRAS54, TaGRAS98, TaGRAS99, TaGRAS122, TaGRAS124, and TaGRAS160) showed expression up to seven folds more while four genes (TaGRAS27, TaGRAS29, TaGRAS53, and TaGRAS123) were expressed more than fifteen folds. While in HS277 genotype, it was observed that three genes (TaGRAS27, TaGRAS29, and TaGRAS123) were found to express more than four folds and four genes (TaGRAS8, TaGRAS53, TaGRAS98, and TaGRAS99) were expressed more than one-folds. After that, we checked the GRAS gene expression in basal condition and the results showed that three genes (TaGRAS8, TaGRAS54, and TaGRAS159) were expressed more than ten folds, two genes (TaGRAS98 and TaGRAS160) were expressed more than twenty folds and three genes (TaGRAS27, TaGRAS29, and TaGRAS53) were expressed more than thirty folds. However, in expression level in HS277 under basal conditions, two genes were expressed more than ten folds, one gene expressed up to five folds, and five genes were expressed more than one-folds (Fig. 6C). Understanding the expression patterns of the TaGRAS gene about stress provides useful information for their functions in managing abiotic stress.
MD simulation analysis
The average change in displacement of a particular set of atoms for a given frame relative to a reference frame is calculated using the Root Mean Square Deviation (RMSD). Tracking the RMSD of the protein throughout the simulations can provide information about protein structural conformation. The RMSD number for the backbone atom of the protein concerning its initial positions increased to 7, 10, and 4 Å for GRAS126, GRAS151, and GRAS161, respectively. The large RMSD value for the GRAS126 and GRAS151 indicates these structures were undergoing large structural changes during simulations. Figure 7A showed that the simulation was equilibrated as the RMSD value stabilized ~ 18 ns which provide an appropriate basis for subsequent investigation.

MDS analysis (a) Root Mean Square Deviation, (b) Root Mean Square Fluctuations, (c) Ribbon shape superimposed structure of GARS_126, (d) GARS_161, (e) GARS_151 are shown, the orange colour structure is the initial phase of the system at 0 ns and the blue colour structure is the final phase of the system after 20 ns of Molecular Dynamics Simulation. GARS domain is shown in red and dark blue for the pre- and post MDS structures generated using the Rosettafold49.
The root mean square fluctuation (RMSF) provides insight into the local changes that occur during the MD simulations in the protein chain. The fluctuations of the major peaks in the RMSF graph were observed majorly at the N-and C-terminal of the proteins. However, no fluctuations were observed in the GRAS motifs in all the proteins. For GRAS151, the backbone residue position between Gly203-Ser206 varies from 6.4 to 7.7 Å, for GRAS126, (Gly396-Ser399) varies from 1.1 to 1.4 Å and for GRAS161, (Gly53-Ser56) it varies from 7 to 10 Å (Fig. 7a,b).
The initial and final frame structure of the proteins was superimposed using DALI server (http://ekhidna2.biocenter.helsinki.fi/dali/) and their deviations are measured in RMSD (Table 3). The secondary structure analysis shows changes in the frequencies, as the helix was found to decrease in the post-MD structure in all GRAS proteins. Furthermore, there was a slight decline in the sheet structure in the GRAS 126, GRAS161 and GRAS151 (Fig. 7c–e). Whereas, change in the secondary structure of GRAS domain was observed only in the GRAS161, (Gly53-Ser56Pre-MD, TTTG; Post-MD, TTTT) (Table 3; Fig. 7d).
Discussion
Plant genome sequencing has been used to investigate genes involved in various developmental stages and stress tolerance in a wide range of crops. Crops whose genomes have not been sequenced yet are receiving benefits from model plant genomes such as A. thaliana and O. sativa. Many studies have shown that transcription factors like the GRAS gene family play a crucial role in resisting abiotic, biotic, and individual plant growth development10, 69. However, the GRAS gene family analysis was previously reported in barley (53) cucumber (35), tomato (53), tea (52), soybean (117), M. sativa (51), foxtail millet (57), wheat (117), and cotton (15)70,71,72,73,74. Nevertheless, few studies have been conducted on wheat too73, 74. However, Liu et al.67 used transcriptomic data for in-silico analysis of 180 GRAS genes using IWGSC RefSeq 1.0 assembly, while Kumar et al.68 identified 183 GRAS genes in wheat using the Gramineae database. Both the studies lack in experimental validation of GRAS genes and protein–protein interaction. Here, we performed a comprehensive analysis of the 177 GRAS members in wheat using the latest assembly (IWGSC_v2.1), including their phylogenetic relationships, gene structure, conserved motifs, chromosomal positions, molecular dynamics simulation, and expression profile under abiotic stresses.
