
Abstract
POU4F3, a member of the POU family of transcription factors, commonly causes autosomal dominant deafness. Exome sequencing was used to identify four novel variants in POU4F3 (NM_002700.2), including c.564dupA: p.Ala189SerfsTer26, c.743T > C:p.Leu248Pro, c.879C > A:p.Phe293Leu, and c.952G > A:p.Val318Met, and diverse aspects of the molecular consequences of their protein expression, stability, subcellular localization, and transcriptional activity were investigated. The expression of three mutant proteins, encoded by missense variants, was reduced compared to the wild-type protein, demonstrating that the mutants were unstable and vulnerable to degradation. Additionally, all the mutant proteins had distinct subcellular localization patterns. A mutant protein carrying p.Ala189SerfsTer26, in which both mono- and bi-partite nuclear localization signals were disrupted, showed abnormal subcellular localization. Resultantly, all the mutant proteins significantly reduced the transcriptional activity required to regulate the downstream target gene expression. Furthermore, we identified the altered expression of 14 downstream target genes associated with inner ear development using patient-derived lymphoblastoid cell lines. There was a significant correlation of the expression profile between patient-derived cells and the cochlear hair cells, which provided a breakthrough for cases where the collection of human cochlear samples for transcriptome studies was unfeasible. This study expanded the genotypic spectrum of POU4F3 in DFNA15, and further refined the molecular mechanisms underlying POU4F3-associated DFNA15.
Similar content being viewed by others


Introduction
POU4F3 (NM_002700; NP_002691) comprises two exons located on chromosome 5q321, encoding a member of the POU family of transcription factors with two highly conserved POU domains: a POU-specific domain and a POU homeodomain2. These domains bind a target DNA octamer that displays a conserved consensus sequence2. The function of POU4F3 as a transcriptional regulator depends on the intact function of both POU domains2,3. Specifically, POU4F3 protein is highly expressed in cochlear and vestibular hair cells in the inner ear, where it is involved in the maturation, differentiation, and survival of these cells4,5. It was recently reported that POU4F3 heterozygous knock-in (Pou4f3Δ/+) and knock-out (Pou4f3−/+) mice developed progressive hearing loss concomitant with the apparent loss of outer hair cells, mirroring the human dominant form of deafness (DFNA15)6. In humans, POU4F3 is one of the most common autosomal-dominant deafness-associated genes, variants of which cause post-lingual onset, progressive non-syndromic hearing loss (NSHL)7,8,9,10,11,12,13. Previous studies suggested that POU4F3 is one of the signature gene associated with mid-frequency hearing loss, showing a down-sloping configuration over time10,12. More than 30 POU4F3 heterozygous variants, mostly in their DNA-binding POU domains, have been reported to cause DFNA15 in the literature, with the phenotypic variability in terms of age of onset, severity, and audiogram configuration7,8,9,10,11,12,13. Nonetheless, evidence on genotype–phenotype correlation and molecular mechanisms remains limited, despite the clinical and scientific importance of POU4F3 hair-cell specific transcriptional factor in the context of NSHL.
In this study, we identified four novel POU4F3 variants segregating as a dominant trait in four unrelated Korean families. We characterize all four novel POU4F3 variants functionally via computational structural modeling and functional studies, revealing diverse pathogenic mechanisms underlying POU4F3-associated hearing loss. Furthermore, we investigate whether POU4F3 variants can impact the expression of downstream target genes, potentially affecting inner ear development, using patient-derived cell lines.
Results
Identification and classification of novel POU4F3 variants
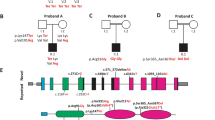
Exome sequencing revealed four novel POU4F3 variants (NM_002700.2) not previously reported in the literature, segregating as a dominant trait in the four unrelated Korean families: one frameshift variant produced a premature termination codon in the POU-specific domain (c.564dupA:p.Ala189SerfsTer26) that lacked both mono- and bi-partite nuclear localization signals (NLSs); two missense variants (c.743T > C:p.Leu248Pro and c.879C > A:p.Phe293Leu) in which the alterations were located within the POU-specific domain and POU homeodomain, respectively, but outside the NLSs; and one missense variant (c.952G > A:p.Val318Met) in which the alteration was located within the bipartite NLS (Fig. 1). Co-segregation of the variants with the phenotypes of the family members, including both parents, was confirmed. The novel frameshift variant (p.Ala189SerfsTer26) produced a truncated protein, which was predicted to escape the nonsense-mediated mRNA decay (NMD). The variant was not present in any population databases. The Ala189 residue produced by POU4F3 orthologs across various species is well preserved (http://genome.ucsc.edu/) (Fig. 1). Furthermore, in silico analyses, including combined annotation dependent depletion (CADD) phred (https://cadd.gs.washington.edu/) and rare exome variant ensemble learner (REVEL) (https://sites.google.com/site/revelgenomics/) scores, consistently indicated that this variant is disease-causing. Next, a novel missense variant (p.Leu248Pro), located in the C-terminus of the POU-specific domain, was extremely rare, meeting the PM2 criteria as per the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines for hearing loss. Moreover, the remaining two novel missense variants (p.Phe293Leu and p.Val318Met), located in the POU-specific domain and POU homeodomain, respectively, were absent from population databases. These three missense variants were consistently predicted to be disease-causing by in silico analyses, as evidenced by CADD phred and REVEL scores, and the corresponding residues in proteins encoded by POU4F3 orthologs between several species are highly conserved. Accordingly, based on ACMG/AMP guidelines for hearing loss, our novel POU4F3 variants were classified as “pathogenic” (p.Ala189SerfsTer26) and “variant of uncertain significance” (p.Leu248Pro, p.Phe293Leu, and p.Val318Met) (Table 1).

Four novel POU4F3 variants within the functional DNA-binding domains. Two (p.Ala189SerfsTer26 in SB218 and p.Leu248Pro in SB307) were located in the POU-specific domain, while the remaining two (p.Phe293Leu in SB438 and p.Val318Met in SB347) were in the POU-homeodomain. Conservation of the affected residues among species was documented for all POU4F3 variants identified in the study.
