
Abstract
This study aimed to find any ambiguous genetic outlier for “oligodendroglioma, IDH-mutant and 1p/19q-codeleted (O_IDH_mut)” and “astrocytoma, IDH-mutant (A_IDH_mut)” and to redefine the genetic landscape and prognostic factors of IDH-mutant gliomas. Next-generation sequencing (NGS) using a brain tumor-targeted gene panel, methylation profiles, and clinicopathological features were analyzed for O_IDH_mut (n = 74) in 70 patients and for A_IDH_mut (n = 95) in 90 patients. 97.3% of O_IDH_mut and 98.9% of A_IDH_mut displayed a classic genomic landscape. Combined CIC (75.7%) and/or FUBP1 (45.9%) mutations were detected in 93.2% and MGMTp methylation in 95.9% of O_IDH_mut patients. In A_IDH_mut, TP53 mutations were found in 86.3% and combined ATRX (82.1%) and TERTp (6.3%) mutations in 88.4%. Although there were 3 confusing cases, NOS (not otherwise specified) category, based on genetic profiles, but they were clearly classified by combining histopathology and DKFZ methylation classifier algorithms. The patients with MYCN amplification and/or CDKN2A/2B homozygous deletion in the A_IDH_mut category had a worse prognosis than those without these gene alterations and MYCN-amplified A_IDH_mut showed the worst prognosis. However, there was no prognostic genetic marker in O_IDH_mut. In histopathologically or genetically ambiguous cases, methylation profiles can be used as an objective tool to avoid a diagnosis of NOS or NEC (not elsewhere classified), as well as for tumor classification. The authors have not encountered a case of true mixed oligoastrocytoma using an integrated diagnosis of histopathological, genetic and methylation profiles. MYCN amplification, in addition to CDKN2A/2B homozygous deletion, should be included in the genetic criteria for CNS WHO grade 4 A_IDH_mut.
Similar content being viewed by others

Introduction
Astrocytoma, isocitrate dehydrogenase (IDH)-mutant (A_IDH_mut) and oligodendroglioma, IDH-mutant and 1p/19q-codeleted (O_IDH_mut) are the 2nd and 3rd most common diffuse infiltrating gliomas in adults after glioblastoma, IDH-wildtype (GBM). According to a meta-analysis, the 10-year progression-free survival (PFS) rates of O_IDH_mut and A_IDH_mut are 62.5% ~ 71.7% and 30.4% ~ 38.3%, respectively1. The genomic and molecular alterations of IDH_mut oligodendroglioma and IDH_mut astrocytoma were well described by The Cancer Genome Atlas in 2015 and Ceccarelli et al. in 20172,3 and introduced in neuropathological diagnosis. As a result, the world health organization (WHO) classification was revised, and these alterations became important biomarkers for the diagnosis of diffuse gliomas4,5,6.
Prior to the era of genetics-integrated diagnosis, astrocytomas and oligodendrogliomas were often confused with mixed oligoastrocytomas, but confusion including interobserver variability has now been greatly reduced, with A and O_IDH_mut being well-defined brain tumors. O_IDH_mut characteristically carries IDH mutation and 1p/19q codeletion, with additional mutations of capicua transcriptional repressor (CIC) and/or far upstream element binding protein 1 (FUBP1), telomerase reverse transcriptase promoter (TERTp) mutation, and O6-methylguanine methyltransferase promoter (MGMTp) methylation. IDH-mutant astrocytomas usually show alpha-thalassemia/mental retardation syndrome X-linked (ATRX) and tumor protein 53 (TP53) mutations5. However, it is unknown how often these gene variants occur in O_IDH_mut and A_IDH_mut. CIC, FUBP1, and ATRX variants are relatively limited, as these tumor-suppressor gene alterations are scattered throughout exons and introns. Although genetics-integrated diagnostics have greatly improved pathological diagnostic accuracy and significantly reduced interobserver variability2,3, there are still cases of morphological and genetic ambiguities, and the existence of mixed oligoastrocytoma is questionable. In 2014, loss of CIC and FUBP1 expression was reported as a potential indicator for shorter relapse times in oligodendroglial tumors7. Nevertheless, this conjecture may lose credibility if CIC and/or FUBP1 mutations are ubiquitous in oligodendrogliomas. First, this study aimed to elucidate the exact incidence and mutation types of CIC and FUBP1. Second, we aimed to show whether mixed oligoastrocytoma does exist or not. Third, we investigated the prognostic factors of adult-type diffuse gliomas. Finally, we explored through a literature review why IDH1 mutation is not the only driver of diffuse gliomas but is usually accompanied by mutations in tumor-suppressor genes and telomere maintenance mechanisms.