When compared to previously identified Arabidopsis (33 GRAS) and rice (60 GRAS) genes11, a fairly large number of these genes were discovered in wheat (177 GRAS). These findings may justify the fact that wheat has a large genome and has evolved with a higher ploidy level75. However, five highly conserved domains were shared with the majority of GRAS genes: LHRI, VHIID, LHRII, PFYRE, and SAW motif (Supplementary Fig. 4). These motifs have previously been shown to affect protein–protein and protein-DNA interactions13. The structural study revealed that the majority of the GRAS genes (72%) in the subfamilies (SHR, HAM1, LAS, and SCL4/7) were free from introns in line with studies conducted in Arabiddopsis, rice, Brassicsa, and Medicago5, 76. This exon–intron structure found in TaGRAS genes was comparable to those seen in other species77, 78. Intron genes are abundant in several large gene subfamilies (LISCL, PAT1, and SCR)79. Nonetheless, distinct exon–intron structures have formed in several GRAS genes, indicating that they have likely acquired new specialized roles to adapt to their environment. According to previous studies, the plant GRAS gene family evolved from a prokaryotic genome by horizontal gene transfer, followed by duplication events80. Many TaGRAS genes have major outliers with more than 5 introns, demonstrating the TaGRAS gene high degree of divergence. These increases and losses of GRAS might be consequences of chromosomal rearrangement and fusion, resulting in functional diversity across gene families. Introns can prolong genes and increase the frequency of gene combinations. However, intron-less genes lack these advantages in gene combination yet respond quickly to stress81, 82. As a result, some TaGRAS genes may respond rapidly to environmental signals. The findings in S. bicolor, B. napus, and M. truncatula81, 83 and the classifications of TaGRAS were comparable, but they were distinct from the reports of eight subgroups in tomato and Chinese cabbage84. LISCL had the most GRAS members, which was consistent with previous findings in Arabidopsis, rice, Populus78, sorghum28, and H. vulgare71, implying that the gene family may have strong differentiation abilities in the long-term evolution processes. TaGRAS classifications were further supported by their conserved motifs with close TaGRAS from the same subfamilies having similar motif compositions. Because GRAS transcription factors with diverse activities have been widely documented, it is worth noting that various occur in certain subgroups, hinting that they may have unique roles10, 69. For example, the DELLA domain, which is located at the N-terminus of members of the DELLA subfamily, may interact with the GA receptors to recognise GA signals85. Synteny analysis was also analyzed to assess the relationship between TaGRAS genes and their counterparts from A. thaliana, A. tauschii, T. diccoides, and O. sativa, representative of Brassicaceae and Poaceae families. The number of orthologous genes discovered between wheat and rice was the highest, suggesting a tight evolutionary relationship, following with T. diccoides, A. tauschii, and Arabidopsis. These genes might have originated from a common ancestor28. Furthermore, complex interactions such as single to many TaGRAS genes were discovered, showing that these T. aestivum members may play a significant role in TaGRAS gene evolution. Furthermore, we discovered that certain GRAS genes were only conserved in a few plants; a similar finding was also discovered in sorghum28. These findings may be linked to the evolutionary relationships of wheat and also other plant species. Large-scale duplication events, which occur before the divergence of some plant species, also played a crucial role in the expansion of the GRAS gene family.