Clinical phenotypes
Diverse audiological phenotypes, in terms of degree of severity and audiogram configuration, were observed (Fig. 2). In family SB218, proband SB218–423 (32 years old at ascertainment) initially demonstrated symmetrical moderate to severe NSHL, including predominant mid-frequency hearing loss (symmetric notch at 1 kHz up to 60 dB HL in both ears), with a down-sloping configuration (Fig. 2a). Affected individuals in this family, including the proband’s father and grandfather, had experienced progressive NSHL since their late 20 s. Specifically, the proband's father planned to undergo cochlear implantation due to progressive deterioration of hearing. In family SB307, proband SB307–610 (29 years old at ascertainment) initially demonstrated symmetrical moderate-to-severe NSHL, including a predominant mid- and high-frequency hearing loss, with a down-sloping configuration in the left ear (Fig. 2b). The affected individual (SB307–610) received a middle ear implant, but the hearing loss gradually progressed. Hearing status of the proband’s father was allegedly poor but had not been documented. In family SB438, proband SB438–852 (24 years old at ascertainment) initially demonstrated mild to moderate NSHL, including predominant mid-frequency hearing loss in the right ear (at 1 kHz up to 50 dB HL), with a down-sloping configuration in the left ear (Fig. 2c). She was using hearing aids because of gradually progressive hearing deterioration at middle and higher frequencies. In family SB347, proband SB347–679 (35 years old at ascertainment) unexpectedly demonstrated severe to profound NSHL (more than 70 dB HL across all frequencies), with a flat configuration in the both ears (Fig. 2d). The age of onset was the late teens. Over a 10-year follow-up period, SB347–679 experienced rapid progressive deterioration of bilateral hearing across all frequencies, as well as a decrease in speech discrimination scores. She underwent bilateral cochlear implantation using a slim modiolar electrode (CI632; Cochlear, Australia) via a round-window approach. Compared to the preoperative speech evaluation, speech perception scores improved significantly by 3 and 6 months postoperatively, and the effects were longitudinally sustained. The proband's mother had also experienced progressive bilateral hearing loss since the age of 20 years, and had undergone cochlear implantation 15 years previously. Medical histories and general physical examinations of all affected individuals were otherwise unremarkable. Specifically, none of them complained of vertigo or dizziness. The radiological evaluation, including temporal bone computed tomography and internal acoustic canal magnetic resonance imaging, did not reveal any inner ear pathology or brain lesion that might have precipitated hearing loss in the probands.

Pedigrees of the four families, Sanger sequencing traces of the respective POU4F3 variants, and the audiological phenotypes of the affected individuals. The pedigrees of the Sanger sequence chromatograms of the four families, exhibiting segregation of p.Ala189SerfsTer26 in SB218 (a), p.Leu248Pro in SB307 (b), p.Phe293Leu in SB438 (c), and p.Val318Met in SB347 (d). The audiograms of SB218, SB307, and SB438 were associated with moderate NSHL with mid-specific hearing loss configuration, while that of SB347 indicated severe-to-profound NSHL with flat configuration.
3D protein structure
The DNA binding interface of Alpha-fold generated POU4F3 model structure is depicted in Fig. S1. Alpha-fold generated model structure of POU4F3 was used to examine the effects of the identified variants on POU4F3 protein structure, compared to wild-type POU4F3 protein (Fig. 3a). Leu248 amino acid residue is present in the helix-d of the POU-specific domain. Intra-helical proline substitution at Leu248 causes helical kinks, resulting in dramatic conformational changes in the POU-specific domain (Fig. 3b). Next, Phe293 amino acid residue is present in the helix-b of the POU-homeodomain. The missense variant p.Phe293Leu collapses the interhelical interface (aromatic ring stacking) between Phe293, Trp321, and Phe322 in helix-a (left) by disrupting biochemical interactions between helix-a and helix-b, which in turn destabilize the helical assembly of POU-homeodomain (Fig. 3c). Val318 amino acid residue is present in the helix-a of the POU-homeodomain. The long side chain of the missense variant p.Val318Met collapses the hydrophobic interactions with Ile307, Leu289, and Leu311, and induces hydrogen-bonding, destabilizing the POU-homeodomain helical assembly by causing molecular clashes with the adjacent Ile307 and Leu289 (Fig. 3d). Predicted aligned error (PAE) score, which reflects inter-domain accuracy, also indicated that p.Ala189SerfsTer26 variant-induced premature termination of translation destroys the POU4F3 protein structure, including the DNA-binding functional domains, in turn destabilizing the protein (Fig. S2). Collectively, the POU4F3 variants compromised protein stability, and probably impaired the DNA-binding ability.

Novel POU4F3 variants destabilize the inter-helical interactions, impairing the transcriptional activity of POU4F3. (a) Sideview of Alphafold generated model structure of POU4F3. (Jumper et al.37 Highly accurate protein structure prediction with AlphaFold., Nature). POU homeodomain (green) and POU-specific domain (cyan) assembled with DNA binding cleft (orange circle) in between. Val318 and Phe293 are present in the Helix-a and Helix-b of the homeodomain (green), respectively, while Leu248 is in the Helix-d of the POU-specific domain (cyan). All the mutant residues are facing intra-helical spaces, not directly interacting with DNA. (b) Intra-helical proline substitution at Leu248 causes helical kinks. A 27-amino acid long helix-d has a natural kink (black dotted line) driven by Pro246 in the middle. Additional proline substitution induces the formation of an additional kink (red dotted line) starting from p.Leu248Pro (red dot), causing dramatic conformational changes in the POU-specific domain. (c) Phe293 forms aromatic ring stacking (black dotted line) with Trp321 and Phe322 in helix-a (left), stabilizing the interhelical interface. The p.Phe293Leu variant largely disrupts biochemical interactions between helix-a and helix-b, destabilizing helical assembly of POU-homeodomain. (d) Key amino acid residues of intramolecular hydrophobic cavity of POU homeodomain. Val318 forms hydrophobic interactions with Ile307, Leu289, and Leu311. The Val318Met mutant with long side chain clashes (red polygons), with the adjacent Ile307 and Leu289, changing the distance between helices. L Leu, V Val, P Pro, F Phe, W Trp, I Ile, M Met.
