
Abstract
An important target in the treatment of type 2 diabetes is α-glucosidase. Inhibition of this enzyme led to delay in glucose absorption and decrease in postprandial hyperglycemia. A new series of phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamides 11a–n were designed based on the reported potent α-glucosidase inhibitors. These compounds were synthesized and screened for their in vitro inhibitory activity against the latter enzyme. The majority of the evaluated compounds displayed high inhibition effects (IC50 values in the range of 45.26 ± 0.03–491.68 ± 0.11 µM) as compared to the positive control acarbose (IC50 value = 750.1 ± 0.23 µM). Among this series, compounds 11j and 11i represented the most potent α-glucosidase inhibitory activities with IC50 values of 45.26 ± 0.03 and 46.25 ± 0.89 µM. Kinetic analysis revealed that the compound 11j is a competitive inhibitor with a Ki of 50.4 µM. Furthermore, the binding interactions of the most potent compounds in α-glucosidase active site were studied through molecular docking and molecular dynamics. The latter studies confirmed the obtained results through in vitro experiments. Furthermore, in silico pharmacokinetic study of the most potent compounds was also performed.
Similar content being viewed by others
Introduction
Glucosidases are an important group of digestive enzymes that are responsible for the hydrolytic cleavage of glycosidic bonds of oligosaccharides and disaccharides1. The catalytic specificity of glucosidases depends on the position of cleavage site, the configuration of the hydroxyl groups, and the number of monosaccharides in substrate2. Two main types of glucosidases are α-glucosidases and β-glucosidases which differ in the orientation of carboxylic acid residues during catalysis process3. Among the latter enzymes, α-glucosidase (EC 3.2.1.20) has received a special attention to the pharmaceutical researchers because of its catalytic activity in the intestine that led to the delay in glucose absorption and the decrease in postprandial hyperglycemia4. Therefore, inhibition of this enzyme is an important protocol in treatment of type 2 diabetes mellitus (T2DM) as the most widespread chronic metabolic disorder related to hyperglycemia5. Acarbose and other available α-glucosidase inhibitors cause various side effects including abdominal discomfort, bloating, diarrhea, pain, and flatulence6. On the other hand, α-glucosidase has also been well appreciated as a valuable therapeutic target for other carbohydrate related diseases including viral infections, cancer, and hepatitis7,8,9. Therefore, development of safe and efficient α-glucosidase inhibitors for treating T2DM and other carbohydrat-related diseases is an important goal for pharmacists.
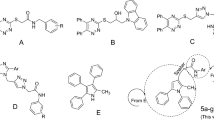
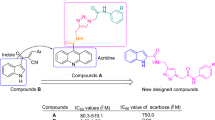
One of the most important heterocycles in the design of new potent synthetic α-glucosidase inhibitors is phthalimide10,11. As can be seen Fig. 1, compounds A–D are derivatives of phthalimide that exhibited high inhibitory activity against α-glucosidase12,13,14,15. On the other hand, several series of phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide derivatives with significant anti-α-glucosidase activity have been reported (Fig. 1, compounds E–F)16,17. Therefore, we concluded that the combination of phthalimide ring and phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide moiety can lead to new potent α-glucosidase inhibitors. In this regards, phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide scaffold designed and fourteen derivatives 11a–n of this scaffold were synthesized and evaluated against α-glucosidase in vitro and in silico.

Design strategy for phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide derivatives 11a–n as new α-glucosidase inhibitors.
Results and discussion
Chemistry
The synthetic route for the synthesis of phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamides 11a–n has been depicted in Scheme 1. This route was started from the reaction of phthalic anhydride 1 and 4-aminophenol 2 in acetic acid at reflux condition to give 2-(4-hydroxyphenyl)isoindoline-1,3-dione 3. Then, compound 3 was reacted with propargyl bromide 4 in the presence of potassium carbonate in acetone and afforded 2-(4-(prop-2-yn-1-yloxy)phenyl)isoindoline-1,3-dione 5. On the other hand, amine derivatives 6a–n were reacted with chloroacetyl chloride 7 in DMF at RT for 30 min to give N-phenyl-2-chloroacetamides 8a–n. The latter compounds and sodium azide 9 were reacted in the mixture of H2O/t-BuOH (1:1) in the presence of triethylamine (Et3N) at RT for 1 h to give azide derivatives 10a–n. Finally, mixture of 2-(4-(prop-2-yn-1-yloxy)phenyl)isoindoline-1,3-dione 5, sodium ascorbate, and copper(II) sulfate (CuSO4) was added to the freshly prepared azide derivatives 10a–n and the reaction was continued at RT for 24–48 h to give the target compounds 11a–n.

Synthesis of phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide derivatives 11a–n.
In vitro inhibitory activity of compounds 11a–n against α-glucosidase
The in vitro enzymatic inhibitory activity of phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide 11a–n was evaluated against yeast α-glucosidase. Acarbose was chosen as positive control and anti-α-glucosidase activities of N-phenylacetamide derivatives 11a–l and N-benzylacetamide derivatives 11m–n are expressed as IC50 values in Table 1.
Structure–activity relationship (SAR) for α-glucosidase inhibitory activity
As evidenced by obtained results, all of the N-phenylacetamide derivatives, with the exception of 2,6-dimethyl derivative 11c and 4-nitro derivative 11j, were more potent than positive control acarbose. In contrast, N-benzylacetamide derivatives 11m–n did not show significant inhibitory activity against this enzyme.
In the N-phenylacetamide derivatives 9a–l, the most potent compound was 4-bromo derivative 11j. Substitution of bromine with nitro group, in case of compound 11k, leads to the complete loss of inhibitory activity and substitution of bromine with ethyl group, in case of compound 11d, leads to a significant decrease in the inhibitory activity.