The phylogenetic tree and cis-element analyses also provided additional evidence in favour of the possible functions of the TaGRAS genes in stress tolerance. Functional characterizations of GRAS genes have shown the conserved roles of probable orthologs in each subgroup84. Previous reports demonstrated that members of this group, PAT1 (AthGRAS6)86 and SCL13 (AthGRAS13), were involved in the phytochrome A and B signal transduction, respectively86, 87. Given that the SHR protein (TaGRAS83-TaGRAS94) from this group may alter root radial patterning and growth, the protein activities of this group may be connected to root development based on alignment with Arabidopsis14, 88. The Arabidopsis root/ meristem's cells must be maintained to allow for the plant tissue's indefinite expansion, and SCR, which is downstream of SHR, is necessary for this14.
SCL14, an Arabidopsis LISCL subgroup member, interacted with TGA-TFs, required for the activation of the stress-inducible promoters4. As a result, several genes in the LISCL subgroup, such as TaGRAS120, TaGRAS121, TaGRAS124, TaGRAS131, TaGRAS151, TaGRAS161 and TaGRAS176 were strongly stimulated by multiple stresses and therefore might be implicated in the regulation of stress response pathways (Fig. 5). Furthermore, various stress- and hormone-associated cis-elements including MBS, LTR, ABR, and TCA elements have been discovered in the promoter of most TaGRAS genes. The findings agreed with earlier studies of GRAS in B. juncea89, C. sativus29, and G. max90.
miRNAs are short noncoding RNAs that regulate cell function at the post-transcriptional and translocation mechanisms. This leads to the degradation of the gene target91. In this study, 14 miRNAs involving sequences of GRAS genes were discovered (Table 1). Among the predicted miRNAs, miR171 controls the expression of the HAM subfamily by directly targeting the HAMs mRNA92, 93. Overexpression of miR171 induced floral transition and spikelet morphological defects in various species, including rice and barley, comparable to Arabidopsis94, 95. This provides indirect evidence for the conservation of HAM and its upstream regulator miR171in crops such as barley. The likely targets of wheat miR171 were identified as TaGRAS82, TaGRAS81, TaGRAS80, TaGRAS79, TaGRAS77, TaGRAS78, TaGRAS74, TaGRAS76, TaGRAS75, and TaGRAS73.
The wheat TaGRAS genes are predicted to be involved in stress tolerance or growth and development through a complex protein-interaction network by the STRING database68. The conserved GRAS domain is essential for the dimerization of GRAS members and other proteins. In Arabidopsis, the homologous gene GAI from wheat TaGRAS27, TaGRAS28, TaGRAS29, and TaGRAS30 have been reported to be involved in reducing ROS accumulation in response to stress, and GAI may interact with several GRAS proteins, including PAT1 (TaGRAS106, TaGRAS108, and TaGRAS176), SCL3 (TaGRAS6, TaGRAS10, and TaGRAS14), and RGA1 (TaGRAS31, TaGRAS32, TaGRAS33 and TaGRAS34), indicating that their counterparts in wheat might tend to form similar proteins complexes. The role of GRAS transcription factor in modifying plant responses in a variety of adverse environmental conditions has been extensively documented67, 77, indicating that GRAS is an interesting candidate for enhancing plant stress tolerance through molecular breeding. OsGRAS23 over-expression, for example, increased rice drought and oxidative stress tolerance through modulating stress-responsive genes96, while PeSCL7 over-expression in Arabidopsis demonstrated drought and salt tolerance27. Tomatoes silenced by SiGRAS6 showed lower tolerance to drought stress97. The relevance of wheat GRAS genes in modulating stress response is currently unknown. In this study, transcriptome data and qRT-PCR results demonstrated that the majority of the identified TaGRAS genes exhibited significant differential expression under a variety of abiotic stressors, indicating that wheat TaGRAS genes can also play critical and diverse roles in response to environmental pollutants. For example, the expression of many TaGRAS genes, including TaGRAS106, TaGRAS108, and TaGRAS176, were significantly elevated. The findings show that these TaGRAS genes might be important players in the stress response. Previously, it was discovered that B. rapa GRAS TF BrGRAS is implicated in drought stress tolerance via an ABA-dependent signaling cascade18. Furthermore, at least two abiotic stresses simultaneously could be upregulated by the transcription of multiple TaGRAS genes indicating that they may have conserved functions in response to these stresses but additional experimental verification is needed.