Protein expression and stability
The western blot analysis demonstrated that the wild-type and the three mutant proteins carrying missense variants (p.Leu248Pro, p.Phe293Leu, and p.Val318Met) were expressed as a single band corresponding to the correct molecular weight (36 kDa), indicating that the staining was derived explicitly from the tagged POU4F3 proteins (Fig. 4a). Compared to the wild-type protein, the three mutant proteins carrying missense variants had weaker intensities, probably due to protein instability (Fig. 4b). Additionally, the p.Ala189SerfsTer26 POU4F3 protein was stably expressed with a smaller molecular weight (21 kDa) due to premature termination of the POU4F3 protein. Interestingly, the expression of a truncated protein (p.Ala189serfs*26) was stronger than the wild-type protein (Fig. 4a,b). To determine whether POU4F3 variants destabilize POU4F3 protein, we performed cycloheximide (CHX) chase assays to block protein synthesis. HEK293T cells were transfected with wild type and four mutant POU4F3 vectors for 24 h, followed by treatment with CHX (80 µg/ml) for 1, 2, and 3 h, respectively. Three missense variants decreased the stability of POU4F3 protein compared with wild-type protein. Conversely, the half-life of the truncated mutant protein (p.Ala189SerfsTer26) lacking both mono- and bi-partite NLSs showed a longer trend compared with the wild-type protein, suggesting the mutant protein (p.Ala189SerfsTer26) was more stable than the wild-type protein (Fig. 4c). The original immunoblots (uncropped, full-length membranes with membrane edges visible) were provided in Fig. S3.

Western blot analysis for POU4F3 wild-type, frameshift, missense variants by transient transfection at HEK293T cell. (a) Expressions of POU4F3 wild-type and mutants were detected by western blotting in HEK 293T cells. Molecular weight of wild-type and mutant proteins (p.Leu248Pro, p.Phe293Leu, and p.Val318Met) are 36 kDa, whereas molecular weight of truncated mutant protein (p.Ala189SerfsTer26) is 21 kDa. The immunoblots are representative of independent repetitive experiments. LacZ is used as a transfection control. (b) The bands intensity was quantified by Image J. The band intensity was normalized to β-actin. Intensity data was presented as means ± standard deviations from two independent plots in a triplicate manner. Consequently, the sample size for each experiment was six. (c) Comparison of the stability of wild-type and mutant POU4F3 using protein stability assays in the transient overexpression system. HEK293 cells, overexpressing POU4F3, were treated with cycloheximide (80 µg/ml) for up to 3 h to block the general translation. The CHX chase assay experiment was performed once, with three measurements taken during the process. As a result, the sample size for each experiment was three. WT, wild type; LacZ, transfection control; β-actin, loading control; ns, no statistical significance; *p < 0.05, ***p < 0.001, one-way ANOVA with Bonferroni comparisons. The original immunoblots (uncropped, full length membranes with membrane edges visible, and standard protein size markers and expected molecular weight labeled) matched to the cropped versions (this figure) were all provided in Fig. S3.
Transcriptional activity
To investigate the transcriptional activities of the mutant POU4F3 proteins, an in vitro luciferase reporter assay incorporating the SNAP-25-Luc reporter construct was performed (Fig. S4). HEK293T cells were transfected with six pCMV6 plasmid constructs encoding Myc-DDK only (negative control), wild-type POU4F3, mutant POU4F3 (p.Ala189SerfsTer26), mutant POU4F3 (p.Leu248Pro), mutant POU4F3 (p.Phe293Leu), and mutant POU4F3 (p.Val318Met) (Fig. 5). We then compared the fold changes in the luciferase activities of mutant POU4F3 proteins to those of the wild-type protein, normalized to those of the internal control (Myc-DDK). The experimental condition yielding optimal wild-type POU4F3-induced transcription efficiency was determined to be 2 μg SNAP-25-Luc, to minimize the influence of the ceiling effect (Fig. 5a). While the wild-type POU4F3 increased the luciferase activity approximately four-fold changes, the mutant POU4F3 showed only two-fold increase in luciferase activity, demonstrating a significantly poorer transcriptional activity of mutant POU4F3 (p < 0.001) to elicit transcription of the downstream target genes of POU4F3 (Fig. 5b,c).

Transcriptional activity of novel POU4F3 variants. (a) Luciferase activities measured under specific conditions. To minimize the ceiling effect, the condition (i.e., empty‐Luc 2 µg and SNAP25‐Luc 2 µg) was determined as the luciferase vector system. (b,c) The transcriptional activities of the wild-type and mutant POU4F3 proteins in the SNAP25-Luc vector were normalized to that of the internal control (Myc-DDK). Using the luciferase vector system, the transcriptional activity in the wild-type and the four POU4F3 variants were analyzed. All variants exhibited significantly reduced transcriptional activities compared to the wild type. Experiments were conducted in duplicate, and measurements were taken three times to determine luciferase activity. Consequently, the sample size for each experiment was six. The four mutant constructs corresponding to each variant differentiated by employing distinct colors for clear visualization. *, a statistical significance by one-way ANOVA and Bonferroni’s multiple comparison test.