Materials and methods
A total of 169 cases of O_IDH_mut (n = 74) involving 70 patients and A_IDH_mut (n = 95) involving 90 patients were obtained from the archives of the Seoul National University Hospital (SNUH) from July 2018 to September 2022. Primary IDH-mutant astrocytomas were 18% more common than primary oligodendrogliomas (59% vs. 41%) over the last 3 years at a single institute (SNUH) when recurrent cases were excluded. Next-generation sequencing (NGS) using a customized FiRST (“Friendly integrated Research-based Smart Trustworthy”) brain tumor-targeted gene panel (BTP) was performed in all cases. A list of genes included in the FiRST BTP is provided in Supplementary Table 1. Two pathologists (SI Kim and SH Park) reviewed the genetics-integrated diagnoses with NGS results. Genetics-integrated diagnoses were made according to the 5th edition of the WHO classifications of the CNS published in 20215. When the genetic features of A_IDH_mut and O_IDH_mut coexisted, methylation profile-based classification using the Deutsches Krebsforschungszentrum (DKFZ) algorithm was performed using Infinium MethylationEPIC 850K BeadChip array platforms.
The epidemiologic features of the samples are summarized in Table 1. Among the 74 O_IDH_mut cases, CNS WHO grades 2 and 3 were seen in 28 (37.8%) and 46 (62.2%) cases, respectively. Among the recurrent O_IDH tumors, 7 (9.5%) were grade 2 and 22 (29.7%) were grade 3. The male-to-female ratio was 1:1.1, and the median age was 45 years (range 19–68 years). The most common site was the frontal lobe in 56.8%, followed by multiple lobes (24.3%), the temporal lobe (8.1%), the insula (4.1%), the parietal lobe (4.1%), and the cingulate cortex (2.7%).
Of the 95 A-IDH cases, CNS WHO grades 2, 3 and 4 were assigned to 16 (16.8%), 43 (45.3%), and 36 (37.9%), respectively. The male-to-female ratio was 1.6:1, and the median age was 41 years (range 14–75 years). The most common site was the frontal lobe (47.4%), followed by multiple lobes (26.3%), the temporal lobe (10.5%), the parietal lobe (7.4%), the insula (4.2%), the occipital lobe (1.1%), the central lobule (1.1%), the lateral ventricle (1.1%), and the cerebellum (1.1%).
Immunohistochemical (IHC) study
Tissue sections were stained with an anti-ATRX polyclonal antibody (1: 300 dilution, ATLAS Antibodies AB, Bromma, Sweden), anti-GFAP (6F2) monoclonal antibody (1: 200 dilution, DAKO, Glostrup, Denmark), anti-IDH1 R132H (H09) monoclonal antibody (1:300 dilution, Dianova, Hamburg, Germany), anti-Ki67 (MIB-1) antibody (1:100 dilution, DAKO), anti-H3K27 M polyclonal antibody (1:700 Millipore, Temecula, USA), anti-p53 monoclonal antibody, DO-7 code M7001 (1:100 dilution, DAKO), anti-PHH3 antibody (1:100 dilution, Cell Marque, Rocklin, CA, USA) and anti-vimentin antibody (1:500, DAKO) (Supplementary Table 2). IHC staining was performed using a standard avidin–biotin-peroxidase method with a BenchMark ULTRA system (Roche Diagnostics, Indianapolis, US). The positive control was known positive tissue, and entrapped positive cells were used. For the negative control, the primary antibody was omitted.
ATRX gene mutations were scattered throughout exons and introns, making some sites difficult to capture using NGS. Since the initial version of our customized brain tumor panel could not detect full variants of ATRX mutations, we relied on ATRX IHC, but upgrade version of BTP panel could detect all the mutations.
