
Abstract
The present investigation aimed to identify genome wide SNPs and to carry out diversity and population structure study using ddRAD-seq based genotyping of 58 individuals of six indigenous milch cattle breeds (Bos indicus) such as Sahiwal, Gir, Rathi, Tharparkar, Red Sindhi and Kankrej of India. A high percentage of reads (94.53%) were mapped to the Bos taurus (ARS-UCD1.2) reference genome assembly. Following filtration criteria, a total of 84,027 high quality SNPs were identified across the genome of 6 cattle breeds with the highest number of SNPs observed in Gir (34,743), followed by Red Sindhi (13,092), Kankrej (12,812), Sahiwal (8956), Tharparkar (7356) and Rathi (7068). Most of these SNPs were distributed in the intronic regions (53.87%) followed by intergenic regions (34.94%) while only 1.23% were located in the exonic regions. Together with analysis of nucleotide diversity (π = 0.373), Tajima’s D (D value ranging from − 0.295 to 0.214), observed heterozygosity (HO ranging from 0.464 to 0.551), inbreeding coefficient (FIS ranging from − 0.253 to 0.0513) suggested for the presence of sufficient within breed diversity in the 6 major milch breeds of India. The phylogenetic based structuring, principal component and admixture analysis revealed genetic distinctness as well as purity of almost all of the 6 cattle breeds. Overall, our strategy has successfully identified thousands of high-quality genome wide SNPs that will further enrich the Bos indicus representation basic information about genetic diversity and structure of 6 major Indian milch cattle breeds which should have implications for better management and conservation of valuable indicine cattle diversity.
Similar content being viewed by others


Introduction
The Indian subcontinent is home to a mega diverse Bos indicus cattle breeds of the world1. The Indian zebu cattle (Bos Indicus) is believed to have originated from wild aurochs Bos primigenius nomadicus2,3 and studies based on mitochondrial DNA markers analysis indicated that Bos indicus separated from Bos taurus between 110,000 and 850,000 years ago4,5. Worldwide, I1 and I2 are the two major mitochondrial DNA (mtDNA) haplogroups have been reported for Bos indicus. The I1 which is predominant haplogroup is believed to have originated from India–Pakistan, whereas haplogroup I2 has a complex diversity pattern making it difficult to resolve its origin6,7,8. Although recent findings have identified a new sub-haplogroup I1a, in the I1 haplogroup within the Bos indicus lineage9. On the other hand, the Y chromosome diversity found in Bos indicus cattle is characterized by a single haplogroup Y3, in contrast to two different haplogroups Y1 and Y2 found in Bos taurus. In addition, two distinct sub-haplogroups within each of the Y2 (Y2a and Y2b) and Y3 (Y3a and Y3b) haplogroups have been identified. The Y3 haplogroup was observed to be inimitable to Bos indicus and findings have shown that the sub-haplogroup Y3a dominated the cattle from South China, whereas the sub-haplotype Y3b was found in the Bos indicus breeds of Indian origin10. With a population of 192.49 million cattle, India has 13.1% of world’s cattle population11. Further, India holds the first rank in milk production in the world with a total production of 198.4 million tons of milk production during 2019–202012. The Indian zebu cattle is an important member of the Bovidae family and is a major resource for milk and drought power in the Indian subcontinent. At present there are 53 well defined Indigenous cattle breeds in India which can be differentiated as dairy, dual or draft purpose breeds on the basis of their utility. The dairy cattle breeds on the average produced more than 1600 kg of milk per lactation, the dual-purpose breeds yield about 150–500 kg per lactation while the draft breeds are mainly used for agricultural work. The major dairy breeds of India include Gir (GIC), Rathi (RAC), Red Sindhi (RSC), Sahiwal (SAC) and Tharparkar (THC), the dual-purpose breeds comprise of Badri, Belahi, Deoni, Gaolao, Hariana, Kankrej, Konkani, Ladakhi, Malnad Gidda, Mewati, Ongole while the remaining breeds are classified as draft breeds.
The Indian native cattle breeds (Bos indicus) are well adapted to withstand the harsh climate and still perform efficiently. Besides, these are suited to low input production system with lower maintenance and management requirements. However, under the present production system, which is mostly focusing on increased milk production, the population size of indigenous cattle is in declining phase due to (1) modernization of agriculture and (2) cross breeding with exotic breeds to maximize the overall production and economic profit. The negligence of superior characters of Indian cattle like adaption to diverse climate and survival on low input system over production is resulting in loss of breeds or overall genetic diversity. Hence mitigation measures to characterize and conserve genetic diversity is key to evade further loss of important gene/gene pool and loss in the variability, which is very important for achieving higher genetic gain in economic traits of the indigenous dairy cattle breeds. In depth characterization and evaluation of genetic diversity among cattle breeds is of great importance to ensure long-term genetic improvement, facilitate rapid adaptation to changing climate and for efficient management and conservation of animal genetic resources13,14.
Genome-wide studies focused on population genetics, phylogeography and conservation biology have been greatly facilitated by quick advances in high-throughput sequencing technologies15. In recent years, reduced representation sequencing method such as double-digest restriction site associated DNA (ddRAD) approaches has received worldwide attention due to their capacity to identify genome-wide variations at relatively low cost. Genome wide SNPs based diversity and population structure analysis using ddRAD have been carried out in different livestock species like buffalo, yak, horse and camel16,17,18,19. ddRAD being a restriction digestion based reduced representation Next Generation Sequencing method fragments a target genome with both frequent and rare cutting restriction enzymes and such a strategy minimizes the hassles of uninformative and repetitive sequences, sequence assembly and SNP calling that accompanies with Whole genome sequencing (WGS). Evidently, ddRAD has been employed for the discovery of species-specific genome wide SNPs in economic, production and adaptation traits related candidate genes20,21,22. Furthermore, reduced representation methods based on whole genome sequencing of single individuals solve the problem of ascertainment bias23. Previously, array-based SNP chips has been widely used in genetic studies of livestock, including genome-wide association studies (GWAS)24,25, selection signature studies26,27, diversity and population structure analysis28,29,30. However, SNP chips commonly include SNPs that were previously discovered by DNA sequencing. These SNPs may not be geographically representative and tend to be at higher frequency than random SNPs and most importantly impair identification of casual mutations. Hence, population genetic parameters such as diversity, population structure and recombination estimates may be biased23,31. Therefore, in this study we have applied ddRAD sequencing approach, to overcome ascertainment biasness for discovering genome wide SNPs and undertake diversity studies in indicine cattle.