Subcellular localization
Subcellular localization of transcriptional factors in the nucleus is necessary for its transcriptional activity that regulates target gene expression. All the mutant POU4F3 proteins showed significantly reduced reporter gene expression compared to the wild-type protein. The HEK293T cells transfected with the empty vector (negative control) demonstrated no cytoplasmic or nuclear fluorescence, confirming that the small tags attached to empty vectors did not induce any additional trafficking of the cloned protein to the target cells and organelles (Fig. 6a,b). Further, HEK293T cells were transfected with constructs encoding wild-type and mutant proteins fused to C-terminal Myc-DDK tags. Notably, the mutant POU4F3 (p.Ala189SerfsTer26), which lacks both mono- and bi-partite NLSs, localized to both the cytoplasm and the nucleus, but the nuclear proportion (approximately 2%) was significantly lower than that of the cytoplasm (approximately 98%) (Fig. 6a,b). In contrast, all other mutant POU4F3 proteins (p.Leu248Pro, p.Phe293Leu, and p.Val318Met) localized exclusively to the nuclei (approximately 98%), consistent with the localization of the wild-type protein (Fig. 6c).

Immunofluorescence of the wild-type and mutant POU4F3 proteins. (a) Cells were immuno-stained with anti-Myc (green) and phalloidin (red). The nuclei were stained with DAPI (blue). (b) Cells were immuno-stained with anti-DDK (green) and Rhodamine-phalloidin (red). Rhodamine-phalloidin (red) staining was used to label F-actin and stabilize actin filaments in vitro. The nuclei were stained with DAPI (blue). Upon examination through confocal microscopy, the region where the green fluorescence (representing the target protein) and the blue fluorescence (DAPI-stained nuclei) overlap appears as a turquoise color. (c) Quantitation of cytoplasmic and nuclear localization of POU4F3, depending on the variants.
RNA sequencing and bioinformatic analyses
We performed RNA sequencing analysis to investigate comprehensively the molecular pathways affected by POU4F3 variants identified from the hearing-loss families. We used patient-derived lymphoblastoid cell lines (LCLs) to mimic the molecular regulatory programs of the original pathogenic circumstances, which were altered, at least in part, by these mutations. Figure 7a depicts the experiment and analysis flow. For subsequent sequencing analysis, we read over 100,000,000 reads from eight RNA-sequencing libraries. Around 95% of the reads passed quality inspection (Q > 30), and an average of 95% reads were successfully mapped to the human reference genome (GRCh37), covering approximately 20,000 human genes (Table S1).


RNA sequencing analysis. (a) Schematic diagram of the analysis flow. (b) Volcano plot of significantly different genes (n = 630). Upregulation (red dot) and downregulation (blue dot) gene numbers were summarized as a pie graph (inlet). (c) Heatmap analyses of differential gene expression. The higher expression level was shown as red color while the lower expression was shown as blue. (d) (Upper) Revigo visualization of the top 30 gene ontology (GO) data. Clustered terms were listed in each box. (Bottom) Top 10 GO terms in biological process. The red dot box showed top2 GO terms, including cell differentiation and cellular developmental process. (e) Ancestor chart view of the QuickGO. In each GO term, enriched gene number was shown in the dark green pie while term size was shown as green pie with the adjust p value calculated by hypergeometric test and multiple testing correction (FDR). The colored arrow showed the relationship between the Ancestor term and the Child term. The black arrow showed “is a” and the blue arrow showed “part of”, which is explained in the index box. 14 genes which belong to the GO-term (GO: 0048839) were significantly enriched (adjusted p value = 0.01). 14 transcripts were significantly enriched for the GO term (GO: 0048839) (adjusted p value = 0.01). To verify their dysregulation, each FPKM value of 14 genes from the health group (gray, n = 4) and the hearing loss group (white, n = 4) was compared as fold change with standard error of the means (SEM). *p < 0.05 was the result of a statistical significance test using the Student's t-test (bottom).
Prior to the transcriptome analyses, we performed a correlation analysis to determine whether these cells exhibited the molecular features of the cochlea. We used mouse transcriptome data, including adult cochlear inner and outer hair cells, and early postnatal cochlea, because public RNA-sequencing data for human cochlear tissues were unavailable. We used adult mouse testis transcriptome data as negative controls. Table S2 summarizes the public data used in this study. The Spearman's correlation coefficients ranged from 0. 4 to 1, indicating a positive correlation (Fig. S5a,b). Statistically significant p values were found in two reference sets: 0.0399 for postnatal day 4 cochlea transcriptomes with patient samples and 0.0404 for postnatal day 7 cochlea transcriptome with patient samples (Fig. S5a,c). There is a moderate correlation between the reference transcriptome and the patient cell transcriptome in each case, as shown by the Spearman's correlation coefficients of 0.53 and 0.521. Of note that both postnatal day 4, and postnatal day 7 cochlear transcriptome have a slightly stronger correlation with adult cochlear transcriptome with an average coefficient of roughly 0.75, suggesting that these transcriptome favorably share similar molecular pathways. However, there was no correlation between the sample transcriptome, adult testis transcriptome, and the other reference transcriptome (Fig. S5). Overall, our RNA-seq data would reflect more of the molecular signature of the early postnatal cochlea (postnatal day 4 and day 7) rather than other response pathways reported in adult tissues.
After observing that RNA-seq data contain more early postnatal cochlear regulatory molecular pathways, we analyzed differential gene expression between wild-type and hearing-loss groups. 630 genes had statistically significant expression variations, which were represented using a volcano plot and a heatmap split into upregulated (n = 203) and downregulated (n = 427) groups (Fig. 7b,c). We used GO (Gene Ontology) analysis to elucidate the biological processes underpinning the observed dysregulation. Approximately 360 GO terms associated with biological processes were substantially enhanced. Revigo was used to further visualize the top 30 GOterms, displaying them as representative. The most distinguishable groups consisted of cellular differentiation, cellular developmental processes, and other GO keywords pertaining to development (Fig. 7d). This trend was also seen in the top ten GO-terms. Two of the most enriched GO-terms (p = 1.18E−11 and 1.8E−11, respectively) were cell differentiation (GO:0030154) and cellular developmental processes (GO:0048869) (Fig. 7d). We further enhanced our studies by showing an ancestor-child view of the enriched GO terms (Fig. 7e). Developmental process is an ancestor term, and inner ear developmental GO-term is its child term (GO:0048839). Noteworthy is the fact that 14 genes are differentially expressed in GO:0048839 in the hearing loss group, with a p value of 0.01. Its other ancestor GO-term is either ear development (GO: 0043583) or anatomical structure development (GO:0048856), for which 15 or 236 genes are enriched in each term, respectively. Using this ancestor chart, we determined that inner ear development is the most enriched child GO-term in the hearing-loss population. We next examined the differential expression of the 14 genes in the inner ear developmental GO-term (Fig. 7e, bottom bar graph). In comparison to the control group, the hearing-loss group demonstrated repressive patterns.
