
Abstract
Regarding the important role of the urease enzyme as a virulence factor in urease-positive microorganisms in this study, new series of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives were designed and synthesized. All compounds evaluated against urease enzyme exhibiting IC50 values of 0.87 ± 0.09 to 8.32 ± 1.21 µM as compared with thiourea as the positive control (IC50 = 22.54 ± 2.34 µM). The kinetic evaluations of 6a as the most potent derivative recorded a competitive type of inhibition. Molecular dynamic simulations of the 6a derivative were also conducted, showing that 6a occupied the active site with closed state. Antimicrobial activities of all derivatives were performed, and 6f (R = 3-Cl), 6g (R = 4-Cl), and 6h (R = 3,4-diCl) analogs demonstrated significant antifungal activities with MIC values of 1, 2, and 0.5 µg/mL compared with fluconazole with MIC = 2 µg/mL. Synthesized analogs also exhibited potent urease inhibitory activities against C. neoformans (IC50 = 83.7–118.7 µg/mL) and P. mirabilis (IC50 = 74.5–113.7 µg/mL), confirming their urease inhibitory potential. The results demonstrated that the designed scaffold could be considered a suitable pharmacophore to develop potent urease inhibitors.
Similar content being viewed by others
Introduction
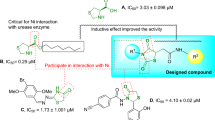
Urease (EC 3.5.1.5) is the first known nickel-containing enzyme found in a wide variety of plants, algae, fungi, and bacteria that catalyzes the hydrolysis of urea to carbamic acids which are further hydrolysis to ammonia and carbon dioxide1,2. Urease is known as a virulence factor found in various pathogenic microorganisms in which the increase in the pH in the medium caused by the accumulation of NH3 results in the development of urolithiasis, pyelonephritis, hepatic encephalopathy, hepatic coma urolithiasis, and urinary catheter encrustation3,4,5. Also, the urease activity of Helicobacter pylori results in the survival of this Gram-negative bacterial in an acidic environment, such as the stomach (pH = 1–2), which plays an important role in the pathogenesis of gastric and peptic ulcer and increases the risk of gastric adenocarcinoma and gastric lymphoma6,7. Importantly, the urease knockout mutants can't colonize in the stomach, suggesting urease's critical role in the survival of H. pylori in the stomach8,9. In agriculture, high urease activity releasing abnormally large amounts of ammonia causes significant environmental and economic problems10.
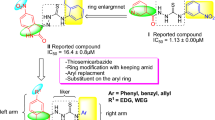
As a result, inhibition of urease activity can be regarded as a favorable strategy to mitigate the negative effect of ureolytic microorganisms. Triazole ring is known as a unique pharmacophore in several pharmaceuticals and natural products as antimalarial11, antiviral12, antibacterial13 as well as antifungal agents14. Triazole rings' favorable properties, including moderate dipole properties, hydrogen bonding capability, and rigidity, are responsible for their enhanced biological activities15,16. Triazole-containing derivatives show promising anti-urease activities that prevent the drug's resistance and have attracted great interest in searching for new anti-urease agents17,18. Compounds A19, B20 and C21 (Fig. 1) are some reports of triazole derivatives with inhibitory potencies against urease.

Rationalization of the newly synthesized [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives with already reported triazole and thiadiazole analogs.
The biological activities of various thiadiazole derivatives and their N-bridged heterocyclic analogs have also been extensively studied. Regarding that thiadiazole is the bioisostere of pyrimidine and oxadiazole22, analogous of this moiety exhibits a wide range of pharmacological properties, including antiviral, antibacterial, antifungal, antiparasitic, anti-inflammatory, and anticancer activities23. More importantly, its relative lipophilicity attributed to the presence of the sulfur atom provides the optimum situation to cross the cellular membranes and induce its effect24.
Khan et al. reported a series of 2,5-disubstituted-1,3,4-thiadiazoles as urease inhibitors, and compound D was found to be the best potencies among thiadiazole analogs25. In 2019, the urease inhibitory activities of triazole, thiadiazole, and thiosemicarbazide linked to quinazoline-4(3H)-one was evaluated, and among them, compound bearing thiadiazole (E, Fig. 1) recorded the best urease inhibitory potencies with IC50 = 1.88 ± 0.17 µg/mL. According to their in silico study, thiadiazole ring effectively interacted with the urease binding site26. In a recent study, the most potent derivative of 5-nitrofuran-2-yl-thiadiazole derivative (F, Fig. 1) showed a tenfold improvement in the inhibitory potency against urease compared to thiourea as a positive control with the non-competitive mode of inhibition27.
Triazolo-thiadiazole is an interesting heterocyclic compound synthesized by fusing 1,2,4-triazole and 1,3,4-thiadiazole rings. Triazolo-thiadiazole is an important analog in many biologically active compounds that showed antibacterial, antifungal, and anti-inflammatory activities28,29. However, limited research was conducted on the anti-urease properties of triazole-thiadiazole derivatives, and compounds G30 and H31 (Fig. 1) are a few examples with improved inhibition compared to the parental structures.
As a result, in the current study, new series of novel triazole-thiadiazole were designed, synthesized, and the anti-urease properties of newly designed compounds were examined against Jack bean urease. Next, the antimicrobial, as well as antiurease properties of these derivatives were evaluated against urease-positive microorganisms. Also, the molecular dynamics simulations of the most potent derivative were performed to get an insight into its behavior within the binding site.