Considering the valuable contribution of the native dairy cattle in supporting the livelihood of many Indians for many generations, few efforts have been made to evaluate the genetic diversity and relationship in Indian cattle using genome wide SNPs. Comprehensive characterization on within-and between-breed genetic diversity of the Indian native cattle breeds to facilitate an effective and rational management is lacking. Exploration of the genetic diversity including population structure and admixture can expedite appropriate conservation programs. Deep and thorough understanding of indigenous genes/gene pool will help to understand the mechanism underlying important functional traits and help to meet the future production demands of the local people. The present investigation was undertaken to identify genome wide SNPs and assess the within and between breed genomic diversity and establish breed relationships and to assess their population structure.
Results
Quality control, alignment and SNP calling
The ddRAD sequencing based genotyping of 58 individuals belonging to six native cattle breeds; Gir, Sahiwal, Tharparkar, Rathi, Red Sindhi and Kankrej cattle with their geographical and ecological distribution (Fig. 1) including the productive purpose, coat colour, representative agroclimatic zone, breeding tract, the geographical co-ordinate of each breeding tract along with animal ID and sex of each individual presented in Supplementary Table S1; resulted in 138.59 million raw reads that corresponded to 23 million reads per breed and 2.2 million reads per animal. After initial filtering on the basis of read quality and adaptor removal, majority of the reads (138.58 million reads; 99.9%) were retained (Supplementary Table S2). A high percentage of reads (94.53%) were mapped to the Bos taurus (ARS-UCD1.2) reference assembly (Supplementary Table S2). In this study, the effort was made to analyze only the SNPs across different cattle breeds, therefore all other variants were not considered in subsequent analysis. The number of SNPs in 6 cattle breeds ranged between 8,42,768 and 3,81,966 after individual variant calling. Maximum number of SNPs were observed in SAC (8,42,768), followed by GIC (8,34,780), KAC (8,10,279), RAC (8,05,020), RSC (6,72,632) and THC (3,81,966) (Table 1). The combined data set across 6 cattle breeds produced a total of 43,47,445 SNPs. Subsequently, the VCF file was processed in a stepwise manner to filter out low quality SNPs. Firstly, the SNPs were filtered at read depth of 2 (RD 2), read depth of 5 (RD 5) and read depth of 10 (RD 10). For further analysis, the data set of 9,82,174 SNPs identified at RD of 5, were utilized for subsequent analysis (Table 1). All those SNPs that were present at low coverage (RD < 5) were removed from the data set. The SNPs that were identified at RD of 5 were further filtered using various criteria’s such as proportion of missing genotypes, minor allele frequency and Hardy Weinberg Equilibrium (HWE). The series of filtering resulted in a total of 84,027 high quality SNPs. Post filtering, the number of SNPs across breeds varied considerably. The highest number of SNPs was observed in GIC (34,743), followed by RSC (13,092), KAC (12,812), SAC (8956), THC (7356) and RAC (7068) (Table 2).

Geographical distribution of six cattle breeds included in this study (The map was generated using websites Map Chart https://www.mapchart.net/ and Paint Maps https://paintmaps.com/).
Functional annotation of variants
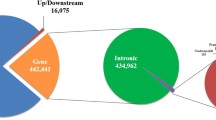
The merged high-quality SNPs dataset of all the 6 milch breeds was annotated to Bos taurus (ARS-UCD1.2) reference genome. With respect to their distribution in the genome, a large number of annotated SNPs were predicted to be in the intronic region (41,372 SNPs, 53.87%) followed by intergenic regions (26,834 SNPs, 34.94%). There were only 948 SNPs (1.23%) that were distributed in the exonic regions. Further, there were 3497 SNPs (4.55%) located within the 5 Kb region upstream and 3661 SNPs (4.77%) in the downstream of transcription start site. The analysis also resulted in 93 SNPs (0.121%) located in 5’UTR, 293 SNPs (0.38%) in 3’UTR region. A total of 8 SNPs (0.01%) were predicted to cause premature stop codon were also identified (Fig. 2).

Overall partitioning of SNPs with respect to genomic distribution for all the breeds.
On the basis of the impact of SNPs on protein coding genes, the SNPs were categorized as having high impact (10 SNPs; 0.01%), moderate impact (298 SNPs; 0.39%), and low impact, (697 SNPs; 0.91%). Majority of the SNPs (75,801; 98.69%) were identified as modifier (Supplementary Table S3). Additionally, high proportion of SNPs (65.74%) were silent in nature, followed by missense (33.37%) and nonsense (0.89%), with an average missense/silent ratio of 0.507 (Supplementary Table S4). In addition, amongst all genotypes substituted identified in the present study, C/T and G/A genotypes were found to be predominant, whereas A/T genotype was found to be in lowest proportions (Supplementary Table S5). For individual breed, the annotation results are summarized in Fig. 3 and Supplementary Table S6. In GIC, highest number of SNPs 32,283 (53.96%) were predicted to be in the intronic region followed by intergenic region 20,395 (34.09%). Only 777 (1.3%) were detected in the exonic region. Similar to GIC, the highest number of SNPs were distributed in intronic region followed by intergenic and exonic region in all other cattle breeds. For example, in SAC, 53.87% of SNPs (8429) were predicted in the intronic region followed by intergenic region 33% (5163 SNPs) and only 1.75% (273 SNPs) in exonic region. A similar trend was observed for RAC, RSC, KAC and THC cattle breeds with 6834 (55.63%), 11,147 (52.12%), 8429 (53.87%), 6374 (52.58%) SNPs, respectively in the intronic region, 4186 (34.08%), 8192 (38.30%), 5163 (33%), 4507 (37.18%) SNPs respectively, in the intergenic region and only 142 (1.16%), 266 (1.24%), 273 (1.75%), 123 (1.02%) were predicted in the exonic regions (Fig. 3). The number of synonymous variants identified in GIC, KAC, RAC, RSC, SAC and THC were 570, 190, 101, 172, 213 and 87 respectively. On the other hand, the number of non-synonymous variants detected for the 6 cattle breeds were 165, 64, 31, 82, 53 and 30 respectively. The TS/TV ratio observed in GIC, KAC, RAC RSC SAC and THC was 2.55, 2.64, 2.33, 2.43, 2.51 and 2.19 respectively (Supplementary Table S6).