Protein-DNA interactions are critical in translating genetic information to biological function. Because protein recognition of specific DNA sequences is very complicated, experimental attempts to predict how certain proteins interact with DNA are challenging. As a result, the use of time- and cost-effective computational approaches such as MD simulations and docking studies are essential at this point to accelerate knowledge recovery and limit the search process for experimental protocols. The results of three predicted GRAS proteins using MD simulations revealed complete dynamic and structural information about GRAS domain-DNA interactions. However, there is no structural base information reported to date for GRAS variants and their mechanism of interaction with DNA. However, except of one GRAS protein (GRAS161), there was no change in the protein secondary structure and the GRAS motif did not vary significantly across all three GRAS protein studied. Furthermore, the current results of stable structures will pave the way for researchers to investigate the interaction between the GRAS domain and DNA and discover critical residues involved in maintaining the interaction. Our findings showed the importance of selecting a sequence to develop newer transgenic plants that would be increasingly tolerant to stress conditions.
Conclusion
177 wheat GRAS genes were identified and phylogenetically divided into 14 subfamilies. We discovered that tandem and segmental duplication played a role in the growth of the TaGRAS family. We also discovered that miRNA171, which had previously been reported to have a regulatory function in GRAS member expression, has target genes in T. aestivum, all belonging to the HAM subgroup. Finally, qRT-PCR expression results demonstrated that GRAS member’s interacted during response against drought, salt, and heat stress. TaGRAS27 might be useful for abiotic stress tolerance for breeding in wheat. This interaction study between the GRAS domain and DNA will identify the key residues important for stabilising the interaction. These findings were critical in understanding the molecular and evolutionary processes of GRAS-mediated plant growth development in wheat.
Data availability
All data generated or analysed during this study are included in this published article [supplementary files].
References
Huang, W., Xian, Z., Kang, X., Tang, N. & Li, Z. Genome-wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biol. 15, 1–18 (2015).
Ceccon, E. Plant productivity and environment. Science 218, 443–448 (1982).
Guttikonda, S. K. et al. Overexpression of AtDREB1D transcription factor improves drought tolerance in soybean. Mol. Biol. Rep. 41, 7995–8008 (2014).
Fode, B., Siemsen, T., Thurow, C., Weigel, R. & Gatz, C. The arabidopsis GRAS protein SCL14 interacts with class II TGA transcription factors and is essential for the activation of stress-inducible promoters. Plant Cell 20, 3122–3135 (2008).
Liu, W. et al. Strigolactone biosynthesis in Medicago truncatula and rice requires the symbiotic GRAS-type transcription factors NSP1 and NSP2. Plant Cell 23, 3853–3865 (2011).
Di Laurenzio, L. et al. The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell 86, 423–433 (1996).
Peng, J. et al. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev. 11, 3194–3205 (1997).
Silverstone, A. L., Ciampaglio, C. N. & Sun, T. P. The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell 10, 155–169 (1998).
Pysh, L. D., Wysocka-Diller, J. W., Camilleri, C., Bouchez, D. & Benfey, P. N. The GRAS gene family in Arabidopsis: Sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 18, 111–119 (1999).
Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 218, 683–692 (2004).
Tian, C., Wan, P., Sun, S., Li, J. & Chen, M. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 54, 519–532 (2004).
Itoh, H., Ueguchi-Tanaka, M., Sato, Y., Ashikari, M. & Matsuoka, M. The gibberellin signaling pathway is regulated by the appearance and disappearance of slender rice1 in nuclei. Plant Cell 14, 57–70 (2002).
Li, S. et al. Crystal structure of the GRAS domain of SCARECROW-LIKE7 in oryza sativa. Plant Cell 28, 1025–1034 (2016).
Helariutta, Y. et al. The SHORT-ROOT gene controls radial patterning of the Arabidopsis root through radial signaling. Cell 101, 555–567 (2000).
Cui, H. et al. An evolutionarily conserved mechanism delimiting SHR movement defines a single layer of endodermis in plants. Science 316, 421–425 (2007).
Li, X. et al. Control of tillering in rice. Nature 422, 618–621 (2003).