The second most potent compound was 2,6-dichloro derivative 11i. 2,3-Dichloro derivative 11g and 2,4-dichloro derivative 11j as regioisomers of compound 11i showed moderate anti-α-glucisidase activity in comparison to this compound. The third potent compound among the newly synthesized compounds was 3-chloro derivative 11f. Replacing of chloro substituent with fluorine atom, as in case of compound 11e (the fourth potent compound), caused to a moderate decrease in the anti-α-glucosidase activity. The fifth potent compound was un-substituted compound 11a. Placing two methyl substituents on the pendant phenyl group of compound 11a depending on their positions, has interesting effects on the inhibitory effect of this compound: 2,3-dimethyl derivative 11b was 1.8-fold less potent than compound 11a while 2,6-dimethyl derivative 11c was inactive. Another interesting point that can be observed about the effect of methyl substitution in the obtained inhibitory activities is that the 4-nitro derivative is an inactive compound, but by adding a methyl on 2-position of the 4-nitrophenyl group, potent compound 11l is obtained.
As can be seen in Table 1, N-benzylacetamide derivatives 11m–n did not show a significant inhibitory against α-glucosidase.
Comparison of the new compounds 11 with template compounds E and F
The comparison of IC50 values of the new phthalimide derivatives 11 with their corresponding analogs of benzimidazole derivatives E revealed that with the exception of 2,6-dichloro and 4-bromo derivatives, benzimidazole analogs were more potent than their corresponding analogs of phthalimide series (Scheme 2).The mentioned trend is also observed in the comparison between phthalimide derivatives 11 and quinazolinone derivatives F (Scheme 2)17.

Comparison of IC50 values of phthalimide derivatives 11 against α-glucosidase with their corresponding analogs of benzimidazole derivatives E and quinazolinone derivatives F.
Kinetic study
To gain further insight into the mechanism of α-glucosidase inhibition of the title class of compounds, a kinetic study was performed on compound 11j as the most potent α-glucosidase inhibitor. For this purpose, the reaction rates in the presence of different concentrations of compound 11j were measured in various concentrations of substrate (p-nitrophenyl-a-D-glucopyranoside). Graphs of different concentrations of inhibitor were drawn by the Lineweaver–Burk plot (Fig. 2a). As the concentrations of inhibitors increased, Vmax values were not affected, but Km values gradually decreased, thereby indicating that compound 11j was a competitive inhibitor against α-glucosidase (Fig. 2a). The Ki value was calculated directly by plotting the slope of each line in the Lineweaver–Burk plots into the different concentrations of compound 11j (Fig. 2b). The results proved that Ki value of compound 11j was 50.4 µM.

(a) Lineweaver–Burk plot of the kinetics of α-glucosidase inhibition by 11j. (b) Secondary re-plot of Lineweaver–Burk plots between the slopes of each line on Lineweaver–Burk plot versus various concentrations of 11j.
Molecular docking study
In order to explain interactions of the most potent compounds 11j, 11i, and 11f. in α-glucosidase active site, molecular docking simulation was carried out18. The superposed structure of acarbose and the title new compounds in the active site of target enzyme is shown in Fig. 3. Interaction modes of the positive control acarbose and selected compounds 11j, 11i, and 11f. were showed in the Fig. 3 and details of their interactions in the active site of target enzyme were listed in Table 2.

Acarbose (cyan) and the most potent compounds 11j, 11i, and 11f. (blue) superimposed in the α-glucosidase active site.
The 2D interaction mode of acarbose demonstrated that this positive control created eight hydrogen bonds with active site residues Thr307, Asn241, Glu304, Pro309, Ser308, Thr301, Arg312, and Gln322 (Fig. 4). Acarbose also formed a hydrophobic interaction with residue His279, two non-classical hydrogen bonds with residues Val305 and His239, and two unfavorable interactions with residues Thr307 and Arg312. Moreover, BE of acarbose was -4.04 kcal/mol.

2D interaction modes of the positive control acarbose and the most potent compounds 11j, 11i, and 11f in the α-glucosidase active site.
The most potent compound 11j created three hydrogen bonds with residues Asn241, His239, and Arg312 via oxygen atom of phenoxy moiety, 1,2,3-triazole ring, and carbonyl unit of acetamide moiety, respectively (Fig. 4). Phthalimide moiety of this compound established three interaction with active site: a π-anion interaction with Glu304 and two hydrophobic interactions with Pro309. Two π-cation interactions were also observed between phenyl ring of phenoxy moiety with residue His279 and 1,2,3-triazole ring with His239. 4-Bromophenyl ring of compound 11j established two hydrophobic interactions with Phe158 and Arg312 via bromo substituent and phenyl ring, respectively. The second potent compound 11i established two hydrogen bonds with residues Asn241 and Phe157 via 1,2,3-triazole ring and NH unit of acetamide moiety, respectively (Fig. 4). The latter amino acid also interacted with carbonyl unit of acatamide moiety via a non-classical hydrogen bond. Compound 11i formed a π-cation interaction and a π-anion interaction with residues His279 and Asp408. This compound also established several hydrophobic interactions with residues Val305, Pro309, His239, Phe311, Tyr313, Arg312, and Phe157.
The third potent compound 11f. created three classical hydrogen bonds with residues Asn241 and Asp408 via acetamide moiety and two non-classical hydrogen bonds with residues Phe311 and Arg312 via 1,2,3-triazole ring (Fig. 4). Phenyl ring of phenoxy moiety established a π-anion interaction with Glu304. Compound 11f. also formed an unfavorable interaction with residue Asp408 via carbonyl unit of acetamide moiety.