Genomic distribution of SNPs across the genome of six Indian milch cattle breeds.
The numbers of intergenic SNPs were 4,639,873 (68.1%) and 1,676,710 (24.6%) were intronic. There were 230,365 (3.4%) SNPs located within 5 kb upstream and 197,827 (2.9%) in downstream of a transcription start site; 12,428 SNPs were located in the 5′ UTR and 2613 in the 3′ UTR. A total of 4356 SNPs were located in splice sites of 2966 genes: 142 were in splice-donor sites, 142 were splice-acceptor sites and 4072 were within the region of the splice site. We identified 45,776 SNPs affecting the coding sequences of 11,538 genes. There were 221 SNPs predicted to cause premature stop codon and 17 to cause gain in coding sequence. The numbers of SNPs predicted to be non-synonymous were 20,828. The numbers of intergenic SNPs were 4,639,873 (68.1%) and 1,676,710 (24.6%) were intronic. There were 230,365 (3.4%) SNPs located within 5 kb upstream and 197,827 (2.9%) in downstream of a transcription start site; 12,428 SNPs were located in the 5′ UTR and 2613 in the 3′ UTR. A total of 4356 SNPs were located in splice sites of 2966 genes: 142 were in splice-donor sites, 142 were splice-acceptor sites and 4072 were within the region of the splice site. The numbers of intergenic SNPs were 4,639,873 (68.1%) and 1,676,710 (24.6%) were intronic. There were 230,365 (3.4%) SNPs located within 5 kb upstream and 197,827 (2.9%) in downstream of a transcription start site; 12,428 SNPs were located in the 5′ UTR and 2613 in the 3′ UTR. A total of 4356 SNPs were located in splice sites of 2966 genes: 142 were in splice-donor sites, 142 were splice-acceptor sites and 4072 were within the region of the splice site. We identified 45,776 SNPs affecting the coding sequences of 11,538 genes. There were 221 SNPs predicted to cause premature stop codon and 17 to cause gain in coding sequence. The numbers of SNPs predicted to be non-synonymous were 20,828. The numbers of intergenic SNPs were 4,639,873 (68.1%) and 1,676,710 (24.6%) were intronic. There were 230,365 (3.4%) SNPs located within 5 kb upstream and 197,827 (2.9%) in downstream of a transcription start site; 12,428 SNPs were located in the 5′ UTR and 2613 in the 3' UTR. A total of 4,356 SNPs were located in splice sites of 2966 genes: 142 were in splice-donor sites, 142 were splice-acceptor sites and 4072 were within the region of the splice site. We identified 45,776 SNPs affecting the coding sequences of 11,538 genes. There were 221 SNPs predicted to cause premature stop codon and 17 to cause gain in coding sequence. The numbers of SNPs predicted to be non-synonymous were 20,828.
Within breed diversity
The nucleotide diversity (π) was highest in THC (π = 0.458) followed by RSC (π = 0.364), SAC (π = 0.363), GIC (π = 0.356), KAC (π = 0.348) and RAC (π = 0.347). The mean nucleotide diversity value was 0.373 (Table 3). The Tajima’s D values were negative for 4 cattle breeds viz., RSC, RAC, SAC and THC except for GIC and SAC where positive D values was observed. The highest negative Tajima’s D value was observed in THC (-1.194) followed by RSC (− 1.088), RAC (− 0.295) and KAC (− 0.279).
The observed heterozygosity (HO) values ranged from 0.464 to 0.551 while the expected heterozygosity (HE) ranged from 0.448 to 0.535. The highest observed heterozygosity values were observed in THC (HO = 0.551) followed by RAC (HO = 0.523), RSC (HO = 0.5184), SAC (HO = 0.5180), GIC (HO = 0.499) and KAC (HO = 0.464) (Table 4). The average FIS (inbreeding coefficient) ranges from -0.253 in THC to 0.0513 in KAC. The FIS estimate amongst the six cattle breeds was highest in THC (FIS = − 0.253) followed by RAC (FIS = − 0.105), whereas the lowest FIS estimate was observed in KAC (FIS = 0.0513) followed by GIC (FIS = − 0.00063). The overall FIS analysis revealed excess of heterozygosity for all the cattle breeds except for KAC (Table 4). The heterozygosity and FIS estimates indicated presence of sufficient diversity within the six cattle breeds.
Between breed diversity
The genetic differentiation on the basis of fixation index (FST) ranged from 0.2840 to 0.3905, indicating sufficient between breed diversity. The highest divergence was observed between RAC-SAC pair (FST = 0.3905), followed by RSC-RAC breed pair (FST = 0.3790), RSC-SAC breed pair (FST = 0.3751). The least divergence was observed for KAC-THC breed pair (FST = 0.2840) (Table 5). Neighbour Joining (NJ) based tree constructed, grouped the individual animals of 6 cattle breeds as per their breed affiliations with GIC and RSC being the most diverse breed amongst the 6 studied cattle breeds. The phylogenetic relationship at individual level is shown in Fig. 4. The breed wise NJ tree depicted in Fig. 5, more or less corroborated with the individual level tree. Furthermore, UPGMA based phylogenetic tree was constructed at breed level using “phangorn” package in R platform with 100 bootstrap values. The bootstrap values of each node were close to 100% indicating high robustness of the constructed tree. UPGMA based phylogenetic tree reflected similar genetic relationship as revealed by NJ based genetic differentiation (individual wise and at breed level) where GIC and RSC appeared as the most distinct breeds. GIC appeared on major node and clustered as one group while the other populations formed two groups with RSC clustered on one node and RAC, THC, SAC and KAC formed other sub clusters (Fig. 6).