Results and discussion
Chemistry
Synthesis of the title compounds 6a–o was schematically described in Scheme 1. It was initiated by the reaction of carbon disulfide and benzohydrazide (compound 1) in ethanol in the presence of catalytic amounts of potassium hydroxide. The reaction mixture was stirred for 16 h at room temperature. The product was filtered off and washed with ether to remove excess carbon disulfide. Next, compound 2 and hydrazine monohydrate was refluxed for 4 h in water. The mixture was cooled to room temperature, diluted with water and the white solid of the required triazole was precipitated out. Next to the solution of 4-amino-5-phenyl-4H-pyrazole-3-thiol (compound 3) in MeOH, potassium hydroxide was added, and the mixture was sired at room temperature for 30 min, followed by dropwise addition of carbon disulfide at 5 °C, and then reflux at 70 °C for 8–12 h. Finally, aryl halides (compound 5a–n) were added to the 3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole-6-thiol (compound 4) in DMF in the presence of potassium carbonate at room temperature. The structure of final products 6a–n was confirmed using NMR, IR, ESI–MS, and elemental analysis.

Synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives 6a–n.
Urease inhibition and structure–activity relationships
All newly synthesized compounds (6a–n) were examined for their inhibitory potentials against the urease enzyme, the results were presented in Table 1, and the structure–activity relationships (SARs) were constructed. From the experimental data, it appears that all of the synthesized compounds have a significant inhibitory effect with IC50 values in the range of 0.87 ± 0.09 to 8.32 ± 1.21 µM as compared with thiourea as the positive control (IC50 = 22.54 ± 2.34 µM).
The parent compound 6a exhibited the best inhibitory activity against urease with an IC50 value of 0.87 µM. This derivative showed around 27 times improvement in the activity vs positive control.
Evaluation of the fluorine derivatives as halogen-substituted analogs revealed that they exhibited good potencies against urease, with no significant differences observed among them. However, the 4-F derivative (6d) demonstrated better results with an IC50 value of 1.01 µM. It was followed by the 3-F derivative (6c) with an IC50 value of 1.40 µM, and the 2-F derivative (6b) with an IC50 value of 1.51 µM. These findings indicate that the presence of fluorine substitutions enhances the inhibitory activity against urease, and the position of the fluorine atom can influence the potency of the compound.
Detailed evaluations of the chlorine-substituted compounds revealed that the 2-Cl derivative (6e) exhibited inferior activity and recorded the worst results within this group, with an IC50 value of 8.32 µM. However, it is worth noting that even though this derivative had reduced potency, it still displayed better inhibitory activity compared to the positive control. Interestingly, when the position of the chlorine atom was changed from ortho to meta (6f) and para (6g), potency was significantly improved. The IC50 values for these derivatives were 1.25 µM and 1.68 µM, respectively, indicating a substantial enhancement in inhibitory activity. Additionally, the compound with 3,4-diCl substitution (6h) exhibited an IC50 value of 1.41 µM, further demonstrating its potent inhibitory effect against urease. These results suggest that the position and number of chlorine substitutions play a crucial role in modulating the potency of the compounds, with meta and para positions leading to improved activity compared to the ortho position.
In the cases of bromine derivatives as a large electron withdrawing group (6i–k), 3-Br compounds showed the best result (IC50 = 1.05 µM) followed by 4-Br ≥ 2-Br. Substitution of 4-nitro moiety (6l) as hydrophilic and high electron withdrawing group at R position deteriorated the potencies compared to all derivatives except 6e.
In comparison to the electron-withdrawing group, compounds containing electron-donating moieties (6m and 6n) also exhibited good potency against urease. Similar to the previous derivatives, no statistical differences were observed between the ortho and meta analogs. These findings suggest that introducing electron-donating groups can contribute to good inhibitory activity against urease, and the position of these groups did not significantly affect the potency.
Literature reviews were conducted to assess the trend of SAR in the current study concerning previously reported articles. A study with the thiazolidinone derivatives showed that although the main backbone is active, electron-donating groups exhibited slightly superior activity32. Another study indicated that the branched analogs of thiazolidine ester are less active than their straight-chain counterparts, potentially due to the steric bulk of the branched-chain substituents33. In another study evaluating the anti-urease potency of coumarin-thiazolotriazole, although a straightforward SAR analysis was not reported, the following trend was observed: 3-NH2 > 3-MeO-4-OH > phenyl34. Additionally, diindolylmethane bearing thiadiazole was designed as a potent urease inhibitor. The study reported that the backbone itself exhibited high potency against urease, and the SAR analysis demonstrated that analogs with electron-withdrawing groups on the phenyl ring displayed greater potential compared to analogs with electron-donating groups35.
Considering that the basic structure of the compounds is somewhat different, it is impossible to extract a general rule, but it is clear that the presence of triazolo-thiadiazole has a positive effect on the anti-urease potency and further studies have to be conducted to comprehensively extract the SAR. Overall, in the current study, the highest potency was observed in the unsubstituted derivative 6a, suggesting that, similar to the previous study, the designed backbone possesses considerable inhibitory activity against urease. In most cases, no significant differences were observed between the types and positions of substituted moieties. However, one exception to this trend was observed in 6e with an IC50 value of 8.32 µM. The results support our hypothesis that the designed backbone exhibits high potency, regardless of the specific type of substitutions. This emphasizes the robustness and effectiveness of the designed structure as a potential inhibitor of urease.
Enzyme kinetic studies
According to Fig. 2a, the Lineweaver–Burk plot showed that 6a is a competitive-type inhibitor. Furthermore, the plot of the Km versus different concentrations of inhibitor gave an estimate of the inhibition constant, Ki of 1.37 µM (Fig. 2b).

Kinetics of urease inhibition by 6a. (a) The Lineweaver–Burk plot in the absence and presence of different concentrations of sample 6a; (b) The secondary plot between Km and various concentrations of 6a.
Molecular dynamics study
The molecular dynamic study was conducted based on the comparison between Jack bean urease in complex with thiourea as the natural physiologic ligand and in complex with compound 6a at the most potent synthesized compound. The root means square deviation (RMSD) of backbone atoms is an indicator of complex steadiness, the RMSD of both complexes during the simulation course of 100 ns showed in Fig. 3.

The RMSD of Jack bean urease in complex with thiourea (blue) and compound 6a (red).
As shown in Fig. 3, the simulation time was adequate for both complexes to reach acceptable steadiness (fluctuations under 3 Å). Moreover, the deviation value of compound 6a complex was notably lower than the thiourea complex, which could be interpreted as the higher stability of Jack bean urease in complex with the synthesized compound rather than the natural state.