To determine if these genes and POU4F3 are physically and functionally associated, we performed STRING analyses under the assumption that mRNA expression level is linearly correlated with translation. We observed possible protein associations in three groups (Fig. S6). Notable was the association of 14 dysregulated genes with the Notch pathway (DLL1), BMP pathway (BMP2), and Wnt (SDC4, PTK7, and CELSR1) pathway, which are connected to Sox9. These pathways have been reported as critical regulators to produce hair cells and dysregulations of them could lead to the hearing loss14. Initially, misregulation of BMP pathway was validated by measuring BMP2 expression level quantitatively (Fig. S7).
Interestingly, POU4F3 clustered together with MYO6 and DFNA5, mutations or dysregulations of which have been found in hearing loss studies1,15,16,17. Since we observed MYO6 expression significantly dysregulated in the hearing-loss group, we further validated by RT-qPCR. Even though marginal, quantitative analyses reached statistical significance, suggesting the POU4F3 variant regulates MYO6 expression in the hearing loss pathology (Fig. S7). In the case of AHI1, even though we did not see the association in the STRING analyses, we further confirmed its expression level by RT-qPCR (Fig. S7) since it has been associated with non-syndromic deafness18. Overall, we confirmed dysregulation of BMP2, MYO6 and AHI1 by RT-qPCR. We concluded that the POU4F3 variations might regulate Wnt, Notch, and/or BMP pathways, specifically leading to the misregulation of BMP2, MYO6, and AHI1 in the pathogenesis of hearing loss.
In addition to the 14 enriched genes involved in the development of the inner ear, we also analyzed known POU4F3 target genes, including Lhx3, Gfi1, Bdnf, Ntf3, MYO6, Caprin1, and Nr2f2. The majority of these genes, with the exception of Lhx3 and Ntf3, were expressed based on transcriptome analysis, with average read-counts of 6224.024 and an average normalized value of 677.85 (Fig. S8a). Statistically significant downregulation was seen in Bdnf and MYO6 in the hearing loss group, compared to the hearing loss group. The expression level of Bdnf was repressed by 93 percent, representing the most significant reduction. A reduced pattern was observed except for Gfi1 and Caprin1, but the p value exceeded 0.05 (Fig. S8). Overall, the dysregulation of POU4F3 downstream targets was reaffirmed.
After that, we explored whether the four variants had distinct transcriptome signatures. Spearman’s correlation analysis revealed that SB218 was less correlated to the other three cases (Fig. S9a). Notably, the SB218 variant (p.Ala189SerfsTer26) altered the subcellular location. DEseq2 was used to pinpoint the dysregulation between the SB218 variant and the others. We used fold-changes and expression levels following the visualization of the MA-plot (Fig. S9b). These patterns of expression clearly demonstrated upregulation or downregulation (Fig. S9c). Then, we studied the enriched biological processes to observe numerous categories, including cellular processes, synthesis, stimuli responses, cellular process regulation, metabolic process regulation, and cellular localization (Fig. S9d). Nuclear import (GO: 0051170) was one of the significantly enriched GO-terms (Fig. S9e). We narrowed them down further to 51 differentially expressed genes. In general, the SB218 variant (p.Ala189SerfsTer26) exhibited a diminished pattern (Fig. S9f). This further confirmed that the SB218 variant (p.Ala189SerfsTer26) dysregulated the nuclear import process at a molecular level.
Discussion
POU-specific and POU-homeodomain regions of POU transcription factors have high-affinity DNA binding19,20. The presence of novel POU4F3 variants in key regions encoding DNA-binding sites suggests that mutant proteins may have reduced ability to bind to DNA targets, resulting in insufficient target gene expression. Consistent with previous studies2,3, Protein modeling and structure analysis reveal that missense variants disrupt the interhelical interface of DNA-binding functional domains, reducing protein expression and stability. Weaker bands of the mutant proteins are detected in western blot analysis due to greater instability compared to wild-type protein, as confirmed by the CHX chase assay. As such, tertiary protein structure changes in the interhelical interface caused by POU4F3 variants may affect DNA-binding ability, leading to reduced transcriptional activity for regulating downstream target gene expression related to inner ear hair cell function. Resultantly, we identified a significant reduction of the transcriptional activity necessary to induce regulation of downstream target gene expression.
Several downstream targets of POU4F3, including Lhx321, Gfi122, Bdnf23, Ntf323, MYO624, Caprin125, and Nr2f226, were known to regulate inner ear hair cell functions. We further identified altered expression of 14 downstream target genes associated with inner ear development using patient-derived LCLs. The alteration of genes that affect inner ear development could cause not infrequently only subclinical and silent phenotype until the accumulated effect inherent from the developmental defect leads to an obvious hearing loss later in adulthood. Unless the developmental sequela caused by the gene is very severe, the cumulative effect of the sequela can manifest the phenotype only after a certain point in such deafness cases. Enlarged vestibular aqueducts, one of the most common inner ear developmental anomalies, showed varying degrees of progressive hearing loss frequently involving their adulthood and even mid ages. Specifically, we observed that POU4F3 variants downregulated the expression of MYO6, essential for maintenance of stereocilia of the hair cells, which is responsible for auditory mechanoelectrical transduction24. The altered expression of downstream POU4F3 targets identified herein may support the a mechanistic basis for hearing loss caused by POU4F3 variants. First, PCR and Sanger sequencing confirmed that POU4F3 was obviously expressed in both HEK293T and patient-derived LCLs (Fig. S10), suggesting that these cell lines could at least provide the transcriptional environment to assess POU4F3 function. Next, the downstream targets (Gfi1, Bdnf, MYO6, Caprin1, and Nr2f2) of POU4F3 was identified to be expressed in the LCLs, and some of them were misregulated. Further, POU4F3 was connected functionally and physically with 14 mis-regulated genes identified in the LCLs. More specifically, the Notch, Wnt, and BMP pathways associated with 14 dysregulated genes were functionally linked with Myo6 and DFNA5, which clustered together with POU4F3. Finally, but not least, there was a significant moderate correlation of the expression profile between patient-derived cells and the cochlear hair cells. Collectively, the results provide evidence that patient-derived LCLs can be implicated in the transcriptome study of genetic hearing loss for studying POU4F3 transcriptional function.