Neighbour-Joining based phylogenetic grouping of 58 animals of six Indian milch cattle breeds using Tassel software.

Neighbour-Joining based grouping of 6 Indian milch cattle breeds using “phangorn” package of R platform.

UPGMA based phylogenetic grouping of six Indian milch breed using “phangorn” package of R platform.
Population structure analysis
The admixture analysis was carried out by partitioning the genome of each individual into a predefined cluster. The analysis was performed at K = 3, 4, 5 and 6 (Fig. 7). The individuals could not be grouped at K = 3 as per their respective breed. Only GIC could distinctly be differentiated while the individuals of KAC and SAC appear as one group and RAC, THC and RSC are clustered together indicating their shared ancestry. At K = 4, and even at K = 5, THC, RAC and RSC clustered together indicating their strong shared ancestry, while all other individuals clustered in their own respective breed. The best K in population structure analysis is K = 6, whereby almost all the animals were grouped to their respective breed, clearly indicating their sperate ancestry, with the exception of RSC and THC which still clustered together. The genetic closeness between RSC and THC could be unveiled by further in-depth studies and by increasing the number of samples.

Admixture analysis assuming 3 ≤ K ≤ 6.
The PCA based analysis also clustered 6 cattle breeds separately and reinforces the fact that these are distinct cattle breeds (Supplementary Fig. S1). Individuals of KAC were grouped together in one quadrant, while individuals of SAC RAC, THC and RSC cattle breeds fall in a different quadrant. Individuals of GIC cattle breed appeared as a distinct population.
Discussion
The Indian subcontinent is bestowed with immense richness of zebu (Bos indicus) cattle breeds. Geographical isolation over time has built up a plethora of genetic types/breeds but the magnitude of genetic differentiation has not been well quantified. Genetic variability in indigenous breeds is a major concern considering the necessity of preserving what may be a precious and irreplaceable richness developed as the results of complex interactions between the genotype and the environment. Hence molecular information is crucial for preserving genetic diversity as well as preventing undesirable loss of alleles. In this study genetic diversity and population structure of 6 major Indian milk cattle breeds was estimated using large number of genome wide SNPs generated through ddRAD sequencing.
In the present study, an average of 2.2 million reads per animal were obtained with a high mapping rate of 94.53% to Bos taurus reference genome (ARS-UCD1.2). The total number of SNPs identified in the present study varies when compared to previous reports. Gurgul et al.32 reported 8065 high-confidence SNPs in 48 individuals of different taurine cattle breeds by single enzyme restriction digestion GBS approach. Likewise, by using the same approach Malik et al.33 identified 1,07,488 SNPs in 24 animals belonging to seven Indian cattle breeds viz., Gangatiri, Hariana, Kankrej, Ongole, Sahiwal, Siri and Tharparkar. Furthermore, De Donato et al.34 identified 63,697 SNPs in 47 animals from both taurine and indicine breeds using a single enzyme GBS method. On the other hand, by using 2 enzyme GBS protocol, Brouard et al.35 reported a total of 2,72,103 variants in 48 Canadian dairy cattle. Similarly, RAD sequencing was used to identify 2,38,725 and 84,854 high-confidence SNPs in Sichuan and Liangshan indigenous cattle breeds of China, respectively36,37. Recently, studies carried out using ddRAD in Indian native cattle breed Sahiwal reported a total of 258,231 genome-wide SNPs with a minimum read depth of 2, 10, 232,570 SNPs at read depth 5 and 193,803 SNPs were identified respectively21. The number of high confidence SNPs identified in the present study and other previous studies could be attributed to the levels and stringency of filtering parameters applied while calling for SNPs.
The average Ts/Tv ratio was found to be 2.53. The observed Ts/Tv ratio in the present study was similar to many other reduced representation sequencing studies carried out in yak, buffaloes and cattle17,38,39. Large numbers of SNPs detected were found to be in the intronic and intergenic regions, which was similarly observed in previous studies16,22. Annotation of SNPs in this study also revealed that the G/A and C/T substituted genotypes were mostly found, whereas AT genotype was the least. This observation was similar to the studies carried out by Kumar et al.11 and Wang et al.36.
The nucleotide diversity (π) with an overall value of 0.373 was significantly higher in the 6 Indian native milch breeds when compared to the mean nucleotide diversity (π = 0.18 and π = 0.227) reported for Chinese cattle using RAD sequencing36,37. Furthermore, the nucleotide diversity in the studied cattle breeds was also comparatively high when compared to the nucleotide diversity reported for Eastern Finn cattle, Western Finn cattle, and Yakutian cattle with π values of 1.559 × 10–3, 1.512 × 10–3, and 1.728 × 10–3, respectively40. Similarly, other taurine breeds such as Braunvieh cattle of Switzerland also showed relatively lower nucleotide diversity41. The results of nucleotide diversity strongly suggest that the 6 major milch breed of India are maintaining sufficient degree of within breed genetic variation.
The negative Tajima’s D values observed for the 4 Indian cattle breeds viz., RSC, RAC, KAC and THC except for GIC and SAC signifies the population size expansion and presence of an excess of rare alleles. Further, the negative Tajima’s D values also signifies the occurrence of recent positive selection in these Indian native cattle breeds. This observation was also consistent with selection signals detected in some of the other SNP array-based studies involving Indian native breeds26 On the contrary the positive Tajima’s D values detected for Gir and Sahiwal indicates signals of balancing selection in these breeds. Similar observation was also reported whereby a lower selection signals in 7 Indian native breeds such as Gir, Hariana, Kankrej Ongole, Red Sindhi, Sahiwal and Tharpakar using 50 K bovine SNPchip data was identified22.