DNA extraction and NGS with customized brain tumor-targeted combined DNA/RNA gene panels
Representative areas of tumors with at least 90% tumor cell purity in hematoxylin and eosin-stained formalin-fixed paraffin-embedded (FFPE) sections were outlined for macrodissection. DNA was extracted according to the manufacturer's instructions from serial sections of the macrodissected tumor tissue using Maxwell® RSC DNA FFPE Kit (AS1450; Promega, USA) with a tumor-targeted gene panel (FiRST brain tumor panel and FiRST pancancer panel), as customized and verified by the Department of Pathology of Seoul National University Hospital (SNUH), and used with Hi-Output NextSeq550Dx. Panels 2 and 3 contained 172 and 207 genes, respectively, and 20 and 52 fusion genes, respectively. Sequencing data were analyzed using the SNUH FiRST Brain Tumor Panel Analysis pipeline. First, we performed quality control on the FASTQ file and analyzed only data that passed the criteria. Paired-end alignment to the hg19 reference genome was performed using BWA-men and GATK Best Practice8. After finishing the alignment step, an “analysis-ready BAM” was produced, and a second quality control was performed to determine if further variant calling was appropriate. Single-nucleotide variation (SNV), insertion and deletion (InDel), copy number variation (CNV), and translocation were analyzed using at least two analysis tools, including in-house and open-source software. The open-source tools used were GATK UnifiedGenotyper, SNVer and LoFreq for SNV/InDel detection; Delly and Manta for translocation discovery; THetA2 for purity estimation; and CNVKit for CNV calling, as previously described by Park et al.9. SnpEff was used to annotate the variants detected in various databases, including RefSeq, COSMIC, dbSNP, ClinVar, and gnomAD. Germline variants were then filtered using the population frequency of these databases (> 1% population frequency). Finally, the multidisciplinary molecular tumor board confirmed the variants through a comprehensive review. For RNA sequencing, tumor RNA was extracted from a paraffin block (tumor fraction: > 90%) using Maxwell® RSC RNA FFPE Kit (AS1440; Promega). The library was generated using SureSelectXT RNA Direct Kit (Agilent, Santa Clara, CA, USA) and sequenced using an Illumina NovaSeq 6000 at Macrogen (Seoul, Republic of Korea). Raw sequencing reads were analyzed using three algorithms, namely, DIFFUSE, Fusion catcher, and Arriba (https://github.com/suhrig/arriba/), to detect gene fusions, and the results were compared. FASTQ files were briefly aligned using the STAR aligner on the hg19 reference genome for Arriba analysis. A chimeric alignment file and read-through alignment file were produced, and fusion candidates were generated with a set of filters that detected artifacts based on various characteristic features.
MGMTp methylation based on methylation-specific polymerase chain reaction (MSP) analysis
The MSP technique was used to analyze MGMTp methylation. Prepared DNA was modified via sodium bisulfite treatment using EZ DNA Methylation-Gold Kit (D5005; Zymo Research, Orange, CA, USA). The primer sequences used for MGMTp were as follows: methylated forward, 5′-TTT CGA CGT TCG TAG GTT TTC GC-3′; methylated reverse, 5′-GCA CTC TTC CGA AAA CGA AAC G-3′; unmethylated forward, 5′-TTT GTG TTT TGA TGT TTG TAG GTT TTT GT-3′; unmethylated reverse, 5′-AAC TCC ACA CTC TTC CAA AAA CAA AAC A-3′. The annealing temperature was set to 64 °C. The obtained polymerase chain reaction products were electrophoresed on 2% agarose gels and visualized under UV illumination after staining with ethidium bromide.
Genomic alteration visualization
Clinical information, mutations, and CNVs were summarized using Oncoprint data, which were generated using the R package “ComplexHeatmap” (version 2.7.6.1002)10.
Methylation array analysis and t-SNE clustering with the Illumina 850 K microarray
DNA methylation array analysis was performed for three ambiguous cases of O_IDH_mut using an Infinium MethylationEpic 850 K BeadChip array platform (Illumina, USA). DNA methylation data analysis was performed using R software (R 4.1.3, https://www.r-project.org/). Our samples were analyzed using the methylation classifier of DKFZ with reference to a diffuse glioma cohort of 551 samples from 10 subclasses11. A preprocessing procedure was conducted for raw methylation intensity signals using the R package “methylationArrayAnalysis” (version 1.14.0)12. We utilized the “combinedArray” command to merge the different platforms (450 K, EPIC). After filtering and normalization, 365,201 probes remained for subsequent analysis. The 20,000 most variably methylated probes were selected based on the standard deviation to perform unsupervised nonlinear dimension reduction. The resulting distance matrix was used as the input for t-distributed stochastic neighbor embedding analysis (t-SNE; Rtsne package version 0.15). The nondefault parameters used were distance = TRUE, perplexity = 30, θ = 0, and max_iter = 2000. A t-SNE plot was generated using the “ggplot2” package (version 3.3.3) for effective visualization. IDAT files were uploaded to versions v11b4 and v12.5 of the online CNS tumor methylation classifier (https://www.molecularneuropathology.org) for classification.
Survival analysis
The median overall survival (OS) and progression-free survival (PFS) were obtained using the R package “survminer” (version 0.4.9). Survival analysis was performed in 70 patients with O_IDH_mut and 87 patients with A_IDH_mut. As most patients with O_IDH_mut were alive at the time of analysis during 1 to 244-months (median 34.7 months) follow-up period, overall survival (OS) could not be obtained.
Ethics approval and consent to participate
The institutional review board (IRB) of Seoul National University Hospital (SNUH) approved this study (IRB No: 1906-020-1037) and has therefore been performed under the ethical standards set out in the 1964 Declaration of Helsinki and its subsequent amendments. As this study is a retrospective review of anonymized electronic medical records, pathology, and NGS data utilizing a brain tumor-specific somatic gene panel, IRB of SNUH (IRB No: 1906-020-1037) waived the informed consent for this study under the Korean Bioethics and Safety Act.