Greb, T. et al. Molecular analysis of the LATERAL SUPPRESSOR gene in Arabidopsis. Genes Dev. 17, 1175–1187 (2003).
Li, P. et al. BrLAS, a GRAS transcription factor from brassica rapa, is involved in drought stress tolerance in transgenic arabidopsis. Front. Plant Sci. 871, 1–15 (2018).
Kaló, P. et al. Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science 80(308), 1786–1789 (2005).
Hirsch, S. et al. GRAS Proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula. Plant Cell 21, 545–557 (2009).
Schulze, S., Schäfer, B. N., Parizotto, E. A., Voinnet, O. & Theres, K. LOST MERISTEMS genes regulate cell differentiation of central zone descendants in Arabidopsis shoot meristems. Plant J. 64, 668–678 (2010).
Engstrom, E. M. et al. Arabidopsis homologs of the petunia HAIRY MERISTEM gene are required for maintenance of shoot and root indeterminacy. Plant Physiol. 155, 735–750 (2011).
Ma, Z. et al. Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 10, 20–21 (2014).
Park, H. J. et al. Use of heat stress responsive gene expression levels for early selection of heat tolerant cabbage (Brassica oleracea L.). Int. J. Mol. Sci. 14, 11871–11894 (2013).
Day, R. B., Shibuya, N. & Minami, E. Identification and characterization of two new members of the GRAS gene family in rice responsive to N-acetyl chitooligosaccharide elicitor. Biochim. Biophys. Acta 1625, 261–268 (2003).
Czikkel, B. E. & Maxwell, D. P. NtGRAS1, a novel stress-induced member of the GRAS family in tobacco, localizes to the nucleus. J. Plant Physiol. 164, 1220–1230 (2007).
Ma, H. S., Liang, D., Shuai, P., Xia, X. L. & Yin, W. L. The salt-and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J. Exp. Bot. 61, 4011–4019 (2010).
Fan, Y. et al. Genome-wide investigation of the GRAS transcription factor family in foxtail millet (Setaria italica L.). BMC Plant Biol. 21, 1–19 (2021).
Li, C. et al. Genome-wide characterization of cucumber (Cucumis sativus L.) GRAS genes and their response to various abiotic stresses. Horticulturae 6, 1–18 (2020).
Liu, J., Wu, Y., Dong, G., Zhu, G. & Zhou, G. Progress of research on the physiology and molecular regulation of sorghum growth under salt stress by gibberellin. Int. J. Mol. Sci. 24, 6777 (2023).
Shan, Z. et al. Genome-wide identification and expression of GRAS gene family members in cassava. BMC Plant Biol. 20, 1–16 (2020).
Goodstein, D. M. et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 40, 1178–1186 (2012).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 1–9 (2009).
Letunic, I., Doerks, T. & Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, 302–305 (2012).
Marchler-Bauer, A. et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, 225–229 (2011).
Finn, R. D. et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Bailey, T. L. et al. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 37, 1–7 (2009).
Chen, C. et al. TBtools: An Integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Hu, B. et al. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 31, 1296–1297 (2015).
Voorrips, R. E. Mapchart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 1–14 (2012).
Houb, E. B. The arms race is ancient history in Arabidopsis, the wildflower. Nat. Rev. Genet. 2, 516–527 (2001).
Szklarczyk, D. et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Shannon, P. et al. Cytoscape: A software environment for integrated models. Genome Res. 13, 426 (1971).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327 (2002).
Kozomara, A. & Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1181 (2014).
Dai, X. & Zhao, P. X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr319 (2011).
Katiyar, A. et al. Identification of novel drought-responsive microRNAs and trans-acting siRNAs from Sorghum bicolor (L.) Moench by high-throughput sequencing analysis. Front. Plant Sci. https://doi.org/10.3389/fpls.2015.00506 (2015).
Metsalu, T. & Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570 (2015).
Beak, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Yearb. Paediatr. Endocrinol. 373, 871–876 (2022).
Dubay, K. H. et al. Accurate force field development for modeling conjugated polymers. J. Chem. Theory Comput. 8, 4556–4569 (2012).