The observed (HO) and expected heterozygosity (HE) values for the 6 cattle breeds ranged from 0.464 to 0.551 (HO) and 0.448 to 0.535 (HE), respectively. The maximum heterozygosity value was observed in THC (HO = 0.551) followed by RAC (HO = 0.523), RSC (HO = 0.5184), SAC (HO = 0.5180), GIC (HO = 0.499), while the lowest level of heterozygosity was observed in KAC (HO = 0.464). The diversity estimates in the present study was much higher to what has been reported in seven taurine and indicine cattle breeds (0.064 to 0.197) from the US and Africa using GBS approach34. In addition, lower heterozygosity value (0.22) was also reported in Chinese cattle using restriction site-associated DNA sequencing (RADSeq)37. Further low diversity values were also reported by Malik et al.33 for two Indian Bos indicus cattle breeds; Sahiwal (HO = 0.084) and Kankrej (HO = 0.086) along with Bos taurus Holstein Frisian cattle33 using GBS approach. The explanation for lower heterozygosity in aforesaid mentioned studies could be due to use of single enzyme digestion in RADSeq and GBS approaches.
The overall FIS analysis revealed significant deficit of inbreeding levels in all the cattle breeds under study (FIS ranges from − 0.253 in THC to 0.0513) as high FIS estimates is linked to high degree of inbreeding. The high negative FIS values obtained in the study are similar to the study carried out by Strucken et al. (2021)28 in 13 Indian cattle breeds using 777 k SNP BovineHD Beadchip with exception for Sahiwal where indications of inbreeding are observed.
Sahiwal is an important and by far the best milch cattle breed of India, hence the race for rearing pure line animals with desired economic traits could have resulted in the slight increase in the FIS estimate. However, the overall FIS estimates observed are in contrast to those identified for Indian native cattle by microsatellite markers42,43,44 who reported high FIS values. The depression in the FIS estimate in the present investigation demonstrates the presence of heterozygote excess in the Indian native cattle breeds.
The fixation index (FST) values that ranged from 0.2840 to 0.3905 suggested moderate to substantial genetic differentiation across the 6 cattle breeds. The maximum divergence was observed between RAC-SAC pair (FST = 0.3905), followed by RSC-RAC breed pair (FST = 0.3790), RSC-SAC breed pair (FST = 0.3751). The least divergence was observed for KAC-THC breed pair (FST = 0.2840). In few other studies based on bovine 50 K and 770 K SNP chips, the authors have reported relatively lower FST values for Indian cattle breeds28,29. The overall high between breed genetic differentiation in the present investigation might be attributed to the fact that the blood samples were obtained from individuals that are true to the breed and the studied populations are genetically distinct from each other.
The dendogram analysis on the basis of genetic distance and branch length revealed that all the 6 native cattle breeds clustered separately with GIC and RSC breed appeared to be the most distinct breeds. Our results are similar with the findings of Nayee et al.29 and Strucken et al.28, wherein they reported genetic distinctness of Gir and Red Sindhi from the other Indian native breeds. Furthermore, close groupings of RAC, THC, SAC and KAC were observed in the present study whereby, RAC and THC appeared to share a common evolutionary history.
The admixture analysis showed that all most all the 6 native cattle breeds have maintained their genetic purity with little traces of admixturing. At subpopulations K = 6, though few individuals of KAC and SAC were observed to have little admixturing from THC. The shared and common geographical area of these breeds and lack of pedigree information in the field conditions might have contributed to small extent of admixturing. Interestingly, at K = 6, a shared ancestry was observed between THC and RSC. Although in previous studies, no admixture analysis was carried out in both Tharparkar and Red Sindhi, our observation of genetic closeness between these breeds can be unraveled by further analysis as close geographical origins exists between the two breeds. Overall the admixture analysis in the present study was in agreement with one of our previous microsatellites based genotyping studies where in high proportions of individuals of native cattle have been assigned to their respective breeds45. Recently, Nayee et al.29 using 50 K and 770 K bovine HD SNP chips have also shown assignment of majority of the animals of Sahiwal, Gir, Kankrej to their respective breeds with minimal mixed ancestry. The genetic purity of Gir was also in agreement with one of the recent studies carried out by Strucken et al.28 using bovine HD 770 K SNP chip. Similarly, Dixit et al.26 have shown that genotyping with Illumina 50 K SNP chip resulted in clustering of most of animals (> 76%) of Gir, Sahiwal, Hariana, Ongole, Kangyam, into their respective breeds.
The genetic separation of these 6 native cattle breeds was also supported by principal component analysis. Except one individual of KAC, all the animals of GIC, KAC and SAC were grouped as per their breed affiliations and widely separated from each other. Similarly, the individuals of THC, RAC and RSC were also grouped as per their breed but were placed closed to each other. The phylogenetic, admixture and PCA analysis thus suggested substantial between breed genetic distinctness of the 6 major milch breeds of India. The outcome of the present study has lot of similarity with many other previous reports on Indian native cattle published either using microsatellite or SNP chip markers26,28,29,43,45.
The present study has shown the utility of ddRAD sequencing strategy in identifying thousands of high-quality SNPs in native milch cattle breeds of India. The study has also provided an opportunity to establish a robust methodology as well as bioinformatics pipeline to generate and characterize genome wide SNPs. The SNPs derived from genome of native cattle could also help to enrich the Bos indicus genome database for future exploitation in diversity and genotype: phenotype association studies. Further, the genome wide SNP data set has provided a strong clue that each of the 6 major milch cattle breeds of India has sufficient within breed diversity. The between breed analysis along with phylogenetic, admixture and PCA analysis showed high level of genetic distinctness and purity of each of the 6 cattle breeds. In future similar approach could be extended to rest of the native cattle breeds (Bos indicus) to define the population structure along with their evolutionary relationships. Further, as the native Indian cattle breeds are known for better milk quality, heat tolerance and disease resistance, therefore such data set could also be exploited to understand the signatures of selection with respect to these traits. Such information will be quite helpful to realize the potential of these tropically adapted native germplasm especially in the era of climate change and global warming.