In this study, POU4F3 variants were associated with distinct subcellular localization patterns. The POU homeodomain contains two putative NLSs required for proper POU4F3 trafficking into the nucleus: N-terminal monopartite (amino acids 274–278; RKRKR) and C-terminal bipartite NLS (amino acids 314–331; KKNVVRVWFCNLQRQKQKR). Weiss et al.2 demonstrated that variants affecting monopartite NLS, bipartite NLS, or both, exerted differential effects on abnormal subcellular localization during the nuclear import process. As such, it is conceivable that the frameshift variant producing a truncated protein (p.Ala189SerfsTer26), lacking both the mono- and bi-partite NLSs, localized exclusively in the cytoplasm. Weiss et al.2 previously reported that a truncated POU4F3 protein lacking both the mono- and bi-partite NLSs exhibited significantly aberrant localization in the cytoplasm (˃ 80%), compared to other truncated POU4F3 proteins lacking only the monopartite (approximately 23%) or the bipartite NLSs (approximately 47%). This suggests that both NLSs contribute significantly to nuclear trafficking of POU4F32. In addition, we observed differentially expressed transcripts associated with cellular localization between p.Ala189SerfsTer26 and three other missense variants, suggesting that aberrant nuclear import affects the ability to activate downstream targets. Among 51 DEGs related to nuclear import, expression of Frizzled-7 receptor (FZD7) was the most downregulated. Specifically, FZD7 interacts with the Wnt/β-catenin pathway27,28, which is required for cochlear hair cell differentiation29. Thus, the truncated POU4F3 protein lacking both the mono- and bi-partite NLSs led to the downregulation of FZD7 expression, which might reduce nuclear β-catenin accumulation and, in turn, possibly affect cochlear air cell differentiation. Of note that these genes are expressed in our RNA-seq. data, further supporting that experimental design and conclusion reflect the hearing loss pathology.
Interestingly, western blot analysis showed that the expression of the mutant protein (p.Ala189SerfsTer26) was stronger than the wild-type protein. Moreover, p.Ala189SerfsTer26 mutant was more stable than the wild-type protein upon protein stability assays. This was in agreement with a previous study, which demonstrated that a mutant POU4F3 protein (p.Ile295Thrfs*5) was more stable than the wild-type protein2. It has also been demonstrated that the mutant protein had a significantly greater half-life than the wild-type protein2. Although the exact mechanism remains poorly understood, recent studies have demonstrated that the ubiquitin–proteasome system is interrelated with the bipartate NLS function with regard to regulation of protein stability11,30. The cellular protein quality control system (i.e., ubiquitin–proteasome system) regulates the half-lives of various regulatory proteins and removes misfolded proteins31. NLS defects, including those of lysine ubiquitination sites, could decrease protein degradation while upregulating their half-lives30. In this sense, the p.Ala189SerfsTer26 mutant lacking the bipartite NLS (including lysine ubiquitination sites) would be likely to be more stable than the wild-type protein, similar to the extended half-life of the mutant protein (p.Ile295Thrfs*5). However, this variant might undergo nonsense-mediated mRNA decay in vivo, in contrast with the in vitro situation.
In this study, the predominantly nuclear localization of the p.Val318Met-POU4F3, residing within a bipartite NLS, did not perfectly align with the classical hypothesis. Lin et al.11 demonstrated that the missense variant (p.Lys328Glu), in which bipartite NLS amino acid residues were affected, was associated with aberrant POU4F3 subcellular localization, which was in contrast with the present study. It has been suggested that the substitution of a basic lysine with an acidic glutamate (p.Lys328Glu) may alter bipartite NLS molecular properties, highlighting the importance of correct basic amino acid cluster alignment of bipartite NLS in maintaining the POU4F3 protein localization.11 Indeed, bipartite NLS basic amino acids, the first two (KK) and the last two (KR), are considered essential for nuclear localization of POU4F3.2 The missense variant in this study (p.Val318Met), with conserved bipartite NLS basic amino acids and molecular properties, may therefore show normal nuclear localization, albeit with disrupted hydrophobic interactions.
At this point, the exact mechanism explaining why the NSHL phenotype in affected individuals carrying p.Val318Met is more severe than in those carrying p.Ala189SerfsTer26 remains unclear. Although the p.Ala189SerfsTer26 mutant, which lacks the bipartite NLS, fails to localize to the nucleus, it appears to be more stable than the wild-type protein. Alternatively, this mutant might undergo nonsense-mediated mRNA decay in vivo, leading to haploinsufficiency, which preserves one copy of the gene. On the other hand, the p.Val318Met mutant exhibits normal nuclear localization but has structural instability. If the molecular function of this mutant results in a dominant-negative effect, the NSHL phenotype would be more severe in cases with haploinsufficiency. However, phenotypic variability cannot be solely explained by different genotypes and pathogenic molecular mechanisms. Phenotypic variability in terms of age of onset, degree of severity, and configuration has been documented in previous studies7,8,9,10,11,12,13. Moreover, intrafamilial variations in age of onset and severity of hearing loss have been observed. Additionally, the phenotypic variability and the lack of a definitive genotype–phenotype correlation in DFNA15 might be attributable to a combination of stochastic and epigenetic factors. As such, our study's findings demonstrate that patient-derived LCLs can be effectively utilized in transcriptome studies of genetic hearing loss to investigate POU4F3 transcriptional function. This approach offers a breakthrough for cases where obtaining human cochlear samples for transcriptome studies is not feasible. Nevertheless, it is important to note that POU4F3-expressing inner ear hair cells, such as iPSC-differentiated hair cell-like cells and otic organoids, could potentially provide further insights into the molecular mechanisms underlying POU4F3-associated DFNA15 and phenotype-genotype correlations. In addition, it is essential to move beyond studying the single gene's individual function and instead examine its broader contribution within the molecular networks. Our current knowledge of its full range of binding partners and regulatory elements remains incomplete. Therefore, conducting systemic and network analyses would be necessary to further elucidate the functions and interactions underlying POU4F3-associated DFNA15.