The quaternary structure of Jack bean urease enzyme involves four subunits, the (αβ)8 TIM barrel domain is the enzyme's active site, which consists of two nickel ions coordinated by the unusual amino acid KCX490. Other coordinating residues such as His407, His409, Asp633, KCX490, His519, His545, and Gly550 are vital for the urease enzyme activity. Based on previous studies36, the hydrolysis stage of urea depends on residues Met590–His607. These residues form a helix-turn-helix shape, a dynamic part of the active site pocket called the mobile flap (Fig. 4a). The closed state of mobile flap conformation can restrict access to the active site pocket of the enzyme. The distance between Ile599-Ala440 residues could indicate the active site flap. As demonstrated in Fig. 4, the average distance for compound 6a was 21 Å (close state) compared to thiourea with 30 Å (open state).

(a) Jack bean urease structure and the dynamic flap of the active site pocket (red). (b) The open state of the flap (black) in complex with thiourea (c) The comparison of flap distance through simulation time for thiourea (blue) and 6a (red) complexes (d) The closed state of the flap (black) in complex with 6a.
Root mean square fluctuation (RMSF) displays the fluctuations of backbone atoms and can be interpreted as a mark of stability for residues that have a smaller value (Fig. 5). The strength of interacting residues with compound 6a correlated with the RMSF of these residues compared with corresponding residues in the thiourea complex. Mobile flap residues (590–610) notably showed lower fluctuation in 6a complex. Furthermore, other interacting residues, His407, His409, Ala440, His519, His545, and Asp633, showed less fluctuation in the 6a complex compared with the thiourea complex.

(a) 2D interactions of compound 6a with the active site of Jack bean urease (b) RMSF of thiourea complex (blue) and 5a (red).
Antimicrobial activity and structure–activity relationships study
The antimicrobial activity of the synthesized compounds 6a–n was determined according to the Clinical and Laboratory Standards Institute (CLSI) methods against Cryptococcus neoformans (H99) and Proteus mirabilis. Table 2 shows the minimum inhibitory concentrations (MICs) and minimum fungicidal concentrations (MFCs) values of the target compounds compared with fluconazole and ciprofloxacin as the standard drug.
Assessment against C. neoformans yeast exhibited that just chlorine-containing derivatives, including 6f (R = 3-Cl), 6g (R = 4-Cl), and 6h (R = 3,4-diCl) derivatives, recorded significant antifungal activities with MIC values of 1, 2, and 0.5 µg/mL, respectively compared with fluconazole with MIC value of 2 µg/mL. These derivatives also exhibited promising MFC with values of 2, 4, and 0.5 µg/mL. Similar to enzymatic assessments, 6e entry bearing 2-Cl substitution was almost inactive under the tested concentrations. It was understood that the presence of chlorine substitutions at meta and para portions of the benzyl ring significantly improved the fungicidal activities of the designed scaffold.
Evaluations of the Gram-negative bacterium P. mirabilis showed that all derivatives behaved similarly and demonstrated MIC values of 256, confirming the backbone's potencies regardless of the type of derivitizations.
Urease inhibitory activities against urease-positive microorganisms
To properly evaluate the mechanism of 6a–n derivatives, their potencies to reduce the urease activities were also assessed against urease-positive microorganisms (Table. 3). The results were reported in terms of IC50. Evaluation against C. neoformans revealed that the unsubstituted analog had an IC50 value of more than 123.7 µg/mL. Any substitutions on the benzyl ring were in favor of inhibition. Assessments of the halogen-substituted groups at different positions exhibited that fluorine substitution at any position of the benzyl ring (6b, 6c, and 6d) as a small and strong electron withdrawing group slightly improved the potencies vs 6a. Noteworthy, 6e, 6f, and 6g containing chlorine group exhibited promising potency with IC50 values of 101.9, 87.5, and 93.3 µg/mL, respectively. Multi-substitution of chlorine (6h, IC50 = 83.7 µg/mL) also improved potency compared to mono-substituted derivatives.
Compounds 6i, 6j, and 6k containing bromine as a large electron withdrawing group did not demonstrate significant differences compared to chlorine counterparts. The same trend was seen in the electron-donating group, which slightly improved the activity as compared to 6a.
P. mirabilis revealed IC50 value in the 73.2 ± 5.5 to 113.7 ± 4.6 µg/mL range. The best results came back to 6f (R = 3-Cl), 6h (R = 3,4-diCl), and 6g (R = 4-Cl). No significant differences exist among the other derivatives bearing fluorine, nitro, and methyl derivatives.
Further evaluations of the site of derivatization did not display statistically significant differences among ortho, meta, and para positions. Overall, the best results came back to compound 6l bearing 3,4-diCl analog. This could be due to the favorable effects of lipophilicity and steric hindrance.
Conclusions
In summary, in this study, a series of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives was developed and evaluated as urease inhibitors. All derivatives exhibited outstanding urease inhibition in the enzymatic assessments with IC50 ranging from 0.87 ± 0.09 to 8.32 ± 1.21 µM as compared with thiourea as the positive control with IC50 value of 22.54 ± 2.34 µM. The SARs analysis through the different types of substitutions displayed that unsubstituted compound 6a recorded the best urease inhibition of the series. The kinetic studies of the most active compound disclosed that this compound exhibited a competitive inhibitor with Ki of 1.37 µM. MD study showed that the compound 6a exhibited pronounced interaction with essential urease active site and mobile flap residues through the [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole moiety by coordinating toward the metal bi-nickel complex. Antimicrobial activities of all analogs with CLSI methods displayed that chlorine derivatives, including 6f (R = 3-Cl), 6g (R = 4-Cl), and 6h (R = 3,4-diCl) were highly potent against C. neoformans with MFCs of 2, 4, and 0.5 µg/mL, respectively. On the other hand, all derivatives recorded MICs value of 256 against P. mirabilis. Overall, the urease activities of all derivatives against urease-positive microorganisms showed that 6h was regarded as a potent derivative against P. mirabilis and C. neoformans with IC50 values of 74.5 ± 4.7 and 83.7 ± 3.2 µg/mL.