Methods
Sample source and DNA extraction
To identify the genome wide SNPs, the blood samples of 58 unrelated animals belonging to Gir (GIC, n = 12), Sahiwal (SAC, n = 12), Kankrej (KAC, n = 12), Rathi (RAC, n = 11), Red Sindhi (RSC, n = 7), and Tharparkar (THC, n = 4) cattle breeds were collected by visiting their respective breeding tracts. However, the samples of Red Sindhi animals were collected from Hosur farm of Krishnagiri district of Tamil Nadu state as this breed is only available in organized cattle farms. The blood samples were collected as per the guidelines of Institutional Animal Ethics Committee (IAEC). Further, all the details related to animals’ experiments were as per the ARRIVE guidelines and all the procedures were approved by the animal ethics committee of ICAR-NBAGR, Karnal. The geographical and ecological distribution of the cattle breeds is shown in Fig. 1. The utility type, coat colour, representative agroclimatic zone, breeding tract and geographical co-ordinates of each breeding tract is presented in Supplementary Table S1. Fresh blood samples (8–9 ml) collected in EDTA vacutainer tubes by jugular vein puncture were stored at − 20 °C until genomic DNA extraction. Genomic DNA was isolated from whole blood using phenol–chloroform extraction method46 followed by purification through RNAse treatment and Qiaquick Nuclease Removal Kit (Qiagen, Valencia, CA) to eliminate any RNA related impurities. The quality of DNA was checked on agarose gel (1%) electrophoresis, and the quantity of DNA was measured using a Nanodrop Spectrophotometer (Nanodrop ND-1000).
ddRAD library preparation and sequencing
For DNA library preparation, each sample was digested with two Restriction enzymes (REs); a 6 cutter EcoR1 (G/AATTC) and a 4 cutter Mse1 (T/TAA) (New England Biolabs, Ipswich,MA, USA) as determined by in-silico simulation using SimRAD package47. Briefly, 0.3–0.6 μg of genomic DNA of each animal was digested with the optimized restriction enzyme set. After digestion each end of digested fragment was ligated to EcoRI-specific P1 and the MseI-specific P2, barcoded adapters with a T4 DNA Ligase (New England Biolabs, Ipswich, MA, USA). The ligation reaction consisted of overnight incubation (> 12 h) at room temperature (approx. 21 °C) and heat deactivation of the enzyme at 65 °C for 10 min. In order to eliminate unincorporated adapters and small DNA fragments, ligation reactions were purified using with 0.8X volume of Agencourt AMPure XP SPRI magnetic beads (Beckman Coulter Life Sciences, Indianapolis, USA). A unique combination of the dual-indexed barcodes was attached to purified fragments with 14 cycles of PCR. Indexed PCR products were pooled in equal volumes and size selected using Agencourt AMPure XP SPRI magnetic beads. The amplification protocol involved initial denaturation at 95 °C for 3 min; 25 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s; followed by a final extension at 72 °C for 5 min.
In the present study, a total of 2 sequencing libraries viz., FGBS20H000717-1a, FGBS20H000718-1a which included all the samples of different cattle breeds were constructed for sequencing on Illumina HiSeq™ 2000 sequencing platform. The concentration of each library was checked using Qubit® 2.0 fluorometer (Thermo Fisher, Waltham, MA, USA). Each library was diluted to 1 ng/ul and the insert size was assessed and quantified using the Agilent high-sensitivity DNA kit in a 2100 Bioanalyzer (Agilent Technologies, CA, USA). Quantitative real-time PCR (qPCR) was performed to detect the effective concentration of each library. Finally, the libraries with appropriate insert size and effective concentration of more than 2 nM were sequenced on Illumina HiSeq™ 2000 and more than 100 bp end reads were generated.
Quality checking, filtering of raw reads and SNPs identification
The raw paired-end FASTQ sequencing files were quality checked using FASTQC software48. The raw reads with less than Q20 were removed from the data set using PRINSEQ software49. Both the adaptors were removed using Cutadapt 1.1550. Finally, the filtered reads with read length of 144 bp were retained for subsequent analysis. The filtered reads were aligned to Bos taurus reference genome ARS-UCD1.2 using Bowtie2 tool51,52. The aligned SAM files were converted to BAM files using SAMtools53 and subsequently sorted using Picard tool. All the duplicate reads were flagged and tagged using MarkDuplicatesWithMateCigar module of Picard tools 54. The reads were re-calibrated using GATK-BQSR tool with default parameters54. In order to discover genome wide SNPs in each animal, GATK Haplotypecaller was run in ERC GVCF mode54. The breed wise cohort GVCF file was created by combining all the individual GVCF files using CombinedGVFs of GATK tool. The cohort GVCF files were converted to VCF using GenotypeGVCF command of GATK tool. The insertions and deletions (INDELS) were discarded using GATKSelect Variants. The SNPs were annotated with reference to 1000 Bull Genome data using BCFTools 55. After annotation, all those SNPs located on X, Y chromosomes as well as mitochondrial DNA were removed using VCFtools56 and only those SNPs which are present in autosomes were retained for further analysis. The SNPs were also filtered at a minimum read depth level of 2, 5 and 10 (RD) and SNPs identified at RD of 5 were further filtered having minimum quality score of GQ30 using VCFtools56. Finally, three rounds of filtering for minor allele frequency (MAF < 0.05), missing genotypes (0.8), and HWE deviation (P < 0.001) was carried out using PLINK 1.9 57 to retain the high-quality SNPs for downstream analysis.