Docking study on the inactive compounds 11c and 11m showed that this compounds formed only a hydrogen bond with the active site of target enzyme. As can be seen in Fig. 5, compound 11c formed a hydrogen bond with Asn241, a π-cation interaction with His279, two non-classical hydrogen bonds with Thr301 and Arg312, an unfavorable interaction with Thr307, and several hydrophobic interactions with Val305, His239, His279, Pro309, and Arg312. Inactive compound 11m established a hydrogen bond with Phe157, two π-anion interactions with Glu304, two non-classical hydrogen bonds with His239 and His279, and several hydrophobic interactions with Pro309 and Arg312.

2D interaction modes of the inactive compounds 11c and 11 m in the α-glucosidase active site.
Molecular dynamics
Interaction of a ligand with a protein is a dynamic phenomenon which takes place on a very small time scale. Molecular dynamic simulation helps to grasp the interaction between ligand and protein and evaluate the stability and flexibility of the resulting complex. To this end, dynamics of the protein–ligand complex is simulated in an environment very similar to the natural environment that includes water and ions. Based on in vitro studies compound 11j has the most potential to inhibit α-glucosidase. Therefore the complex of α-glucosidase-11j was simulated in an explicit hydration environment by molecular dynamics simulation to evaluate the stability, flexibility and intermolecular interactions between α-glucosidase and this compound19. Moreover molecular dynamics of acarbose as a standard inhibitor in complex with α-glucosidase was simulated in an explicit hydration environment to have a decent reference for comparison. In this study, molecular dynamic simulation was performed in two steps. A 10 ns simulation at the first step to investigate if the ligands i.e. acarbose and 11j were stable at their binding site on α-glucosidase. After confirming the stability of ligands in their binding site, simulation time was extended for another 10 ns to gain a better comprehension of the behavior of these compounds in the active site of α-glucosidase. Stability of 11j and α-glucosidase was confirmed in this step too. For further evaluation the trajectory file was analyzed by several tools. To evaluate the stability of the complexes, root-mean-square deviation (RMSD) and radius of gyration (Rg) of all structures of the trajectory were calculated and the related graphs were drawn. To assess the residual flexibility and the flexibility of ligand atoms, the root mean square fluctuation (RMSF) of backbone atoms of α-glucosidase and heavy atoms of ligands were calculated.
Figures 6 and 7 show the result of RMSD calculations. According to Fig. 6 that shows the RMSD of backbone atoms of α-glucosidase in complex with 11j and acarbose, RMSD of α-glucosidase does not change very much and is less than 0.25 nm in all the simulation trajectory. RMSD less than 0.3 nm indicates a stable structure. The average RMSD values of α-glucosidase in the complex with acarbose and/or 11j were 0.17 and 0.16 nm, respectively. RMSD less than 0.25 nm was observed for acarbose and/or 11j in the complex with α-glucosidase too that shows they had the least conformational changes and were completely stable during the simulation time (Fig. 7). The average RMSD values of acarbose and/or 11j in complexes with α-glucosidase were 0.14 and 0.14 nm, respectively.

Superimposed RMSD of Cα atoms of α-glucosidase in complex with 11j (red) and acarbose (indigo).

Superimposed RMSD of 11j (red) and acarbose (indigo) in complex with α-glucosidase.
All atoms and consequently residues in a protein have some fluctuations. The fluctuation of α-glucosidase residues in complexes with acarbose and 11j are depicted in Fig. 8. According to this figure RMSF of α-glucosidase residues in the complex of this enzyme with acarbose and/or 11j are very similar and almost match. α-Glucosidase is a big protein with 579 residues and several domains with different structure and functions. As it could be seen from Fig. 8 the fluctuation of different parts of this big protein are not the same. Active site of this enzyme is located in a cleft between two domains i.e. A domain and B domain and the residues of these domains that contribute to the non-bond interactions with ligands have the lowest fluctuations. On the other hand, as is to be expected, residues located in the loop regions including B domain loop and active site lid have more fluctuations. RMSF of heavy atoms of acarbose and 11j are depicted in Fig. 9. As can be seen in this figure, the RMSF of all heavy atoms in these ligands is less than 0.2 nm. This low RMSF indicates that these compounds have a stable structure in complex with α-glucosidase and intermolecular interactions limit their fluctuations. Among the heavy atoms of acarbose and 11j those that were part of a ring had the lowest RMSF. Rings usually have less fluctuations as atoms of the ring limit their movements and make stable non-bond interactions such as π-Anion, π-Cation, π-Alkyl, π-π T-shaped, π-sigma, and hydrogen bonds with binding site residues of the protein. The radius of gyration (Rg) of α-glucosidase was calculated for evaluation of protein compactness during simulation (Fig. 10). The average Rg of α-glucosidase was 2.530 and 2.54 nm in the complex of α-glucosidase with acarbose and 11j, respectively. The Rg value of α-glucosidase in complexes with both acarbose and 11j was only in the narrow range of 2.45 to 2.52 nm and did not show a significant upward or downward trend during the simulation time that shows stable protein structures.

(A) RMSF graph of the Cα atoms of α-glucosidase in complex with acarbose (indigo) and 11j (red). (B) Close-up representation of α-glucosidase active site.

RMSF graph of the heavy atoms of 11j (A) and acarbose (B) in complex with α-glucosidase. Structure of these compounds and parts of these molecules with greatest fluctuations are illustrated.

Time dependence of the radius of gyration (Rg) graph of α-glucosidase in complex with 11j (red) and acarbose (indigo).