Based on the information provided, it appears that [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives have shown high activity against urease enzyme and urease-positive microorganisms, indicating their potential as urease inhibitors. Additionally, in silico studies have provided supportive results. The proposed mechanism for the activity of these derivatives suggests that they might inhibit the urease enzyme. However, it is important to note that further studies are required to fully evaluate and understand the mechanism of action of these compounds.
Material and methods
Synthesis
6-(benzylthio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6a)
Brown solid; Yield: 81%; MP = 217–219 °C; IR (KBr, vmax, 3030 (CH Aromatic), 2975 (CH Aliphatic), 1451,1435 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.16 (d, J = 7.50 Hz, 2H, H2, H6), 7.65–7.47 (m, 5H, H Ar), 7.41–7.25 (m, 3H, H3,, H4, H5), 4.65 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 168.36, 157.68, 146.49, 143.04, 139.25, 137.48, 135.29, 134.41, 132.06, 131.25, 130.90, 129.26, 128.93, 128.78, 128.59, 128.43, 127.56, 127.43, 127.19, 126.62, 125.23, 122.92121.36, 115.09, 36.73 ppm; ESI–MS (C16H12N4S2): calculated m/z 324.05 [M+H]+, observed m/z 324.08 [M+H]+; Anal. Calcd C16H12N4S2, C, 59.23; H, 3.73; N, 17.27; Found; C, 59.44; H, 3.95; N, 17.47.
6-((2-fluorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6b)
Brown solid; Yield: 71%; MP = 190–192 °C; IR (KBr, vmax, 3040 (CH Aromatic), 2950 (CH Aliphatic), 1452,1438 (C=N) Cm−1; 1H NMR (250 MHz,DMSO-d6) δ 8.15 (d, J = 7.10 Hz, 2H, H2, H6), 7.61–7.51 (m, 4H, H Ar), 7.40–7.30 (m, 1H, H Ar), 7.27–7.13 (m, 2H, H Ar), 4.67 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 133.72, 132.78, 132.65, 131.79, 130.59, 127.27, 126.79, 117.08, 116.74, 38.20 ppm; ESI–MS (C16H11FN4S2): calculated m/z 342.04 [M+H]+, observed m/z 342.15 [M+H]+; Anal. Calcd C16H11FN4S2, C, 56.12; H, 3.24; N, 16.36; Found; C, 56.29; H, 3.49; N, 16.61 (Supplementary file).
6-((3-fluorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6c)
Brown solid; Yield: 75%; MP = 216–218 °C; IR (KBr, vmax, 3045 (CH Aromatic), 2975 (CH Aliphatic), 1452,1430 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.13 (d, J = 6.10 Hz, 2H, H2, H6), 7.60–7.49 (m, 3H, H Ar), 7.42–7.29 (m, 3H, H Ar), 7.11 (t, J = 7.50 Hz, 1H, H4), 4.65 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 132.06, 131.93, 131.78, 130.54, 127.26, 126.71, 117.58, 117.22, 116.30, 115.96, 38.32 ppm; ESI–MS (C16H11FN4S2): calculated m/z 342.04 [M+H]+, observed m/z 342.10 [M+H]+; Anal. Calcd C16H11FN4S2, C, 56.12; H, 3.24; N, 16.36; Found; C, 56.30; H, 3.45; N, 16.50.
6-((4-fluorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6d)
Brown solid; Yield: 77%; MP = 199–201 °C; IR (KBr, vmax, 3035 (CH Aromatic), 2965 (CH Aliphatic), 1451,1436 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.15 (d, J = 7.30 Hz, 2H, H2, H6), 7.65–7.46 (m, 5H, H Ar), 7.17 (t, J = 8.5 Hz, 2H, H3,, H5), 4.64 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 133.72, 132.78, 132.65, 131.79, 130.59, 127.27, 126.79, 117.08, 116.74, 38.20 ppm; ESI–MS (C16H11FN4S2): calculated m/z 342.04 [M+H]+, observed m/z 342.09 [M+H]+; Anal. Calcd C16H11FN4S2, C, 56.12; H, 3.24; N, 16.36; Found; C, 56.32; H, 3.45; N, 16.57.
6-((2-chlorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6e)
Brown solid; Yield: 72%; MP = 224–226 °C; IR (KBr, vmax, 3030 (CH Aromatic), 2940 (CH Aliphatic), 1456,1434 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.15 (d, J = 8.20 Hz, 2H, H2, H6), 7.72–7.53 (m, 4H, H Ar), 7.50–7.44 (m, 1H, H Ar), 7.42–7.29 (m, 2H, H Ar), 4.65 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 168.36, 157.68, 146.49, 143.04, 139.25, 137.48, 135.29, 134.41, 132.06, 131.25, 130.90, 129.26, 128.93, 128.78, 128.59, 128.43, 127.56, 127.43, 127.19, 126.62, 125.23, 122.92121.36, 115.09, 36.73 ppm; ESI–MS (C16H11ClN4S2): calculated m/z 358.01 [M+H]+, observed m/z 358.013 [M+H]+; Anal. Calcd C16H11ClN4S2, C, 53.55; H, 3.09; N, 15.61; Found; C, 53.72; H, 3.14; N, 15.81.
6-((3-chlorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6f)
Brown solid; Yield: 85%; MP = 216–218 °C; IR (KBr, vmax, 3025 (CH Aromatic), 2950 (CH Aliphatic), 1456,1434 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.16 (d, J = 6.60 Hz, 2H, H2, H6), 7.67–7.48 (m, 5H, H Ar), 7.37–7.29 (m, 2H, H Ar), 4.74 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 168.36, 157.68, 146.49, 143.04, 139.25, 137.48, 135.29, 134.41, 132.06, 131.25, 130.90, 129.26, 128.93, 128.78, 128.59, 128.43, 127.56, 127.43, 127.19, 126.62, 125.23, 122.92121.36, 115.09, 36.73 ppm; ESI–MS (C16H11ClN4S2): calculated m/z 358.01 [M+H]+, observed m/z 358.06 [M+H]+; Anal. Calcd C16H11ClN4S2, C, 53.55; H, 3.09; N, 15.61; Found; C, 53.70; H, 3.10; N, 15.75.