Annotation of SNP sites
The high-quality SNPs identified in each breed was annotated using SnpEff Ver. 4.3 software58. The VCF file and annotation data of the Bos taurus reference genome were used to partitioned the SNPs as per their genomic location such as exonic, intronic, upstream/downstream regions, splicing sites and intergenic regions. The SNPs were also categorized based on their functional impact on protein coding genes such as high, moderate, modifier, missense, nonsense and silent.
Diversity and Population structure analysis
Nucleotide diversity (π), TajimaD in each of the 6 cattle breeds was computed using TASSEL software (v. 5.0)59 software by selecting 500-SNP sliding window with step size of 100-SNP. VCF tools was employed to calculate observed heterozygosity (HO), expected heterozygosity (HE), inbreeding co-efficient (FIS) and Wright's FST estimates. For phylogenetic relationship, amongst the studied cattle populations, TASSEL software (v. 5.0)59 and phangorn60 softwares available in R language were used. A bootstrap value of 100 was used to draw the tree based on UPGMA and Neighbor joining (NJ) algorithms. The SNPs that were in strong Linkage Disequilibrium (LD) (r2 > 0.5) in a 5000-Kb sliding windows with 50 SNPs were pruned using PLINK v.1.957. The admixture analysis was performed using the pruned SNPs data by applying the ADMIXTOOLS of admixr-R package61. The admixture analysis was performed by assuming different numbers of sub-populations K = 6 in order to identify the optimal number of ancestral populations by detecting the lowest value of cross-validation error. Similarly, the Principal component analysis (PCA) was performed using pruned data by employing “adegent” software and the results were plotted using ggplot.
Animal ethics
All the experimental procedure was done in accordance with the ARRIVE guidelines and regulations of Institutional Animal Ethics Committee (IAEC), ICAR-National Bureau of Animal Genetic Resources (ICAR-NBAGR), Karnal, Haryana, India.
References
Loftus, R. T., MacHugh, D. E., Bradley, D. G., Sharp, P. M. & Cunningham, P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. 91(7), 2757–2761 (1994).
Jarrige, J. F. & Meadow, R. H. The antecedents of civilization in the Indus Valley. Sci. Am. 243(2), 122–137 (1980).
Meadow, R. H. The origins and spread of agriculture and pastoralism in northwestern South Asia. The origins and spread of agriculture and pastoralism in Eurasia, pp. 390–412 (1996).
Bradley, D. G., MacHugh, D. E., Cunningham, P. & Loftus, R. T. Mitochondrial diversity and the origins of African and European cattle. Proc. Natl. Acad. Sci. 93(10), 5131–5135 (1996).
MacHugh, D. E., Shriver, M. D., Loftus, R. T., Cunningham, P. & Bradley, D. G. Microsatellite DNA variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus). Genetics 146(3), 1071–1086 (1997).
Chen, S. et al. Zebu cattle are an exclusive legacy of the South Asia Neolithic. Mol. Biol. Evol. 27(1), 1–6 (2010).
Jia, S. et al. A new insight into cattle’s maternal origin in six Asian countries. J. Genet. Genom. 37(3), 173–180 (2010).
Manomohan, V. et al. Legacy of draught cattle breeds of South India: Insights into population structure, genetic admixture and maternal origin. PLoS ONE 16(5), e0246497 (2021).
Xia, X. et al. Abundant genetic diversity of Yunling Cattle based on mitochondrial genome. Animals 9(9), 641 (2019).
Chen, N. et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9(1), 1–13 (2018).
Ganguly, I. et al. Y-chromosome genetic diversity of Bos indicus cattle in close proximity to the centre of domestication. Sci. Rep. 10(1), 1–9 (2020).
20th Livestock Census. All India report: ministry of agriculture, department of animal husbandry, dairying & fisheries, Krishi Bhawan, New Delhi, India. 2019. [Cited 13 May 2020]. https://dahd.nic.in/sites/default/filess/20th%20Livestock%20census-2019%20All%20India%20Report_0.pdf.
Annual Report 2020–21. Government of India. Ministry of Ministry of Fisheries, Animal Husbandry and Dairying. Department of Animal Husbandry, Dairying: Krishi Bhavan; New Delhi 2021. https://dahd.nic.in/sites/default/filess/Annual%20Report%20English.pdf
Hanotte, O., Dessie, T. & Kemp, S. Time to tap Africa’s livestock genomes. Science 328(5986), 1640–1641 (2010).
Groeneveld, L. F. et al. Genetic diversity in farm animals–a review. Anim. Genet. 41, 6–31 (2010).
Van Dijk, E. L., Auger, H., Jaszczyszyn, Y. & Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 30(9), 418–426 (2014).
Kumar, D. R. et al. Genomic diversity and selection sweeps identified in Indian swamp buffaloes reveals it’s uniqueness with riverine buffaloes. Genomics 112(3), 2385–2392 (2020).
Sivalingam, J. et al. Genomic divergence reveals unique populations among Indian Yaks. Sci. Rep. 10(1), 1–7 (2020).
Tezuka, A., Takasu, M., Tozaki, T. & Nagano, A. J. The ability of ddRAD-Seq to estimate genetic diversity and genetic introgression in endangered native livestock. bioRxiv, p. 454108 (2018).
Liu, C., Chen, H., Yang, X., Zhang, C. & Ren, Z. Exploring the genomic resources of seven domestic Bactrian camel populations in China through restriction site-associated DNA sequencing. PLoS ONE 16(4), e0250168 (2021).
Devadasan, M. J. et al. Reduced representation approach for identification of genome-wide SNPs and their annotation for economically important traits in Indian Tharparkar cattle. 3 Biotech 10(7), 1–6 (2020).
Vineeth, M. R. et al. Genome-wide discovery of SNPs in candidate genes related to production and fertility traits in Sahiwal cattle. Trop. Anim. Health Prod. 52(4), 1707–1715 (2020).
Mishra, D. C. et al. Identification and characterization of trait-specific SNPs using ddRAD sequencing in water buffalo. Genomics 112(5), 3571–3578 (2020).
Zhan, B. et al. Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genom. 12(1), 1–20 (2011).