In silico pharmacokinetic and toxicity studies
Pharmacokinetic (ADME) and toxicity predictions of the positive control acarbose and the most potent compounds 11j and 11i were performed by PreADMET online software (Table 3). As can be seen in Table 3, acarbos did not follow of Lipinski 'Rule of five' while compounds 11j and 11i followed of this rule. Acarbose and compounds 11j and 11i had poor permeability to Caco-2. Permeability to blood brain barrier (BBB) and skin for the title compounds is in the acceptable range. Compounds 11j and 11i had high human intestinal absorption (HIA) while acarbose did not have HIA. Prediction of mutagenicity of acarbose and compounds 11j and 11i demonstrated that these compounds are mutagen. Moreover, this study predicted that acarbose had carcinogenic effect on mouse and did not have this effect on rat while compound 11j has carcinogenic effect on rat and mouse. Unlike the compound 11j, compound 11i has not carcinogenicity on the latter animals. Cardiotoxicity (hERG inhibition) of the positive acarbose is ambiguous while compounds 11jand 11i in term of this type of toxicity have medium risk.
Conclusion
In conclusion, we designed and synthesized a novel series of phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide 11a–n. The synthesized compounds 11a–n were evaluated against α-glucosidase because were designed based on active pharmacophores of potent reported α-glucosidase inhibitors. The majority of the title compounds displayed high α-glucosidase inhibitory activity. Among them, compounds 11j and 11i represented the most potent α-glucosidase inhibitory activities with IC50 values of 45.26 ± 0.03 and 46.25 ± 0.89 µM as compared to the positive control acarbose (IC50 = 750.1 ± 0.23 µM). In addition, kinetic analysis demonstrated that compound 11j behaves as a competitive inhibitor with a Ki value of 50.4 µM. The binding interactions of the most potent compounds and two inactive compounds at α-glucosidase active site were studied through molecular docking. The obtained results revealed that potent compounds formed significant interactions with the active site in comparison to inactive compounds. Molecular dynamics study of the most potent compound 11j and positive control acarbose also demonstrated that our new compound showed an appropriate state in the α-glucosidase active site. Furthermore, in silico pharmacokinetic study predicted that the most potent new compounds 11j and 11i have satisfactory pharmacokinetics after the oral admission as drug candidates.
Experimental
General
Melting points of compounds 11a–n were measured with a Kofler hot stage apparatus. IR spectra of these compounds were recorded with a Nicolet Magna FTIR 550 spectrophotometer (KBr disks). 1H and 13C NMR spectra of the title phthalimide derivatives were obtained with a Bruker FT-400 (TMS was used as an internal standard). Mass spectrometry results were obtained with an Agilent Technology (HP) mass spectrometer (Ionization potential: 70 eV). Elemental analysis was determined with an Elementar Analysen system GmbH VarioEL CHNS mode.
Synthesis of 2-(4-hydroxyphenyl)isoindoline-1,3-dione 3
A mixture of phthalic anhydride 1 (10 mmol) and 4-aminophenol 2 (10 mmol) in acetic acid (50 mL) was stirred at reflux condition for 5 h. Then, cold water was added to the reaction mixture and appeared participates were separated by filtration to give pure 2-(4-hydroxyphenyl)isoindoline-1,3-dione 3.
Synthesis of 2-(4-(prop-2-yn-1-yloxy)phenyl)isoindoline-1,3-dione 5
A mixture of 2-(4-hydroxyphenyl)isoindoline-1,3-dione 3 (1 mmol), propargyl bromide 4 (1.2 mmol, 0.15 mL), and K2CO3 (1.2 mmol) in acetone (10 mL) was stirred at RT for 12 h. After completion of the reaction (checked by TLC), the reaction mixture poured into cold water. Subsequently, the precipitated product was filtered off to give pure 2-(4-(prop-2-yn-1-yloxy)phenyl)isoindoline-1,3-dione 5.
General procedure for the synthesis of azide derivatives 10a–n
Azide derivatives 10a–n were synthesized in situ according to our pervious reported work16.
General procedure for the synthesis of phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamide 11a–n
A mixture of 2-(4-(prop-2-yn-1-yloxy)phenyl)isoindoline-1,3-dione 5 (1 mmol), sodium ascorbate, and CuSO4.5H2O (7 mol %) was added to the prepared azide derivatives 10a–n, and the reaction mixture was stirred at RT for 24–48 h. After that, the reaction mixture was poured into crushed ice and appeared participates 11a–n were filtered off, washed with cold water, and purified by recrystallization in ethyl acetate.
2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (11a)
Yield: 77%. White crystal. M.p. 218–220 °C. IR (KBr): 3428, 1680, 1367, 1198 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.34 (s, 1H), 7.92 (ddt, J = 20.0, 5.5, 3.2 Hz, 4H), 7.62 (dd, J = 8.8, 4.9 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.33–7.10 (m, 5H), 5.38 (s, 2H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.59, 158.11, 143.06, 135.25, 135.06, 132.03, 129.24, 126.96, 125.17, 123.79, 121.54, 121.46, 115.87, 115.34, 61.71, 52.66 ppm. EI-MS (70 eV): m/z (%) = 453.2 (M+). Anal. Calcd for C25H19N5O4: C 66.22; H 4.22; N 15.44; Found: C 66.46; H 4.38; N 15.68.