Victor, R. Berendsen and Nose-Hoover thermostats Temperature in MD MD at constant Temperature: NVT ensemble. Unknown 1–4 (2007).
Martyna, G. J., Klein, M. L. & Tuckerman, M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643 (1992).
Pettersen, E. F. et al. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Kaur, A., Gupta, O. P., Meena, N. L., Grewal, A. & Sharma, P. Comparative temporal expression analysis of micrornas and their target genes in contrasting wheat genotypes during osmotic stress. Appl. Biochem. Biotechnol. 181, 613–626 (2017).
Ye, J. et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134. https://doi.org/10.1186/1471-2105-12-134 (2012).
Sreedharan, S. P. et al. Primer design and amplification efficiencies are crucial for reliability of quantitative PCR studies of caffeine biosynthetic N-methyltransferases in coffee. 3 Biotech 8, 1–8. https://doi.org/10.1007/s13205-018-1487-5 (2018).
Wei, L. et al. Abscisic acid enhances tolerance of wheat seedlings to drought and regulates transcript levels of genes encoding ascorbate-glutathione biosynthesis. Front Plant Sci. 6, 1–112015. https://doi.org/10.3389/fpls.2015.004588 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods https://doi.org/10.1006/meth.2001.1262 (2001).
Geng, Y. & Zhou, Y. HAM gene family and shoot meristem development. Front. Plant Sci. 12, 1–6 (2021).
Wen, C. K. & Chang, C. Arabidopsis RGL1 encodes a negative regulator of gibberellin responses. Plant Cell 14, 87–100 (2002).
Dill, A., Jung, H. S. & Sun, T. P. The DELLA motif is essential for gibberellin-induced degradation of RGA. Proc. Natl. Acad. Sci. U. S. A. 98, 14162–14167 (2001).
Lee, S. et al. Gibberellin regulates Arabidopsis seed germination via RGL2, a GAI/RGA-like gene whose expression is up-regulated following imbibition. Genes Dev. 16, 646–658 (2002).
Piskurewicz, U. & Lopez-Molina, L. The GA-signaling repressor RGL3 represses testa rupture in response to changes in GA and ABA levels. Plant Signal. Behav. 4, 63–65 (2009).
Llave, C., Xie, Z., Kasschau, K. D. & Carrington, J. C. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297, 2053–2056 (2002).
Hakoshima, T. Structural basis of the specific interactions of GRAS family proteins. FEBS Lett. 592, 489–501 (2018).
Zhang, H. et al. Genome-wide identification of gras gene family and their responses to abiotic stress in Medicago sativa. Int. J. Mol. Sci. 22, 7729 (2021).
To, V. T. et al. Genome-wide analysis of the GRAS gene family in barley (Hordeum vulgare L.). Genes 11, 1–22 (2020).
Wang, T. T. et al. Genome-wide analysis of the gras gene family and functional identification of GmGRAS37 in drought and salt tolerance. Front. Plant Sci. 11, 604690 (2020).
Liu, Y. & Wang, W. Characterization of the GRAS gene family reveals their contribution to the high adaptability of wheat. PeerJ 9, e10811 (2021).
Kumar, B. & Bhalothia, P. Evolutionary analysis of GRAS gene family for functional and structural insights into hexaploid bread wheat (Triticum aestivum). J. Biosci. 46, 45 (2021).
Walkowiak, S. et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 588, 277–283 (2020).
Song, X. M. et al. Genome-wide analysis of the GRAS gene family in chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 103, 135–146 (2014).
Wang, Z. et al. GRAS-domain transcription factor PAT1 regulates jasmonic acid biosynthesis in grape cold stress response. Plant Physiol. 186, 1660–1678 (2021).
Liu, X. & Widmer, A. Genome-wide comparative analysis of the GRAS gene family in populus, arabidopsis and rice. Plant Mol. Biol. Rep. 32, 1129–1145 (2014).
Jain, M., Khurana, P., Tyagi, A. K. & Khurana, J. P. Genome-wide analysis of intronless genes in rice and Arabidopsis. Funct. Integr. Genom. 8, 69–78 (2008).
He, S., Zou, M. & Guo, B. The roles and evolutionary patterns of intronless genes in deuterostomes. Comp. Funct. Genom. 2011, 1–8 (2011).