Xiang, R. et al. Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations. Nat. Commun. 12(1), 1–13 (2021).
Liu, L. et al. GWAS-based identification of new loci for milk yield, fat, and protein in Holstein cattle. Animals 10(11), 2048 (2020).
Dixit, S. P. et al. Genome analyses revealed genetic admixture and selection signatures in Bos indicus. Sci. Rep. 11(1), 1–11 (2021).
Saravanan, K. A. et al. Genomic scans for selection signatures revealed candidate genes for adaptation and production traits in a variety of cattle breeds. Genomics 113(3), 955–963 (2021).
Strucken, E. M. et al. Genetic diversity and effective population sizes of thirteen Indian cattle breeds. Genet. Sel. Evol. 53(1), 1–17 (2021).
Nayee, N. et al. Suitability of existing commercial single nucleotide polymorphism chips for genomic studies in Bos indicus cattle breeds and their Bos taurus crosses. J. Anim. Breed. Genet. 135(6), 432–441 (2018).
Dash, S. et al. Evaluation of bovine high-density SNP genotyping array in indigenous dairy cattle breeds. Anim. Biotechnol. 29(2), 129–135 (2018).
Pérez-Enciso, M. & Ferretti, L. Massive parallel sequencing in animal genetics: Wherefroms and wheretos. Anim. Genet. 41(6), 561–569 (2010).
Gurgul, A. et al. Genotyping-by-sequencing performance in selected livestock species. Genomics 111(2), 186–195 (2019).
Malik, A. A. et al. Analysis of genetic relatedness among Indian cattle (Bos indicus) using genotyping-by-sequencing markers. Anim. Genet. 49(3), 242–245 (2018).
De Donato, M., Peters, S. O., Mitchell, S. E., Hussain, T. & Imumorin, I. G. Genotyping-by-sequencing (GBS): A novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS ONE 8(5), e62137 (2013).
Brouard, J. S., Boyle, B., Ibeagha-Awemu, E. M. & Bissonnette, N. Low-depth genotyping-by-sequencing (GBS) in a bovine population: Strategies to maximize the selection of high quality genotypes and the accuracy of imputation. BMC Genet. 18(1), 1–14 (2017).
Wang, W. et al. Genome-wide SNP discovery and evaluation of genetic diversity among six Chinese indigenous cattle breeds in Sichuan. PLoS ONE 13(8), e0201534 (2018).
Mao, C. et al. Genome-wide SNP discovery and genetic diversity evaluation of Liangshan cattle in China. Anim. Biotechnol. 32(6), 671–675 (2021).
Surya, T. et al. Genomewide identification and annotation of SNPs in Bubalus bubalis. Genomics 111(6), 1695–1698 (2019).
Iqbal, N. et al. Genomic variants identified from whole-genome resequencing of indicine cattle breeds from Pakistan. PLoS ONE 14(4), e0215065 (2019).
Weldenegodguad, M. et al. Whole-genome sequencing of three native cattle breeds originating from the northernmost cattle farming regions. Front. Genet. 9, 728 (2019).
Bhati, M., Kadri, N. K., Crysnanto, D. & Pausch, H. Assessing genomic diversity and signatures of selection in Original Braunvieh cattle using whole-genome sequencing data. BMC Genom. 21(1), 1–14 (2020).
Mukesh, M., Sodhi, M., Bhatia, S. & Mishra, B. P. Genetic diversity of Indian native cattle breeds as analysed with 20 microsatellite loci. J. Anim. Breed. Genet. 121(6), 416–424 (2004).
Sharma, R. et al. Genetic diversity and relationship of Indian cattle inferred from microsatellite and mitochondrial DNA markers. BMC Genet. 16(1), 1–12 (2015).
Purva, M., Suthar, M. K., Bishnoi, J., Maherchandani, S. & Kashyap, S. K. Genetic diversity and relatedness among different four cattle breeds reared in Rajasthan. Int. J. Livest. Res. 8(12), 93–104. https://doi.org/10.5455/ijlr.20180131043854 (2018).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual 2nd edn. (Cold Spring Harbour Laboratory Press, 1989).
Lepais, O. & Weir, J. T. Sim RAD: An R package for simulation-based prediction of the number of loci expected in RAD seq and similar genotyping by sequencing approaches. Mol. Ecol. Resour. 14(6), 1314–1321 (2014).
Andrews, S. FastQC: A quality control tool for high throughput sequence data [WWW document]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. (2010)
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27(6), 863–864 (2011).
Langmead, B., Wilks, C., Antonescu, V. & Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35(3), 421–432 (2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17(1), 10–12 (2011).
Li, H. et al. 1000 genome project data processing subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21), 2987–2993 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27(15), 2156–2158 (2011).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81(3), 559–575 (2007).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6(2), 80–92 (2012).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23(19), 2633–2635 (2007).
Schliep, K. P. phangorn: Phylogenetic analysis in R. Bioinformatics 27(4), 592–593 (2011).
Petr, M., Vernot, B. & Kelso, J. admixr—R package for reproducible analyses using ADMIXTOOLS. Bioinformatics 35(17), 3194–3195 (2019).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Acknowledgements
The authors duly acknowledge the financial support provided by Indian Council of Agriculture Research, New Delhi under National Fellow Scheme (Grant Number: 27(3)/2010-HRD).
Author information
Authors and Affiliations
Contributions
M.M., M.S. conceived, designed the study, provided critical inputs during the data analysis and manuscript preparation and edited the manuscript. N.M. conducted bioinformatics analysis, drafted the manuscript. D.C., I.P. conducted bioinformatics analysis, data visualisation. P.V., M.T. collected blood samples. P.K. isolated and prepared genomic DNA samples.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masharing, N., Sodhi, M., Chanda, D. et al. ddRAD sequencing based genotyping of six indigenous dairy cattle breeds of India to infer existing genetic diversity and population structure. Sci Rep 13, 9379 (2023). https://doi.org/10.1038/s41598-023-32418-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32418-6
- Springer Nature Limited