N-(2,3-dimethylphenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11b)
Yield: 85%. White crystal. M.p. 206–208 °C. IR (KBr): 3432, 1681, 1336, 1185 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.33 (s, 1H), 7.93 (ddt, J = 21.1, 5.5, 3.2 Hz, 4H), 7.52–7.27 (m, 2H), 7.30–7.12 (m, 3H), 7.14–6.91 (m, 2H), 5.44 (s, 2H), 5.26 (s, 2H), 2.26 (s, 3H), 2.13 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.71, 164.90, 158.13, 142.80, 137.63, 135.72, 135.07, 132.03, 131.51, 129.24, 127.75, 126.87, 125.78, 125.17, 123.79, 123.73, 115.34, 61.72, 52.41, 20.61, 14.50 ppm. EI-MS (70 eV): m/z (%) = 481.2 (M+). Anal. Calcd for C27H23N5O4: C 67.35; H 4.81; N 14.54; Found: C 67.46; H 14.69; N 14.68.
N-(2,6-dimethylphenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11c)
Yield: 88%. White crystal. M.p. 209–211 °C. IR (KBr): 3429, 1681, 1359, 1210 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H), 8.33 (s, 1H), 7.93 (ddd, J = 24.9, 5.5, 3.1 Hz, 4H), 7.45–7.26 (m, 2H), 7.28–7.14 (m, 2H), 7.09 (s, 3H), 5.43 (s, 2H), 5.26 (s, 2H), 2.18 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.50, 158.12, 142.82, 135.56, 135.08, 134.66, 132.04, 129.25, 128.23, 127.24, 126.84, 125.17, 123.80, 115.34, 61.70, 52.14, 18.52 ppm. EI-MS (70 eV): m/z (%) = 481.8 (M+). Anal. Calcd for C27H23N5O4: C 67.35; H 4.81; N 14.54; Found: C 67.58; H 14.77; N 14.36.
2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-ethylphenyl)acetamide (11d)
Yield: 82% (394 mg). White crystal. M.p. 202–204 °C. IR (KBr): 3434, 1683, 1320, 1162 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.34 (s, 1H), 7.92 (ddt, J = 21.1, 5.5, 3.2 Hz, 4H), 7.53 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 7.19 (dd, J = 20.9, 8.3 Hz, 4H), 5.39 (s, 2H), 5.27 (s, 2H), 2.55 (q, J = 7.6 Hz, 2H), 1.15 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.70, 164.39, 158.13, 142.83, 139.67, 136.57, 135.06, 132.01, 129.22, 128.55, 126.89, 125.17, 123.78, 119.78, 115.35, 61.72, 52.72, 28.07, 16.10 ppm. EI-MS (70 eV): m/z (%) = 481.3 (M+). Anal. Calcd for C27H23N5O4: C 67.35; H 4.81; N 14.54; Found: C 67.60; H 5.04; N 14.31.
2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(3-fluorophenyl)acetamide (11e)
Yield: 75%. White crystal. M.p. 204–206 °C. IR (KBr): 3441, 1686, 1328, 1201, 989 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.33 (s, 1H), 7.93 (ddt, J = 20.0, 5.5, 3.2 Hz, 4H), 7.58 (dt, J = 11.6, 2.1 Hz, 1H), 7.46–7.25 (m, 4H), 7.21 (d, J = 8.5 Hz, 2H), 6.93 (td, J = 8.4, 2.5 Hz, 1H), 5.41 (s, 2H), 5.27 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.10, 163.79 (1JC-F = 240 Hz), 158.11, 142.90, 140.61, (3JC-F = 11 Hz), 135.07, 132.03, 131.13, 113.04, 129.25, 126.92, 125.18, 123.79, 115.48, 115.45 (3JC-F = 10 Hz), 110.86 (2JC-F = 21 Hz), 106.67 (2JC-F = 26 Hz), 61.70, 52.71 ppm. EI-MS (70 eV): m/z (%) = 471.3 (M+).
Anal. Calcd for C25H18FN5O4: C 63.69; H 3.85; N 14.86; Found: C 63.48; H 4.69; N 14.99.
N-(3-chlorophenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11f.)
Yield: 71%. White crystal. M.p. 210–212 °C. IR (KBr): 3437, 1687, 1364, 1165, 758 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 8.33 (s, 1H), 8.05–7.84 (m, 4H), 7.79 (s, 1H), 7.54–7.32 (m, 4H), 7.26–7.06 (m, 3H), 5.41 (s, 2H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.13, 158.11, 142.88, 140.28, 135.08, 133.67, 132.04, 131.13, 129.26, 126.93, 125.17, 124.00, 123.80, 119.21, 118.12, 115.35, 61.70, 52.71 ppm. EI-MS (70 eV): m/z (%) = 487.5 (M+). Anal. Calcd for C25H18ClN5O4: C 61.54; H 3.72; N 14.35; Found: C 61.27; H 3.88; N 14.55.
N-(2,3-dichlorophenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11 g)
Yield: 68% (354 mg). White crystal. M.p. 203– 205 °C. IR (KBr): 3444, 1691, 1330, 1171, 763 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.33 (s, 1H), 7.93 (ddt, J = 20.2, 5.5, 3.1 Hz, 4H), 7.75 (dd, J = 8.2, 1.5 Hz, 1H), 7.50 (dd, J = 8.1, 1.4 Hz, 1H), 7.43–7.31 (m, 3H), 7.30–7.09 (m, 2H), 5.51 (s, 2H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.56, 158.11, 142.88, 136.62, 135.08, 132.46, 132.06, 129.25, 128.62, 127.55, 126.94, 125.43, 125.18, 125.18, 123.80, 115.34, 61.69, 52.44 ppm. EI-MS (70 eV): m/z (%) = 521.3 (M+).
Anal. Calcd for C25H17Cl2N5O4: C 57.49; H 3.28; N 13.41; Found: C 57.70; H 3.66; N 13.50.