Fan, Y. et al. Genome-wide identification and expression analysis of the bHLH transcription factor family and its response to abiotic stress in foxtail millet (Setaria italica L.). BMC Genom. 22, 1–21 (2021).
Shabalina, S. A. et al. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 27, 1745–1749 (2010).
Liu, M. et al. A multilayered cross-species analysis of GRAS transcription factors uncovered their functional networks in plant adaptation to the environment. J. Adv. Res. 29, 191–205 (2021).
Guo, P. et al. Genome-wide survey and expression analyses of the GRAS gene family in Brassica napus reveals their roles in root development and stress response. Planta 250, 1051–1072 (2019).
Murase, K., Hirano, Y., Sun, T. P. & Hakoshima, T. Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 456, 459–463 (2008).
Bolle, C., Koncz, C. & Chua, N. H. PAT1, a new member of the GRAS family, is involved in phytochrome A signal transduction. Genes Dev. 14, 1269–1278 (2000).
Torres-Galea, P., Huang, L. F., Chua, N. H. & Bolle, C. The GRAS protein SCL13 is a positive regulator of phytochrome-dependent red light signaling, but can also modulate phytochrome A responses. Mol. Genet. Genom. 276, 13–30 (2006).
Miyashima, S., Hashimoto, T. & Nakajima, K. ARGONAUTE1 acts in arabidopsis root radial pattern formation independently of the SHR/SCR pathway. Plant Cell Physiol. 50, 626–634 (2009).
Li, M. et al. Identification of the GRAS gene family in the Brassica juncea genome provides insight into its role in stem swelling in stem mustard. PeerJ 7, e6682 (2019).
Wang, L., Yang, X., Gao, Y. & Yang, S. Genome-wide identification and characterization of tale superfamily genes in soybean (Glycine max L.). Int. J. Mol. Sci. 22, 1–21 (2021).
Liu, S. R., Zhou, J. J., Hu, C. G., Wei, C. L. & Zhang, J. Z. MicroRNA-mediated gene silencing in plant defense and viral counter-defense. Front. Microbiol. 8, 1–12 (2017).
Han, H. et al. A signal cascade originated from epidermis defines apical-basal patterning of Arabidopsis shoot apical meristems. Nat. Commun. 11, 1–17 (2020).
Huang, W. et al. Overexpression of a tomato miR171 target gene SlGRAS24 impacts multiple agronomical traits via regulating gibberellin and auxin homeostasis. Plant Biotechnol. J. 15, 472–488 (2017).
Curaba, J., Talbot, M., Li, Z. & Helliwell, C. Over-expression of microRNA171 affects phase transitions and floral meristem determinancy in barley. BMC Plant Biol. 13, 1–10 (2013).
Fan, T. et al. Rice osa-miR171c mediates phase change from vegetative to reproductive development and shoot apical meristem maintenance by repressing four OsHAM transcription factors. PLoS ONE 10, 1–24 (2015).
Xu, K. et al. OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol. 15, 1–13 (2015).
Mayrose, M., Ekengren, S. K., Melech-Bonfil, S., Martin, G. B. & Sessa, G. A novel link between tomato GRAS genes, plant disease resistance and mechanical stress response. Mol. Plant Pathol. 7, 593–604 (2006).
Acknowledgements
This work was supported by Indian Council of Agricultural Research -Core Grant, New Delhi, India. We are thankful to ICAR-Indian Institute of Wheat and Barley Research for providing the facility and Germplasm Unit for supplying the seeds of wheat genotypes.
Author information
Authors and Affiliations
Contributions
P.S. conceived the study. S.M. and P.S. analysed the data, B.P., analysed the MD simulation part. S.M., P.S. wrote the first draft. P.S., R.C., G.S. edited the manuscript. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mishra, S., Chaudhary, R., Pandey, B. et al. Genome-wide identification and expression analysis of the GRAS gene family under abiotic stresses in wheat (Triticum aestivum L.). Sci Rep 13, 18705 (2023). https://doi.org/10.1038/s41598-023-45051-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45051-0
- Springer Nature Limited