Results
Molecular study of oligodendroglioma, IDH-mutant and 1p/19q-codeleted
All O_IDH_mut samples (n = 74) showed ATRX expression and were positive for IDH1 (H09) by immunohistochemistry. Vimentin was mostly negative in tumor cells, but variable vimentin-positive glial cells were found in the margin of the tumor, the gray matter with infiltrating tumor cells, and some minigemistocytes.
NGS revealed IDH and TERTp mutations and 1p/19q whole-arm deletion in all O_IDH_mut samples. IDH1 and IDH2 mutations were found in 91.9% (68/74) and 8.2% (6/74) of patients, respectively (Table 2). C228T and C250T mutations in the TERT promoter were detected in 67.6% (50/74) and 32.4% (24/74), respectively, of patients. CIC, FUBP1, and both CIC and FUBP1 mutations were found in 75.7%, 45.9%, and 30.0% of patients, respectively. The numbers of CIC and FUBP1 variants were 70 and 36, respectively.
The CIC gene variants found comprised missense (21.6%), frameshift (23.0%), multihit (20.3%), nonsense (6.8%), and intronic (splicing site) (4.1%) mutations (Table 2). The FUBP1 gene exhibited 14.9% nonsense (stop gained), 10.8% frameshift, 9.5% intronic (splicing), 6.8% multihit, and 2.7% missense mutations. Among these variants, 22.9% (16/70 variants) of CIC and 11.1% (4/36 variants) of FUBP1 were novel, which were found in 19 O_IDH_mut cases (5 WHO grade 2, 14 WHO grade 3). Sixteen and four novel variants of CIC and FUBP1 are listed in Supplementary Table 3.
A total of 13 (86.7%) O_IDH_mut cases with novel CIC variants also showed additional pathogenic gene mutations, such as FUBP1 (23.1%), KRAS (33.1%), NOTCH1 (23.1%), TP53 (15.4%), PIK3CA (7.7%), SETD2 (7.7%), BRAF amplification (7.7%), and PTEN loss (8.3%), but two cases had no additional pathogenic variant. Among the 4 O_IDH_mut with FUBP1 novel variants, one involved a pathogenic mutation in KIT, and the other one involved pathogenic NOTCH1 mutation. The remaining two had no additional pathogenic gene mutations. Two cases of O_IDH_mut also exhibited TP53 pathogenic mutations (3.4%) in addition to 1p/19q-codeletion.
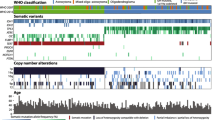
The rare mutations found in O_IDH_mut were ARID1A, CDKN2C, JAK3, MYB, NF1, NOTCH1 and PIK3CA in CNS WHO grade 2 O_IDH_mut and BRIP1, DDX3X, KIT, KRAS, NF1, NOTCH1, PIK3CA, RAD54L, SETD2, and SMARCB1 in CNS WHO grade 3 O_IDH_mut. This genomic landscape of O_IDH_mut is illustrated in Fig. 1A.

(A) The oncomap of IDH-mutant astrocytomas (n = 95) and (B) oligodendrogliomas (n = 74) of this study. The NGS study was performed twice on sequentially removed 18 tumors from 5 patients with A_IDH_mut and 4 patients with O_IDH_mut (#).
Molecular study of astrocytoma, IDH-mutant
In 95 cases of A_IDH_mut, 86.3% carried a TP53 mutation, 82.1% an ATRX mutation, and 6.3% a TERTp mutation (Table 3). The IDH1/2 variants were R132H (96.8%), R132S (1.1%), R132G (1.1%) and IDH2 R172G (1.1%). The variant types of TP53 were missense in 58.9%, multihit in 16.8%, nonsense in 5.3%, frameshift in 3.2%, and intronic in 2.1% of cases.
The ATRX mutation found by NGS and ATRX loss by IHC showed a 100% correlation in our cases. The ATRX mutation was found to be a frameshift in 28.4%, ATRX loss through IHC in 25.3%, nonsense (stop gained) in 16.8%, intronic (splicing) in 7.4%, multihit in 3.2% and deletion in 1.1% of cases.
The other CNVs found in A_IDH_mut are listed in Table 3. PDGFRA gain or amplification was detected in 8.4%, CDK4 amplification in 5.3%, MET gain or amplification in 5.3%, MYCN amplification in 4.2% and GLI1 amplification in 2.1% of cases. Chromosome 19q deletion, 1p deletion, and 1p/19q codeletion were found in 13.7%, 1.1% and 1.1% of cases, respectively.