N-(2,4-dichlorophenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11 h)
Yield: 74% (385 mg). White crystal. M.p. 232–234 °C. IR (KBr): 3435, 1688, 1341, 1210, 734 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.31 (s, 1H), 8.05–7.84 (m, 4H), 7.80 (d, J = 8.8 Hz, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.45 (dd, J = 8.7, 2.4 Hz, 1H), 7.41–7.30 (m, 2H), 7.26–7.00 (m, 2H), 5.49 (s, 2H), 5.25 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.54, 158.11, 142.83, 135.09, 133.85, 132.07, 130.25, 129.55, 129.27, 128.19, 127.57, 127.31, 126.91, 125.17, 123.81, 115.33, 61.68, 52.42 ppm. EI-MS (70 eV): m/z (%) = 521.1 (M+). Anal. Calcd for C25H17Cl2N5O4: C 57.49; H 3.28; N 13.41; Found: C 57.28; H 3.44; N 13.19.
N-(2,6-dichlorophenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11i)
Yield: 66% (343 mg). White crystal. M.p. 202–204 °C. IR (KBr): 3446, 1693, 1389, 1251, 770 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.32 (s, 1H), 7.93 (ddt, J = 20.2, 5.5, 3.1 Hz, 4H), 7.57 (d, J = 8.1 Hz, 2H), 7.39 (dd, J = 8.3, 5.0 Hz, 3H), 7.20 (d, J = 8.6 Hz, 2H), 5.47 (s, 2H), 5.25 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.71, 164.95, 158.11, 142.89, 135.08, 133.91, 132.50, 132.06, 130.06, 129.25, 129.10, 126.93, 125.17, 123.80, 115.33, 61.67, 51.89 ppm. EI-MS (70 eV): m/z (%) = 521.6 (M+). Anal. Calcd for C25H17Cl2N5O4: C 57.49; H 3.28; N 13.41; Found: C 57.36; H 3.14; N 13.72.
N-(4-bromophenyl)-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11j)
Yield: 73%. White crystal. M.p. 237–239 °C. IR (KBr): 3443, 1685, 1344, 1158, 632 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.33 (s, 1H), 7.93 (ddt, J = 20.0, 5.5, 3.1 Hz, 4H), 7.71–7.47 (m, 4H), 7.43–7.30 (m, 2H), 7.28–7.06 (m, 2H), 5.39 (s, 2H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.71, 164.88, 158.11, 142.86, 138.25, 135.07, 132.22, 132.05, 129.25, 126.90, 125.17, 123.80, 121.62, 115.88, 115.34, 61.70, 52.73 ppm. EI-MS (70 eV): m/z (%) = 531.3 (M+). Anal. Calcd for C25H18BrN5O4: C 56.41; H 3.41; N 13.16; Found: C 56.66; H 3.20; N 13.35.
2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-nitrophenyl)acetamide (11 k)
Yield: 66% (328 mg). Light brown crystal. M.p. 260–262 °C. IR (KBr): 3430, 1691, 1552, 1349, 1178 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.34 (s, 1H), 8.29–8.18 (m, 2H), 8.00–7.93 (m, 2H), 7.91 (td, J = 5.6, 5.1, 2.0 Hz, 2H), 7.87–7.77 (m, 2H), 7.45–7.31 (m, 2H), 7.27–6.94 (m, 2H), 5.48 (s, 2H), 5.27 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.82, 158.10, 144.97, 143.05, 142.88, 135.08, 132.06, 129.27, 126.90, 125.60, 125.18, 123.80, 119.50, 115.34, 61.68, 52.82 ppm. EI-MS (70 eV): m/z (%) = 498.2 (M+).
Anal. Calcd for C25H18N6O6: C 60.24; H 3.64; N 16.86; Found: C 60.39; H 3.47; N 17.02.
2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(2-methyl-4-nitrophenyl)acetamide (11 l)
Yield: 72%. Light brown crystal. M.p. 228–230 °C. IR (KBr): 3427, 1690, 1551, 1352, 1205 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.34 (s, 1H), 8.17 (d, J = 2.6 Hz, 1H), 8.09 (dd, J = 8.9, 2.7 Hz, 1H), 8.00–7.81 (m, 5H), 7.43–7.32 (m, 2H), 7.25–7.11 (m, 2H), 5.55 (s, 2H), 5.27 (s, 2H), 2.43 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.73, 158.10, 143.94, 142.88, 142.58, 135.09, 132.05, 131.80, 129.27, 126.92, 126.02, 125.18, 123.80, 123.73, 122.32, 115.34, 61.69, 52.66, 18.32 ppm. EI-MS (70 eV): m/z (%) = 512.5 (M+). Anal. Calcd for C26H20N6O6: C 60.94; H 3.93; N 16.40; Found: C 61.16; H 4.14; N 16.59.
N-benzyl-2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11m)
Yield: 80%. White crystal. M.p. 201–203 °C. IR (KBr): 3445, 1689, 1388, 1220 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (t, J = 5.8 Hz, 1H), 8.28 (s, 1H), 7.93 (ddd, J = 24.4, 5.6, 3.1 Hz, 4H), 7.49–6.82 (m, 9H), 5.24 (s, 2H), 5.23 (s, 2H), 4.35 (d, J = 5.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.90, 158.13, 142.78, 139.17, 135.08, 132.04, 129.25, 128.84, 127.87, 127.48, 126.80, 125.16, 123.80, 115.34, 61.71, 52.12, 42.87 ppm. EI-MS (70 eV): m/z (%) = 467.3 (M+). Anal. Calcd for C26H21N5O4: C 66.80; H 4.53; N 14.98; Found: C 66.68; H 4.74; N 15.15.