6-((4-chlorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6g)
Brown solid; Yield: 87%; MP = 218–220 °C; IR (KBr, vmax, 3020 (CH Aromatic), 2970 (CH Aliphatic), 1457,1432 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.13 (d, J = 7.30 Hz, 2H, H2, H6), 7.64–7.47 (m, 5H, H Ar), 7.41 (d, J = 5.90 Hz, 2H, H3,, H5), 4.63 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 136.67, 132.45, 131.80, 130.58, 130.02, 127.26, 126.75, 38,21 ppm; ESI–MS (C16H11ClN4S2): calculated m/z 358.01 [M+H]+, observed m/z 358.03 [M+H]+; Anal. Calcd C16H11ClN4S2, C, 53.55; H, 3.09; N, 15.61; Found; C, 53.75; H, 3.15; N, 15.80.
6-((3,4-dichlorobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6h)
Brown solid; Yield: 81%; MP = 215–217 °C; IR (KBr, vmax, 3065 (CH Aromatic), 2950 (CH Aliphatic), 1459,1437 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.11 (d, J = 7.30 Hz, 2H, H2, H6), 7.78 (d, J = 5.90 Hz, 2H, H6), 7.62–7.43 (m, 5H, H Ar), 4.60 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 139.11, 132.14, 131.85, 131.74, 130.83, 130.55, 127.23, 126.77, 37.60 ppm; ESI–MS (C16H10Cl2N4S2): calculated m/z 391.97 [M+H]+, observed m/z 391.99 [M+H]+; Anal. Calcd C16H10Cl2N4S2, C, 48.86; H, 2.56; N, 14.24; Found; C, 49.05; H, 2.75; N, 14.45;.
6-((2-bromobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6i)
Brown solid; Yield: 89%; MP = 210–212 °C; IR (KBr, vmax, 3020 (CH Aromatic), 2885 (CH Aliphatic), 1450,1431 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.16 (d, J = 6.50 Hz, 2H, H2, H6), 7.70–7.49 (m, 5H, H Ar), 7.35 (d, J = 7.30 Hz, 1H, H Ar), 7.27 (d, J = 8.00 Hz, 1H, H2, Ar), 4.72 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 133.29, 132.12, 131.76, 130.61, 129.64, 127.25, 126.77, 38.09 ppm; ESI–MS (C16H11BrN4S2): calculated m/z 401.96 [M+H]+, observed m/z 401.99 [M+H]+; Anal. Calcd C16H11Br4S2, C, 47.65; H, 2.75; N, 13.89; Found; C, 47.80; H, 2.92; N, 14.10.
6-((3-bromobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6j)
Brown solid; Yield: 75%; MP = 225–227 °C; IR (KBr, vmax, 3060 (CH Aromatic), 2950 (CH Aliphatic), 1457,1433 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.14 (d, J = 7.30 Hz, 2H, H2, H6), 7.75 (s, 1H, H2), 7.63–7.42 (m, 5H, H Ar), 7.11 (t, J = 7.50 Hz, 1H, H5), 4.64 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 131.80, 130.56, 127.24, 125.10, 71.23, 38.11. ppm; ESI–MS (C16H11BrN4S2): calculated m/z 342.04 [M+H]+, observed m/z 342.15 [M+H]+; Anal. Calcd C16H11BrN4S2 C, 47.65; H, 2.75; N, 13.89; Found; C, 47.79; H, 2.89; N, 14.13.
6-((4-bromobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6k)
Brown solid; Yield: 78%; MP = 212–214 °C; IR (KBr, vmax, 3060 (CH Aromatic), 2970 (CH Aliphatic), 1456,1433 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.12 (d, J = 6.50 Hz, 2H, H2, H6), 7.64–7.50 (m, 5H, H Ar), 7.45 (d, J = 8.20 Hz, 2H, H3, H5), 4.61 (s, 2H, CH2), ppm. 13C NMR (62 MHz, DMSO-d6): δ 137.09, 132.94, 132.75, 131.79, 130.57, 127.25, 126.74, 122.43, 38.25 ppm; ESI–MS (C16H11BrN4S2): calculated m/z 401.96 [M+H]+, observed m/z 401.98 [M+H]+; Anal. Calcd C16H11BrN4S2, C, 47.65; H, 2.75; N, 13.89; Found; C, 47.80; H, 2.92; N, 14.10.
6-((4-nitrobenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6k)
Yellow solid; Yield: 79%; MP = 220–222 °C; IR (KBr, vmax, 3048 (CH Aromatic), 2863 (CH Aliphatic), 1454,1432 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.20 (d, J = 6.3 Hz, 2H, H2, H6), 8.10 (d, J = 8.3 Hz, 2H, H2, H6), 7.79 (d, J = 8.7 Hz, 2H, H3, H5), 7.57 (m, 3H, HAr). 4.78 (s, 2H, CH2) ppm. 13C NMR (62 MHz, DMSO-d6): δ 131.80, 130.56, 127.24, 125.10, 71.23, 38.01 ppm; ESI–MS (C16H11N5O2S2): calculated m/z 401.96 [M+H]+, observed m/z 401.99 [M+H]+; Anal. Calcd C16H11N5O2S2, C, 52.02; H, 3.00; N, 18.96; Found; C, 52.27; H, 3.19; N, 19.14.