Among the 17 cases of A_IDH_mut without ATRX mutation, TERTp mutation was observed in 29.4% (5 cases), TP53 mutation in 76.5% (13 cases), and CNV of genes or chromosomes in 52.9% (9 cases) (Table 4). The amplified genes included CDK4 (n = 3), MYCN (n = 2), PDGFRA (n = 1), and GLI (n = 1). Chromosomal aberrations included 7 gain, 11 gain, 19 deletion, 22 deletion, and 1p/19 codeletion. As expected, the higher the grade was, the higher the occurrence of CNVs was (chi-square test, p < 0.00001) (Supplementary Table 4). This genomic landscape of A_IDH_mut is illustrated in Fig. 1B.
Comparison of the molecular profiles of sequentially developed tumors
When comparing the molecular profiles of the 5 pairs of A_IDH_mut and 4 pairs of O_IDH_mut, no significant difference was noted. However, in one case each of A_IDH_mut and O_IDH_mut, the homozygous deletion of CDKN2A/2B was disappeared in the recurrent tumors that was present in the previous tumors.
Hypermutated astrocytoma, IDH-mutant
There were three hypermutated A_IDH_mut tumors; two MMR-deficient primary astrocytomas and one temozolomide (TMZ)-induced hypermutated recurrent astrocytoma (grade 4).
The first patient was a 28-year-old male who initially had grade 3 A_IDH_mut. He received local RT (50.4 + 10.8 Gy) after initial resection of the tumor and a second RT (45 Gy/25 Fx) at 3 and a half years later when the tumor relapsed as WHO grade 4 A_IDH-mut. He underwent a total of 3 reoperations. This patient was not treated with TMZ. This patient harbored an MLH1 germline mutation (p.Arg687Trp, c.2059C > T), and the tumor exhibited high microsatellite instability (MSI-H), corresponding to Lynch syndrome-associated, MMR-deficient A_IDH_mut. Nevertheless, the patient developed no tumor other than the brain tumor.
The second patient was a 15-year-old boy with A-IDH-mut, WHO grade 4 with MSH6 germline mutation (p.Arg1172fs, c.3514dupA). The tumor showed MSI-H. He underwent CCNU + TMZ (6 cycles). He has had no recurrent brain tumors or other extracranial tumors during the 40-month follow-up.
The third patient initially had grade 3 A_IDH_mut at the age of 32 years. After receiving concurrent chemoradiotherapy (CCRT) with TMZ for 2 years, the tumor recurred, and he underwent a second CCRT. After six months, the recurrent tumor was removed, and A_IDH_mut, grade 4, was diagnosed. NGS of the recurrent tumor revealed hypermutation. Because the patient did not carry a germline mutation in the MMR gene, the case was judged to be TMZ-induced hypermutation.
Study of histologically and genetically confusing cases with DNA methylation profiling
Although most O_IDH_mut cases were not difficult to diagnose, 2.7% (2/74) of O_IDH_mut and 1.1% (1/95) of A_IDH_mut cases in our study showed confounding with respect to genomic profiles and morphology (Figs. 2, 3, and 4). All three cases had TP53 mutations, along with classic genomic signatures of O_IDH_mut; 1p/19q-codeletion, TERTp and CIC, mutations. The first two cases showed histopathological oligodendroglioma-like morphology with minigemistocytic or gliofibrillary oligodendrocytes. Remaining one patient had histopathologically CNS WHO grade 2 astrocytoma as the initial tumor but progressed to CNS WHO grade 3, with an ambiguous appearance with more rounded nuclei in the recurrent tumor (Fig. 4).

TP53-mutant oligodendroglioma, CNS WHO grade 3. (A) The tumor shows fried egg-appearing monotonous rounded cells with fine capillaries. (B) Some tumor cells have eosinophilic cytoplasm, consistent with minigemistocytic and gliofibrillary oligodendrocytes. (C–E) This tumor cells are positive for IDH1 and are retained expression of ATRX, but negative for vimentin. (F) Copy number variation plots obtained from methylation profiles show 1p/19q co-deletions and chromosomes 4, 9, 14, and 18 deletions (1 copy). In addition, FUBP1 (splicing,247-1G>A, VAF 36.8%; splicing 1105-1G>A, 40.99%), TERT promotor mutation (C228T) and MGMT promoter methylation are present (A,G H&E, C ATRX IHC, D vimentin IHC, E CNV plot, under bar size A, C–D 50, B: 25 micrometer).

The second case of TP53 mutant oligodendroglioma, CNS WHO grade 3. (A) Tumor shows fried egg-appearing monotonous round cells with microvascular proliferation. (B) Some tumor cells have pleomorphic and hyperchromic nuclei (arrows) and abundant eosinophilic cytoplasm. This photo is a little bit out of focus because of nearby calcifications. (C,D) These tumor cells are positive for IDH1 (H09) and P53. (E) Copy number variation plots obtained from methylation profiles represent 1p/19q codeletion (one copy deletion). In addition, CIC (p.R1124W, VAF: 85.1%), TERT promoter (C250T), TP53 (p.R175H, VAF: 26.1%) mutations and MGMT promoter methylation are present (A,B H&E, C IDH1 IHC, D p53 IHC, E CNV plot, under bar sizes A–D 50 micrometer).