Materials and methods
Subjects
All procedures in this study were approved by the Institutional Review Boards of Seoul National University Hospital (IRB-H-0905-041-281) and Seoul National University Bundang Hospital (IRB-B-1007-105-402). Written informed consent was obtained from both affected and unaffected members of the families. In the case of pediatric participants, written informed consent was obtained from their parents or guardians. All methods were performed in accordance with the relevant guidelines and regulations. The affected individuals underwent comprehensive phenotypic evaluations, including medical history interviews, physical examinations, imaging, and audiological and vestibular assessments.
Audiological assessments
Pure tone audiometry (PTA) with thresholds of air and bone conduction was performed in all the probands, as well as the affected individuals if possible, according to the standard protocols. The hearing thresholds for six different octaves (0.25, 0.5, 1, 2, 4, and 8 kHz) were evaluated using PTA. The frequency ranges were classified as follows: low frequency, 250–500 Hz; mid-frequency, 1–2 kHz; and high frequency, 4–8 kHz. The mean hearing threshold was calculated as the mean of the thresholds at 0.5, 1, 2, and 4 kHz, and the severity of hearing loss was classified as mild (20–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB), or profound (> 90 dB), as previously reported. The audiogram configurations were classified as down-sloping, rising, or flat. Specifically, the down-sloping configuration on PTA was defined by increasing the thresholds from 0.25 to 8 kHz, while the difference of the thresholds between 0.25 and 8 kHz was > 20 dB HL32.
Molecular genetic testing
Genomic DNA was extracted from peripheral blood using the standard procedure and was subjected to exome sequencing using a Sure Select 50 Mb Hybridization and Capture Kit and a HiSeq2000 platform in four proband samples. The paired-end read length was 100 bp, and the reads were aligned using the University of California Santa Cruz (UCSC) hg19 reference genome browser (https://genome.ucsc.edu/). As described previously,33,34 bioinformatics analysis and strict filtering were performed to retrieve candidate variants of hearing loss. The candidate variants were confirmed through Sanger sequencing, and a segregation study was performed using parental DNA samples whenever available. The pathogenicity of the four novel POU4F3 variants was classified using the ACMG/AMP guidelines for hearing loss.35,36 The in-silico prediction analysis for candidate variants in each family are presented in Table S3.
Structural analysis of POU4F3
AlphaFold Protein Structure Database generated the model structure of POU4F337,38. POU homeodomain and POU-specific domain were assembled with the DNA binding cleft in between. The homeobox protein HOX-B1/DNA ternary complex (PDB ID: 1B72) was aligned to the POU4F3 structure to allow DNA-binding analysis39. The stabilities of truncated POU4F3 variants were evaluated based on the PAE score, which reflects the inter-domain accuracy. All the figures were generated using PyMOL (ver. 2.5.2) software (PyMOL Molecular Graphics System ver. 2.0, Schrödinger Inc., New York, NY, USA).
Molecular study
The series of steps for the molecular work, including plasmid construction, cell culture, transfection, western blot, immunocytochemistry, real-time quantitative reverse transcription-polymerized chain reaction, and luciferase assay, are all described in Table S4.
RNA sequencing and bioinformatic analysis
The detailed information of RNA sequencing and bioinformatic analysis is described in Table S5.
Statistics
The detailed information of statistics conducted in this study is described in each Figure legend.
Data availability
Sequence variations were submitted to Clinvar with the accession code (SCV003926516, SCV003926517, SCV003926518, SCV003926519). The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The data (Individual-level whole-exome sequence data and RNA-sequencing data) that support the findings of this study are available from the corresponding author (B.Y.C., choiby2010@gmail.com) upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
Vahava, O. et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279, 1950–1954 (1998).
Weiss, S. et al. The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Mol. Cell. Biol. 23, 7957–7964 (2003).
Collin, R. W. et al. Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum. Mutat. 29, 545–554 (2008).
Erkman, L. et al. Role of transcription factors a Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 381, 603–606 (1996).
Xiang, M. et al. Essential role of POU–domain factor Brn-3c in auditory and vestibular hair cell development. Proc. Natl. Acad. Sci. 94, 9445–9450 (1997).
Zhu, G.-J. et al. Aldh inhibitor restores auditory function in a mouse model of human deafness. PLoS Genet. 16, e1009040 (2020).
Freitas, É. L. et al. Deletion of the entire POU4F3 gene in a familial case of autosomal dominant non-syndromic hearing loss. Eur. J. Med. Genet. 57, 125–128 (2014).
He, L., Pang, X., Chen, P., Wu, H. & Yang, T. Mutation in the hair cell specific gene POU4F3 is a common cause for autosomal dominant nonsyndromic hearing loss in Chinese Hans. Neural Plast. 2016, 1–6 (2016).
Cai, X. Z. et al. Exome sequencing identifies POU4F3 as the causative gene for a large Chinese family with non-syndromic hearing loss. J. Hum. Genet. 62, 317–320 (2017).
Gao, X. et al. A missense mutation in POU4F3 causes midfrequency hearing loss in a Chinese ADNSHL family. BioMed Res. Int. 2018, 1–7 (2018).
Lin, Y.-H. et al. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci. Rep. 7, 1–6 (2017).
Kitano, T. et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 12, e0177636 (2017).