6-((2-methylbenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6l)
Brown solid; Yield: 83%; MP = 212–214 °C; IR (KBr, vmax, 3035 (CH Aromatic), 2965 (CH Aliphatic), 1452,1433 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.16 (d, J = 7.10 Hz, 2H, H2, H6), 7.64–7.45 (m, 3H, H Ar), 7.26 (d, J = 7.60 Hz, 1H, H3), 7.12–7.02 (m, 2H, H Ar), 6.84 (d, J = 7.90 Hz, 1H, H2), 4.60 (s, 2H, CH2), 3.62 (s, 3H, CH3) ppm. 13C NMR (62 MHz, DMSO-d6): δ 131.77, 131.17, 130.57, 127.27, 126.78, 122.79, 116.25, 114.85, 56.49, 38.89 ppm; ESI–MS (C17H14N4S2): calculated m/z 338.07 [M+H]+, observed m/z 338.13 [M+H]+; Anal. Calcd C17H14N4S2, C, 60.33; H, 4.17; N, 16.55; Found; C, 60.55; H, 4.38; N, 16.74.
6-((3-methylbenzyl)thio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6m)
Brown solid; Yield: 86%; MP = 213–215 °C; IR (KBr, vmax, 3020 (CH Aromatic), 2975 (CH Aliphatic), 1454,1436 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.17 (d, J = 6.60 Hz, 2H, H2, H6), 7.63–7.52 (m, 3H, H Ar), 7.45 (d, J = 6.60 Hz, 1H, H Ar), 7.26–7.10 (m, 3H, H Ar), 4.67 (s, 2H, CH2), 2.36 (s, 3H, CH3) ppm. 13C NMR (62 MHz, DMSO-d6): δ 132.01, 131.79, 131.70, 130.58, 129.82, 127.62, 127.62, 127.31, 126.83, 37.62, 20.31 ppm; ESI–MS (C17H14N4S2): calculated m/z 338.07 [M+H]+, observed m/z 338.15 [M+H]+; Anal. Calcd C17H14N4S2, C, 60.33; H, 4.17; N, 16.55; Found; C, 60.53; H, 4.35; N, 16.75.
6-(methylthio)-3-phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole (6n)
Brown solid; Yield: 81%; MP = 213–215 °C; IR (KBr, vmax, 3010 (CH Aromatic), 2960 (CH Aliphatic), 1456,1431 (C=N) Cm−1; 1H NMR (250 MHz, DMSO-d6) δ 8.17 (d, J = 7.50 Hz, 2H, H2, H6), 7.62–7.42 (m, 3H, H Ar), 2.81 (s, 2H, CH3), ppm. 13C NMR (62 MHz, DMSO-d6): δ 13C NMR (63 MHz, DMSO) δ 131.72, 130.58, 127.24, 17.26.ppm; ESI–MS (C10H8N4S2): calculated m/z 248. 02 [M+H]+, observed m/z 248.11 [M+H]+; Anal. Calcd C10H8N4S2, C, 48.37; H, 3.25; N, 22.56; Found; C, 48.57; H, 3.42; N, 22.49.
Screening of urease inhibitory activities
Urease inhibition effects of the synthesized compounds were determined according to the previously reported procedure37,38,39. Briefly, 100 μL of the synthesized compounds at different concentrations was added to 850 μL of urea as substrate and 50 μL urease (3 mg/mL) in phosphate buffer, pH = 7.4). After 30 min, to 100 μL of the incubated solution, 500 μL of solution I (5.0 g phenol and 25.0 mg sodium nitroprusside in 500 mL water) was added, followed by the addition of 500 μL of solution II (2.5 g sodium hydroxide, 4.2 mL sodium hypochlorite, and 5% chlorine in 500 mL water) which was further incubated at 37 °C for 30 min. The absorbance of blue-colored indophenol of each cell is related to the percentage of enzyme inhibition using the following equation at 625 nm. The IC50 values for all synthesized compounds were calculated using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA).
Kinetic studies
The kinetic study for the inhibition of urease by compound 6a was carried out using four different concentrations of inhibitor. Compound 6a was tested at 0, 0.5, 1, and 28 μM concentrations against urease for the kinetic study. The Lineweaver–Burk reciprocal plot was constructed by plotting 1/V against 1/[S] at variable concentrations of the substrate urea (3.12 to 100 mM). The inhibition constant Ki was calculated by the plot of slopes versus the corresponding concentrations of compound 6a.
In silico studies
Maestro Molecular Modeling platform (version 10.5) by Schrödinger, LLC (Maestro, Schrödinger, LLC, New York, NY, 2021) was used to perform the docking study of compound 6a and thiourea on the jack bean urease enzyme. The corresponding crystallographic structure of jack bean urease was downloaded from http://www.rcsb.com by the PDB (PDB ID:4h9m). preparation wizard40. Afterward, missing used for primary preparation of the receptor for the next stage, missed sidechains and loops were filled utilizing the prime tool41. 2D structure of ligands sketched in ChemDraw (ver. 16) and saved as SDF files. The Ligprep module (LigPrep, Schrödinger, LLC, New York, NY, 2021) was used to prepare ligand molecules with OPLS_2005 forcefield and EPIK program42 at a target pH of 7.0 ± 1. Induced fit docking simulation43 was performed considering AHA as the grid center, with a maximum number of 20 poses for each ligand. Receptor and ligand van der Waals radii were set as 0.7 A and 0.5 Å, respectively. Structures with prime energy levels beyond 30 kcal/mol were eliminated based on standard precision glide docking. Molecular dynamic simulation was performed using maestro desmond44. MD simulation complexes obtained from the stage IFD results. The simulation was conducted in a cubic cell filled with 27869 TI3P model water molecules. 92 sodium atoms and 78 chloride ions were added to neutralize the system electrostatic charge. The NPT ensemble (constant number of atoms; constant pressure, i.e., 1.01325 bar; and constant temperature, i.e.,300 K) was used with default settings. Overall simulation duration set on 100 ns for both urease-thiourea and urease-compound 6a complexes with 100 ps for each trajectory frame. The simulation results were analyzed using the maestro graphical interface.