2-(4-((4-(1,3-dioxoisoindolin-2-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-fluorobenzyl)acetamide (11n)
Yield: 78% (367 mg). White crystal. M.p. 204–206 °C. IR (KBr): 3439, 1685, 1313, 1224, 963 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (t, J = 5.8 Hz, 1H), 8.28 (s, 1H), 7.93 (ddt, J = 19.7, 5.3, 3.1 Hz, 5H), 7.45–7.28 (m, 4H), 7.26–7.08 (m, 4H), 5.24 (s, 2H), 5.22 (S, 2H), 4.33 (d, J = 4.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.93, 162.94 (1JC-F = 241 Hz), 158.12, 142.82, 135.44 (4JC-F = 3 Hz), 135.08, 132.05, 129.92 (3JC-F = 8 Hz), 129.25, 126.83, 126.82, 125.16, 123.80, 115.67 (2JC-F = 22 Hz), 115.33, 61.71, 52.10, 42.14 ppm. EI-MS (70 eV): m/z (%) = 485.3 (M+). Anal. Calcd for C25H18FN5O4: C 63.69; H 3.85; N 14.86; Found: C 63.82; H 4.08; N 15.01.
In vitro α-glucosidase inhibition assay
Saccharomyces cerevisiae (S. cerevisiae) form of α-glucosidase (EC3.2.1.20, 20 U/mg) and p-nitrophenyl glucopyranoside (substrate) were purchased from Sigma-Aldrich. α-Glucosidase solution was produced in potassium phosphate buffer (PPB, pH 6.8, 50 mM) and phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamides 11a–n were dissolved in DMSO (10% final concentration)18. The various concentrations of the latter compounds 11a–n (20 µL), α-glucosidase solution (20 µL) and PPB (135 µL) were added in the 96-well plate and incubated at 37 °C for 10 min. Then, substrate (25 µL, 4 mM) was added to this plate and allowed to incubate at 37 °C for 20 min. The change in absorbance was measured at 405 nm using by a standard spectrophotometer (Gen5, Power wave xs2, BioTek, America). Acarbose and DMSO (10% final concentration) were used as positive and negative controls, respectively.
Kinetic study
The kinetic study was performed on the most potent compound 11j to determine inhibition mode of the newly synthesized compounds. The 20 µL of α-glucosidase solution (1 U/mL) was incubated with different concentrations of compound 11j (0, 12, 23 and 46 µM) for 15 min at 30 °C. The reaction was then started by adding different concentrations of substrate (1–10 mM) and change in absorbance was measured for 20 min at 405 nm using by spectrophotometer (Gen5, Power wave xs2, BioTek, America).
Docking study
Homology model of α-glucosidase was prepared based on the described method by Imran et al.19,20. In the first step, a PDB with high sequence similarity with S. cerevisiae α-glucosidase was searched by SWISS-MODEL and S. cerevisiae isomaltase with PDB code: 3A4A) was selected. This enzyme was 72% identical and had 85% similarity with the S. cerevisiae α-glucosidase. In the second step, S. cerevisiae isomaltase was subjected through sequence alignment and homology model was constructed using by automated homology modeling pipeline SWISS-MODEL (managed by Swiss Institute of Bioinformatics) and the quality of the obtained homology model was verified using PROCHECK21.
In the third step docking study of acarbose and the selected compounds was performed in the active site of modeled α-glucosidase. The 3D structures of the acarbose and selected inhibitors were built by MarvineSketch 5.8.3, 2012, ChemAxon (http://www.chemaxon.com) and converted to pdbqt coordinate using Auto dock Tools. The pdbqt coordinate of enzyme was produced using the same software. Prepared pdbqt files were used as input files for the AUTOGRID program. In this program for each atom type in the selected ligand, maps were calculated with 0.375 Å spacing between grid points and the center of the grid box was placed at x = 12.5825, y = 7.8955, and z = 12.519. The dimensions for the active site box were set at 40 × 40 × 40 Å. Flexible ligand dockings were accomplished for the selected ligands. Each docked system was carried out by 50 runs of the AUTODOCK program search by the Lamarckian genetic algorithm (LGA). The best poses of the selected ligands were selected for analyzing the interactions between target enzyme and the selected ligand. The obtained results were visualized using BIOVIA Discovery Studio v.3.5.
Molecular dynamics
MD simulations were performed using Groningen machine for chemical simulations (GROMACS) 5.1.222. Topology files and other force field parameters of the selected compounds were made by SwissParam server23. Protein topology file was constructed by using the pdb2gmx command and CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins). The protein–ligand complex (in.gro format) was created in Notepad + + and the topology file of the protein was edited to include topology parameters of ligand as well. The resulting complex was centered in a cubic box with a side length of 2.0 nm and the SPC216 water model was used to fill the system. The net negative charge of the protein was neutralized by fifteen Na + ions which replaced the same number of water molecules. The Steepest descent minimization algorithm was used for the minimization of the system in a maximum number of 50,000 steps until the maximum force became less than 10.0 kJ/mol. For NVT equilibration the v-rescale algorithm was used in 300 K with a coupling constant of 0.1 ps and time duration of 500 ps. The last phase in preparation of the system was NPT equilibration. In this step, Berenson pressure coupling algorithm with a coupling constant of 5.0 ps was applied for 1000 ps of NPT simulation. Particle Mesh Ewald (PME) algorithm was used for long-range electrostatics and cut-off method for van der Waals interactions. Cut off distances were set at 1.0 nm for the calculation of the electrostatic and 1.2 nm for van der Waals interactions. Finally, 20 ns MD simulation was performed for the protein–ligand complex.