Bai, X. et al. Identification of two novel mutations in POU4F3 gene associated with autosomal dominant hearing loss in Chinese families. J. Cell. Mol. Med. 24, 6978–6987 (2020).
Müller, U. & Barr-Gillespie, P. G. New treatment options for hearing loss. Nat. Rev. Drug Discov. 14, 346–365 (2015).
Zhu, M. et al. Mutations in the γ-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am. J. Hum. Genet. 73, 1082–1091 (2003).
Van Wijk, E. et al. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genet. 40, 879–884 (2003).
Ahmed, Z. M. et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72, 1315–1322 (2003).
Elsayed, S. M. et al. Non-manifesting AHI1 truncations indicate localized loss-of-function tolerance in a severe Mendelian disease gene. Hum. Mol. Genet. 24, 2594–2603 (2015).
Ruvkun, G. & Finney, M. Regulation of transcription and cell identity by POU domain proteins. Cell 64, 475–478 (1991).
Verrijzer, C. P. et al. The DNA binding specificity of the bipartite POU domain and its subdomains. EMBO J. 11, 4993–5003 (1992).
Hertzano, R. et al. Lhx3, a LIM domain transcription factor, is regulated by Pou4f3 in the auditory but not in the vestibular system. Eur. J. Neurosci. 25, 999–1005 (2007).
Hertzano, R. et al. Transcription profiling of inner ears from Pou4f3 ddl/ddl identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum. Mol. Genet. 13, 2143–2153 (2004).
Clough, R. L. et al. Brn-3c (POU4F3) regulates BDNF and NT-3 promoter activity. Biochem. Biophys. Res. Commun. 324, 372–381 (2004).
Ma, D. B. et al. Inhibition of Myo6 gene expression by co-expression of a mutant of transcription factor POU4F3 (BRN-3C) in hair cells. Mol. Med. Rep. 9, 1185–1190 (2014).
Towers, E. R., Kelly, J. J., Sud, R., Gale, J. E. & Dawson, S. J. Caprin-1 is a target of the deafness gene Pou4f3 and is recruited to stress granules in cochlear hair cells in response to ototoxic damage. J. Cell Sci. 124, 1145–1155 (2011).
Tornari, C., Towers, E. R., Gale, J. E. & Dawson, S. J. Regulation of the orphan nuclear receptor Nr2f2 by the DFNA15 deafness gene Pou4f3. PLoS ONE 9, e112247 (2014).
Gurney, A. et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. 109, 11717–11722 (2012).
Kim, M. et al. Functional interaction between Wnt3 and Frizzled-7 leads to activation of the Wnt/β-catenin signaling pathway in hepatocellular carcinoma cells. J. Hepatol. 48, 780–791 (2008).
Jacques, B. E. et al. A dual function for canonical Wnt/β-catenin signaling in the developing mammalian cochlea. Development 139, 4395–4404 (2012).
An, L. et al. Dual-utility NLS drives RNF169-dependent DNA damage responses. Proc. Natl. Acad. Sci. 114, E2872–E2881 (2017).
Yun, Y. et al. Proteasome activity in the plasma as a novel biomarker in mild cognitive impairment with chronic tinnitus. J. Alzheimer’s Dis. 78, 195–205 (2020).
Lee, S.-Y. et al. No auditory experience, no tinnitus: Lessons from subjects with congenital-and acquired single-sided deafness. Hear. Res. 354, 9–15 (2017).
Lee, S.-Y. et al. Severe or profound sensorineural hearing loss caused by novel USH2A variants in Korea: Potential genotype-phenotype correlation. Clin. Exp. Otorhinolaryngol. 13, 113 (2020).
Lee, S.-Y. et al. Novel KCNQ4 variants in different functional domains confer genotype-and mechanism-based therapeutics in patients with nonsyndromic hearing loss. Exp. Mol. Med. 53, 1192–1204 (2021).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mut. 39, 1593–1613 (2018).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mut. 39, 1517–1524 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2021).
Piper, D. E., Batchelor, A. H., Chang, C.-P., Cleary, M. L. & Wolberger, C. Structure of a HoxB1–Pbx1 heterodimer bound to DNA: Role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell 96, 587–597 (1999).
Acknowledgements
The manuscript was edited and revised by highly qualified native English-speaking editors at Textcheck.
Funding
This work was supported by the grant (Nos. 04-2022-4010 and 04-2022-3070 to S-Y. Lee) from the SNUH Research Fund. This work was supported by the grant (NRF-2023R1A2C1003248 to H. Seok) from NRF. This study is supported by the Basic Science Research Program through the NRF, funded by the Ministry of Education (Grant 2021R1A2C2092038 to B.Y. Choi), Bio Core Facility center program through the NRF-2022M3A9G1014007 to B.Y. Choi and also by the Basic Research Laboratory program through the NRF, funded by the Ministry of Education (Grant RS-2023-0021971031482092640001 to B.Y. Choi).
Author information
Authors and Affiliations
Contributions
The final manuscript has been seen and approved by all the authors, and they have given necessary attention to the manuscript to ensure the integrity of the work. S.Y.L. and B.Y.C. designed and wrote the paper. S.Y.L., M.Y.K., J.H.H., S.S.P., Y.J. performed experiments and analyzed data. S.Y.L. and B.Y.C. collected and analyzed genetic data. S.J. and H.S. performed RNA sequencing and transcriptome analysis. J.J.H. and J.H.L. provided critical revision. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, SY., Kim, M.Y., Han, J.H. et al. Ramifications of POU4F3 variants associated with autosomal dominant hearing loss in various molecular aspects. Sci Rep 13, 12584 (2023). https://doi.org/10.1038/s41598-023-38272-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38272-w
- Springer Nature Limited
This article is cited by
-
Functional pathogenicity of ESRRB variant of uncertain significance contributes to hearing loss (DFNB35)
Scientific Reports (2024)
-
Allelic hierarchy for USH2A influences auditory and visual phenotypes in South Korean patients
Scientific Reports (2023)