In vitro antimicrobial activities
The antimicrobial activities of all derivatives against C. neoformans (H99), and clinical isolate of P. mirabilisas urease positive microorganism, were tested as recommended by the Clinical and Laboratory Standards Institute (CLSI) (M07-A9 for bacteria; M27-A3 for yeasts). The compounds were diluted, and stock solutions of 20 mg/mL in DMSO were prepared. Mueller–Hinton Broth (HiMedia) and RPMI-1640 (Sigma) were prepared as recommended for antimicrobial susceptibility testing of bacterial and fungal strains, respectively. Two-fold dilutions were made in the range of 1–512 μg/mL for tested compounds. The microbroth dilution test was accomplished using a 96-well microtiter plate containing growth control (drug free wellls) and sterility control (only broth media). The antimicrobial susceptibility test was accomplished by adding a cell suspension adjusted to the 0.5 McFarland standard (1–2 × 108 CFU/mL for bacterial strains; 1–5 × 106 cells/mL for yeast) to different concentrations of tested compounds. Following incubation, the minimum inhibitory concentration (MIC) was established as the lowest concentration of compounds that completely inhibits the organism's growth in wells, as detected visually. All experiments were performed in duplicates.
Anti-ureolytic activities against ureolytic microorganisms
The colorimetric microdilution technique using urea broth media (Merck) was used to examine the ureolytic activity of C. neoformans (H99), and clinical isolate of P. mirabilis treated with tested substances (supplemented with glucose; pH = 6 for C. neoformans). Compounds in the concentration range of 1 to 512 μg/mL were exposed to ureolytic microorganisms, and the color of the medium was evaluated visually and spectroscopically at 560 nm after three days for C. neoformans and 24 h for P. mirabilis. The positive control, which included ureolytic bacteria but no drugs, changed color from yellow to dark pink or magenta. This shifts, allowing the determination of the inhibitory activity of compounds against the urease activity of organisms even without a microliter plate reader45,46.
Data availability
The datasets generated and/or analyzed during the current study are available in the Worldwide Protein Data Bank repository with PDB DOI of https://doi.org/10.2210/pdb4H9M/pdb, (https://www.rcsb.org/structure/4h9m).
References
Blakeley, R. L. & Zerner, B. Jack bean urease: The first nickel enzyme. J. Mol. Catal. 23(2–3), 263–292 (1984).
Huma, R. et al. Thermal and spectroscopic studies of some metal complexes with a new enaminone ligand 3-chloro-4-((4-methoxyphenyl)amino)pent-3-en-2-one and their investigation as anti-urease and cytotoxic potential drugs. Arab. J. Chem. 15(3), 103640 (2022).
Jin, D. et al. Differences in ureolytic bacterial composition between the rumen digesta and rumen wall based on ureC gene classification. Front. Microbiol. 8, 385 (2017).
Biglar, M. et al. Novel N,N-dimethylbarbituric-pyridinium derivatives as potent urease inhibitors: Synthesis, in vitro, and in silico studies. Bioorg. Chem. 95, 103529 (2020).
Azizian, H. et al. Large-scale virtual screening for the identification of new Helicobacter pylori urease inhibitor scaffolds. J. Mol. Model. 18(7), 2917–2927 (2012).
Jahantab, M. B., Safaripour, A. A., Hassanzadeh, S. & Yavari Barhaghtalab, M. J. Demographic, chemical, and Helicobacter pylori positivity assessment in different types of gallstones and the bile in a random sample of cholecystectomied Iranian patients with cholelithiasis. Can. J. Gastroenterol. Hepatol. 2021, 3351352 (2021).
Abdullah Al-Mohammadi, J. et al. Synthesis, in vitro evaluation, and molecular docking studies of benzofuran based hydrazone a new inhibitors of urease. Arab. J. Chem. 15(8), 103954 (2022).
Cheok, Y. Y. et al. An overview of Helicobacter pylori survival tactics in the hostile human stomach environment. Microorganisms 9(12), 2502 (2021).
Karkhah, A. et al. Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases. Microbiol. Res. 218, 49–57 (2019).
Kumar, S. Plant ureases: Physiological significance, role in agriculture and industrial applications—a review. South Asian J. Food Technol. Environ. 1(2), 105–115 (2015).
Lopes, F. V. et al. 1,2,3-Triazole derivatives: Synthesis, docking, cytotoxicity analysis and in vivo antimalarial activity. Chemico-Biol. Interact. 350, 109688 (2021).
El-Sebaey, S. Recent advances in 1,2,4-triazole scaffolds as antiviral agents. ChemistrySelect 5(37), 11654–11680 (2020).
Gao, F., Wang, T., Xiao, J. & Huang, G. Antibacterial activity study of 1,2,4-triazole derivatives. Eur. J. Med. Chem. 173, 274–281 (2019).
Brüggemann, R. J. et al. Management of drug–drug interactions of targeted therapies for haematological malignancies and triazole antifungal drugs. Lancet Haematol. 9(1), e58–e72 (2022).
Bozorov, K., Zhao, J. & Aisa, H. A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 27(16), 3511–3531 (2019).
Iraji, A. et al. Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur. J. Med. Chem. 141, 690–702 (2017).
Zhang, S. et al. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 138, 501–513 (2017).
Dheer, D., Singh, V. & Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 71, 30–54 (2017).
Akhtar, T., Hameed, S., Khan, K. M., Khan, A. & Choudhary, M. I. Design, synthesis, and urease inhibition studies of some 1,3,4-oxadiazoles and 1,2,4-triazoles derived from mandelic acid. J. Enzyme Inhib. Med. Chem. 25(4), 572–576 (2010).
Moghimi, S. et al. Synthesis, evaluation, and molecular docking studies of aryl urea-triazole-based derivatives as anti-urease agents. Arch. Pharm. 351(7), 1800005 (2018).
Rezaei, E. B. et al. Design, synthesis, and evaluation of metronidazole-1,2,3-triazole derivatives as potent urease inhibitors. Chem. Pap. 75(8), 4217–4226 (2021).