The 1p/19q-codeleted astrocytoma, IDH-mutant. This tumor recurred twice. (A,B) The initial tumor is a diffuse astrocytoma, IDH-mutant, consisting of hereditary astrocytes and IDH1-positive cells. (C,D) The second recurrent tumor shows morphological changes in the monotonous round shape with IDH1-positive round nuclei in the tumor cell nucleus. (E,F) Recurrent tumor cells are negative for H3K27me3 and vimentin, also reported in oligodendrocytes. (G) Copy number variation plots show 1p/19q-codeletion, chromosome 11 gain and 9p/18/22 deletion. This tumor also has CIC mutations (p.H264R, VAF: 83.0%), TERT promoter mutations (C228T), and TP53 mutations (p.G224*, VAF: 85.7%), and MGMT promoter methylation (Under bar scale, A,C: 25, B,E,F: 50, D: 100 micrometer).
The first two cases (Cases #24 and #26) were matched with O_IDH_mut in two versions of methylation classifier (Case #24: v11b4: score 0.98853, v12.5: score 0.97911, Case #26: v11b4: score 0.79244, v12.5: score 0.78328) (Table 5). The methylation class of the remaining case was different according to the version of the DKFZ methylation classifier. Based on v11b4 DKFZ methylation classifier, it matched O_IDH_mut (score 0.75304) and did not match A_IDH_mut_HG (score 0.16219); however, with the recently updated v12.5 DKFZ algorithm, it matched diffuse glioma, IDH mutant (score: 0.92333), and A_IDH_mut_HG (score: 0.83527) (Table 5).
We performed a dimensional reduction procedure to visualize the results into t-SNE plot using a reference cohort of adult-type diffuse gliomas and these three cases whown in Table 5. Since the reference cohort data of the t-SNE plot was version v11b4 of DKFZ, one case of 1p/19q-codeleted A_IDH_mut clustered with O_IDH_mut, as mentioned above (Supplementary Fig. 1, t-SNE plot by v11b4); unfortunately, the t-SNE plot by v12.5 is not shown because we could not obtain the reference data by v12.5.
Survival analysis
Kaplan–Meier survival analysis of the OS and PFS of patients with A_IDH_mut revealed significantly worse survival with increasing grade (Fig. 5A, B).

(A, B) Overall and progression-free survivals (OS and PFS) were significantly worse with increasing grade of A_IDH_mut. (C, D) OS and PFS were significantly worse in the patients with MYCN-amplified and (E, F) CDKN2A/2B homozygously deleted A_IDH_mut.
Patients with A_IDH_mut with CDKN2A/2B homozygous deletion or MYCN amplification had significantly worse survival than those with A_IDH_mut without these two gene alterations (Fig. 5C–F). These results correspond to previous studies and cIMPACT-NOW updates 5 and 66,13,14,15. However, PDGFRA amplification and PTEN loss did not affect patient survival in the A_IDH_mut group in our study (Fig. 6A–D). The HR and p-values of these parameters are provided in Fig. 7. No parameters for O_IDH_mut had significant hazard ratios.

(A, B) PTEN deletion and (C, D) PDGFRA amplification do not affect the OS and PFS.

The hazard plot of various grade and genetic parameters in A_IDH_mut and O_IDH_mut. In A_IDH_mut, grade and CDKN2A/2B were statistically significant and MYCN-amplified tumor also were statistical significance, however, PTEN loss and PDGFRA amplification did not affect on the biological behavior of A_IDH_mut. In O_IDH_mut, the higher grade the higher HR, but it does not have statistical significance in this study (OS overall survival, N number of cases, HR hazard ratio, MVP microvascular proliferation).
Discussion
A_IDH_mut and O_IDH_mut have characteristic histopathological, genetic and epigenetic profiles. Although it is well known that morphologically ambiguous cases exist, they can be diagnosed as A_IDH_mut or O_IDH_mut through a genetics-integrated diagnosis. This study aimed to determine whether mixed oligoastrocytoma occurs and whether “NOS” cases can be eliminated using genetics-integrated diagnostics and DKFZ methylation classifier.