In silico pharmacokinetic and toxicity predictions
In silico prediction of pharmacokinetic properties and toxicity profile of the positive control acarbose and the most potent compounds 11j and 11i was performed using by the preADMET online server24.
Ethical approval and consent to participate
The ethics code for this work is IR.NIMAD. REC.1400.155 (https://ethics.research.ac.ir/IR.NIMAD.REC.1400.155).
Data availability
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Kashtoh, H. et al. Recent updates on phytoconstituent alpha-glucosidase inhibitors: An approach towards the treatment of type two diabetes. Plants 11, 2722 (2022).
Naumoff, D. G. et al. Hierarchical classification of glycoside hydrolases. Biochem. (Mosc.) 76, 622–635 (2011).
de Melo, E. B. et al. α-and β-Glucosidase inhibitors: chemical structure and biological activity. Tetrahedron 62, 10277–10302 (2006).
Gong, L. et al. Inhibitors of α-amylase and α-glucosidase: Potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci. Nutr. 8, 6320–6337 (2020).
Hedrington, M. S. & Davis, S. N. Considerations when using alpha-glucosidase inhibitors in the treatment of type 2 diabetes. Expert Opin. Pharmacother. 20, 2229–2235 (2019).
Tan, M. H. α-Glucosidase inhibitors in the treatment of diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 4, 48–55 (1997).
Warfield, K. L. et al. Targeting endoplasmic reticulum α-glucosidase I with a single-dose iminosugar treatment protects against lethal influenza and dengue virus infections. J. Med. Chem. 63, 4205–4214 (2020).
Gueder, N. et al. sp2-Iminosugar α-glucosidase inhibitor 1-C-octyl-2-oxa-3-oxocastanospermine specifically affected breast cancer cell migration through Stim1, β1-integrin, and FAK signaling pathways. J. Cell. Physiol. 232, 3631–3640 (2017).
Lazar, C. et al. Treatment of hepatitis B virus-infected cells with α-glucosidase inhibitors results in production of virions with altered molecular composition and infectivity. Antivir. Res. 76, 30–37 (2007).
Dhameja, M. & Gupta, P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 176, 343–377 (2019).
Mbarki, S. et al. 3D-QSAR for α-glucosidase inhibitory activity of N-(phenoxyalkyl) phthalimide derivatives. IJRRAS 11, 395–401 (2012).
Pascale, R. et al. New N-(phenoxydecyl) phthalimide derivatives displaying potent inhibition activity towards α-glucosidase. Bioorg. Med. Chem. 18, 5903–5914 (2010).
Dodo, K. et al. Co-existence of α-glucosidase-inhibitory and liver X receptor-regulatory activities and their separation by structural development. Bioorg. Med. Chem. 16, 4272–4285 (2008).
Sherafati, M. et al. Design, synthesis and biological evaluation of novel phthalimide-Schiff base-coumarin hybrids as potent α-glucosidase inhibitors. Chem. Pap. 74, 4379–4388 (2020).
Sadat-Ebrahimi, S. E. et al. New phthalimide-benzamide-1, 2, 3-triazole hybrids; design, synthesis, α-glucosidase inhibition assay, and docking study. Med. Chem. Res. 29, 868–876 (2020).
Asemanipoor, N. et al. Synthesis and biological evaluation of new benzimidazole-1, 2, 3-triazole hybrids as potential α-glucosidase inhibitors. Bioorg. Chem. 95, 103482 (2020).
Yavari, A. et al. α-Glucosidase and α-amylase inhibition, molecular modeling and pharmacokinetic studies of new quinazolinone-1, 2, 3-triazole-acetamide derivatives. Med. Chem. Res. 30, 702–711 (2012).
Mohammadi-Khanaposhtani, M. et al. New biscoumarin derivatives as potent α-glucosidase inhibitors: synthesis, biological evaluation, kinetic analysis, and docking study. Polycycl. Aromat. Compd. 40, 915–926 (2020).
Imran, S. et al. Synthesis of novel flavone hydrazones: in vitro evaluation of a-glucosidase inhibition, QSAR analysis and docking studies. Eur. J. Med. Chem. 105, 156–170 (2015).
Imran, S. et al. Synthesis, in vitro and docking studies of new flavone ethers as aglucosidase inhibitors. Chem. Biol. Drug Des. 87, 361–373 (2016).
Kiefer, F. et al. The SWISS-MODEL Repository and associated resources. Nucl. Acids Res. 37, D387–D392 (2008).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Zoete, V. et al. A fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 (2011).
Seul, South Corea: Bioinformatics and Molecular Design Research Center; 2004. PreADMET program. http://preadmet.bmdrc.org.
Acknowledgements
The presented work was financially supported by the National Institute for Medical Research Development (NIMAD). Furthermore, the authors thankfully acknowledge the scientific support provided by Research and Technology Empowerment Committee of Babol University of Medical Science.
Funding
The presented work was financially supported by NIMAD (the Grant No.: 4000574).
Author information
Authors and Affiliations
Contributions
M.E. and M.H. performed in silico studies and wrote the main manuscript text, M.M. and M.M-Kh conceived the idea and designed the experiments, A.A. and S.H contributed in the analysis of data, and R.G, A.A.M, S.M., and M.A.F. performed in vitro biological assay. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Emadi, M., Halimi, M., Moazzam, A. et al. Design, synthesis, in vitro anti-α-glucosidase evaluations, and computational studies of new phthalimide-phenoxy-1,2,3-triazole-N-phenyl (or benzyl) acetamides as potential anti-diabetic agents. Sci Rep 13, 10030 (2023). https://doi.org/10.1038/s41598-023-36890-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36890-y
- Springer Nature Limited