Li, Y., Geng, J., Liu, Y., Yu, S. & Zhao, G. Thiadiazole—a promising structure in medicinal chemistry. ChemMedChem 8(1), 27–41 (2013).
Atmaram, U. A. & Roopan, S. M. Biological activity of oxadiazole and thiadiazole derivatives. Appl. Microbiol. Biotechnol. 1–17 (2022).
Szeliga, M. Thiadiazole derivatives as anticancer agents. Pharmacol. Rep. 72(5), 1079–1100 (2020).
Khan, I. et al. Synthesis, antioxidant activities and urease inhibition of some new 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Eur. J. Med. Chem. 45(11), 5200–5207 (2010).
Menteşe, E., Akyüz, G., Emirik, M. & Baltaş, N. Synthesis, in vitro urease inhibition and molecular docking studies of some novel quinazolin-4(3H)-one derivatives containing triazole, thiadiazole and thiosemicarbazide functionalities. Bioorg. Chem. 83, 289–296 (2019).
Asadi, M. et al. Synthesis and in vitro urease inhibitory activity of 5-nitrofuran-2-yl-thiadiazole linked to different cyclohexyl-2-(phenylamino)acetamides, in silico and kinetic studies. Bioorg. Chem. 120, 105592 (2022).
Mathew, V., Keshavayya, J., Vaidya, V. P. & Giles, D. Studies on synthesis and pharmacological activities of 3,6-disubstituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and their dihydro analogues. Eur. J. Med. Chem. 42(6), 823–840 (2007).
Gilani, S. J., Khan, S. A. & Siddiqui, N. Synthesis and pharmacological evaluation of condensed heterocyclic 6-substituted 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazole and 1,3,4-oxadiazole derivatives of isoniazid. Bioorg. Med. Chem. Lett. 20(16), 4762–4765 (2010).
Khan, I. et al. Exploring biological efficacy of coumarin clubbed thiazolo [3,2–b][1,2,4] triazoles as efficient inhibitors of urease: A biochemical and in silico approach. Int. J. Biol. Macromol. 142, 345–354 (2020).
Hanif, M. et al. Synthesis, urease inhibition, antioxidant and antibacterial studies of some 4-amino-5-aryl-3H-1,2,4-triazole-3-thiones and their 3,6-disubstituted 1,2,4-triazolo [3,4-b] 1,3,4-thiadiazole derivatives. J. Braz. Chem. Soc. 23, 854–860 (2012).
Dastyafteh, N. et al. New thioxothiazolidinyl-acetamides derivatives as potent urease inhibitors: Design, synthesis, in vitro inhibition, and molecular dynamic simulation. Sci. Rep. 13(1), 21 (2023).
Rahim, F. et al. Synthesis of 4-thiazolidinone analogs as potent in vitro anti-urease agents. Bioorg. Chem. 63, 123–131 (2015).
Khan, I. et al. Exploring biological efficacy of coumarin clubbed thiazolo[3,2–b][1,2,4]triazoles as efficient inhibitors of urease: A biochemical and in silico approach. Int. J. Biol. Macromol. 142, 345–354 (2020).
Taha, M. et al. Synthesis of diindolylmethane (DIM) bearing thiadiazole derivatives as a potent urease inhibitor. Sci. Rep. 10(1), 7969 (2020).
Balasubramanian, A. & Ponnuraj, K. Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J. Mol. Biol. 400(3), 274–283 (2010).
Asgari, M. S. et al. New 1,2,3-triazole-(thio)barbituric acid hybrids as urease inhibitors: Design, synthesis, in vitro urease inhibition, docking study, and molecular dynamic simulation. Arch. Pharm. 353(9), e2000023 (2020).
Pedrood, K. et al. Arylmethylene hydrazine derivatives containing 1,3-dimethylbarbituric moiety as novel urease inhibitors. Sci. Rep. 11(1), 10607 (2021).
Sedaghati, S. et al. Novel (thio)barbituric-phenoxy-N-phenylacetamide derivatives as potent urease inhibitors: Synthesis, in vitro urease inhibition, and in silico evaluations. Struct. Chem. 32(1), 37–48 (2021).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-aided Mol. Des. 27(3), 221–234 (2013).
Jacobson, M. P. et al. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 55(2), 351–367 (2004).
Greenwood, J. R., Calkins, D., Sullivan, A. P. & Shelley, J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24(6), 591–604 (2010).
Farid, R., Day, T., Friesner, R. A. & Pearlstein, R. A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 14(9), 3160–3173 (2006).
Bowers, K. J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing 84-es (2006).
Knezevic, P., Aleksic Sabo, V., Simin, N., Lesjak, M. & Mimica-Dukic, N. A colorimetric broth microdilution method for assessment of Helicobacter pylori sensitivity to antimicrobial agents. J. Pharm. Biomed. Anal. 152, 271–278 (2018).
Nakamura, Y., Kano, R., Watanabe, S., Takahashi, H. & Hasegawa, A. Susceptibility testing of Cryptococcus neoformans using the urea broth microdilution method: Empfindlichkeitsprüfung von Cryptococcus neoformans in Harnstoff-Bouillon mittels Mikrodilution. Mycoses 41(1–2), 41–44 (1998).
Funding
The authors wish to thank the support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (grant number: IR.SUMS.REC.1401.544).
Author information
Authors and Affiliations
Contributions
M.K.G., M.N., N.D., M.H.S., S.J., and M.A., synthesized compounds and contributed to the characterization of compounds. M.N.M., S.Y., and A.N., B.L. performed biological tests. K.Z. supervised the biological assay. H.B. contribute to the designing of study and manuscript preparation. A.I., CI. performed in silico study and contributed to the manuscript preparation. M.M. supervised all phases of the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khalili Ghomi, M., Noori, M., Nazari Montazer, M. et al. [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives as new therapeutic candidates against urease positive microorganisms: design, synthesis, pharmacological evaluations, and in silico studies. Sci Rep 13, 10136 (2023). https://doi.org/10.1038/s41598-023-37203-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37203-z
- Springer Nature Limited