Function of oncometabolites produced by IDH mutation
The oncometabolite 2-hydroxyglutarate (2-HG), which is produced by IDH1/2 mutations, is a competitive inhibitor of multiple alpha-ketoglutarate (a-KG)-dependent dioxygenases. 2-HG induces a wide range of histone demethylases at the promoter level and the ten eleven translocation (TET) family of dioxygenases of 5-methylcytosine (5mC) at the gene level16. The resulting chromatin compaction promotes the glioma CpG island hypermethylator phenotype (gCIMP), resulting in global silencing of tumor-suppressor genes (TSGs)16,17. Lu et al. reported that impaired histone lysine demethylation by accumulation of 2-HG caused by IDH mutation prevents cellular differentiation18. However, the exact mechanism of action of oncometabolites remains unknown. Unexpectedly, Filipski et al. and Habiba et al. found that IDH1 R132H-mutant O_IDH_mut involves H3K27me3 protein loss in 96% (25/26) and 90% (36/40) of cases, respectively, but non-R132H IDH-mutant O_IDH_mut does not show H3K27me3 loss19,20. As mentioned above, loss of nuclear H3K27me3 is known to increase histone methylation by impairing demethylation of inhibitory histone markers induced by increased 2-HG levels in IDH mutant gliomas18. We believe that 2-HG may play a pivotal role in histone demethylation. CpG island DNA methylation in the nonpromoter region of IDH mutant cells, that is, DNA methylation of the CCCTC-binding factor (CTCF) binding site, results in loss of the insulated site, intercepts the topographically associated domain (TAD) boundary, and leads to an active enhancer in genes, such as PDGFRA21.
Dysfunction of tumor-suppressor genes, TP53 in astrocytoma, CIC and FUBP1 in oligodendroglioma
IDH mutations are an early oncogenic molecular change in IDH-mutant gliomas. Mutation of IDH can cause oncogene (oncometabolite)-induced cellular senescence22. Constitutive activation of RB1, TP53, and CDKN2A is a well-known growth arrest signaling pathway that produces hypoproliferative senescent cells23. Senescent glioma cells adopt a different path to survive. Tumor cells need to block senescence-inducing TSGs, such as TP53, RB1, and CDKN2A/2B, so that can bypass senescence-induced tumor cell apoptosis. These molecular mechanisms may induce simultaneous TP53 mutations in A_IDH_mut and CIC and/or FUBP1 mutations in O_IDH_mut to obtain proliferative activity and to avoid cellular senescence.
CIC mutations are found in approximately 70% of oligodendrogliomas24, and we found them in 75.7% of our O_IDH_mut tumors. CIC is an important tumor suppressor that acts through transcriptional repression of target genes, including the polyoma enhancer activator 3 (PEA3) subfamily of E26 transformation-specific (ETS) transcription factors24. CIC is a transcriptional repressor that recruits histone deacetylations. CIC inactivation by mutations or deletion increases the level of histone acetylation, leading to transcription of EGFR/RAS/MAPK pathway components, promoting mitogen-independent tumor growth24.
FUBP1 mutations are reported in approximately 15% of O_IDH_mut cases25, but they were found in 45.9% of O_IDH_mut cases in our series, including intronic mutations in 9.5%. Only four novel FUBP1 mutations were identified in the present study (Table 2). FUBP1 was first described in 1994 as a single-stranded DNA-binding protein that binds to a noncoding, single-stranded far upstream element (FUSE) 2.5 kb upstream of the MYC promoter. FUBP1 binds to the transcription factor IIH (TFIIH), activates the MYC oncogene, and directly represses p21 in normal hematopoiesis26,27. FUBP1 is also a long-tail cancer driver that cooperates with other tumor-suppressor genes28. FUBP1 is a posttranscriptional regulator of N6-methyladenosine (m6A) RNA methylation, translation, mRNA stability, and splicing. Its loss leads to global changes in RNA splicing and widespread expression of aberrant driver isoforms28. Therefore, somatic alteration of FUBP1 (FUBP1−/−) contributes to neoplastic transformation via aberrant RNA splicing and m6A methylation28. FUBP1 missense, nonsense, silent mutations, whole-gene deletions, frameshift deletions, and insertions have been observed in oligodendrogliomas7.
Telomere lengthening by ATRX and TERTp mutations
Cancer involves rapidly proliferating cells that result in telomere shortening until the maximal number of cell divisions is reached (“Hayflick limit”)22. If replication proceeds, tumor cells gain chromosomal instability and eventually undergo apoptosis. For immortal growth, glioma cells exhibit telomere maintenance mechanisms (TMMs), which are TERTp mutation in O_IDH_mut and IDH-wildtype GBM and ATRX mutation in the majority of astrocytomas and histone-mutant pediatric-type high-grade gliomas29. TERT allows stabilization and elongation of telomeres. Altered length of telomeres (ALT) is another TMM that is induced by dysfunction of the ATRX/death-associated protein 6 (DAXX) complex30. However, TERTp mutation may be a second genetic event following oncogenic activation, such as IDH mutation in diffuse gliomas, BRAF V600E mutation in thyroid cancer, and FGFR3 mutation in urothelial carcinoma29,31,32.
In our study, TERTp mutations were found in 100% of O_IDH_mut and 6.3% of A_IDH_mut cases, while ATRX mutations were present in 82.1% of A_IDH_mut cases but not in oligodendrogliomas. TERT has many functions, both canonical and noncanonical. The most significant canonical function of TERT is telomere lengthening, and the most important noncanonical functions of TERT are reduction of apoptosis, regulation of chromatin structure and gene expression31. Inhibition or inactivation of CIC by mutation is associated with TERTp mutation and increased TERT mRNA expression in O_IDH_mut33. TERTp and ATRX mutations are mutually exclusive, suggesting that they have equivalent TMMs34; however, questions of why O_IDH_mut involves TERTp mutation rather than ATRX mutation and most A_IDH_mut have ATRX mutations, not TERTp mutations, remain. In patients with A-IDH-mut, there was no difference in OS or PFS between the those with ATRX mutation and TERTp mutation (Supplementary Fig. 2A,B). ATRX- or TERTp-wildtype in patients with A_IDH_mut were associated with worse OS (p = 0.046) but did not affect PFS (Supplementary Fig. 2C–F).
MGMTp methylation
DNA methylation patterns are generally stable and unique in differentiated cells; however, methylation profiles can be altered by extrinsic or intrinsic factors35. Both hypermethylation and hypomethylation play essential roles in long-term gene regulation and reactivation of oncogenes, TSG inhibition, deregulation of mRNA expression, mutagenesis, or alteration of functional chromosomal stability in cancers36. Genome-wide DNA methylation patterns can be used to subclassify brain tumors, which correlate with mutational status, DNA copy-number aberrations, and gene expression signatures37,38.
MGMT is a key DNA repair enzyme that removes mutagenic methyl groups from the P6 position of guanine by transferring it to the cysteine acceptor site of the protein itself39. MGMTp methylation is a better prognostic factor for gliomas40 and was eventually confirmed as a predictive marker for alkylating agents by European Organization for Research and Treatment (EORTC)41. MGMTp methylation belongs to the glioma CIMP family and was found in 95.9% of our O_IDH_mut series, and loss of nuclear H3K27me3 can be generated using IDH mutation-induced demethyltransferase blockade. According to Horbinski et al.’s study, the average beta values of O_IDH_mut of all 147 MGMTp CpG sites were significantly higher at 44.9%, and transcriptionally sensitive regions were 69% more methylated in O_IDH_mut. However, none were significantly more methylated in astrocytomas42. It is curious why MGMTp methylation appears to be less frequent (73.7%) in A_IDH_mut than in O_IDH_mut.
Gene dosage effect in gliomas
In this study, higher grade A_IDH_mut showed significantly more CNVs than lower grade A_IDH_mut (Supplementary Table 3). A comprehensive genomic landscape revealed the gene dosage effect of gliomas, with more gene mutations resulting in more aggressive tumors43. A summary of the genomic features and possible effect of the molecular changes in O_IDH_mut and A_IDH_mut in our study is shown in Fig. 8.

The summary of the genetic profiles and its effects in A_IDH_mut and O_IDH_mut.
Conclusion
The combined oncometabolite 2-HG induced by IDH mutations, TSG blockade, and TMM are important drivers and diagnostic hallmark of IDH-mutant gliomas. Despite reliable histological or molecular characteristics of our IDH-mutant gliomas, there were doubts in three cases because of the mixed features of O_IDH_mut and A_IDH_mut. The methylation classifier of DKFZ (v.12.5) matched perfectly with either O_IDH_mut or A_IDH_mut, and no genetically or epigenetically ambiguous IDH-mutant adult-type diffuse gliomas were observed. These valuable features should be recognized in differential diagnosis and genotype-specific therapeutic strategy. Finally, patients with MYCN-amplified A_IDH_mut had the worst prognosis, strongly suggesting that MYCN amplification, in addition to CDKN2A/2B homozygous deletion, should be included in the genetic criteria for grade 4 A_IDH_mut.
Data availability
The datasets generated and/or analysed during the current study are available in the Gene Expression Omnibus (GEO) repository under accession number GSE222423 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222423).
Abbreviations
- A_IDH_mut:
-
Astrocytoma, IDH-mutant
- BTP:
-
Brain tumor-targeted gene panel
- CNS:
-
Central nervous system
- WHO:
-
World health organization
- DKFZ:
-
Deutsches Krebsforschungszentrum
- FFPE:
-
Formalin-fixed paraffin-embedded
- GBM:
-
Glioblastoma, IDH-wildtype
- IHC:
-
Immunohistochemical study, immunohistochemistry
- NGS:
-
Next-generation sequencing
- NOS:
-
Not otherwise specified
- NEC:
-
Not elsewhere classified
- O_IDH_mut:
-
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- SNUH:
-
Seoul National University Hospital
- TMMs:
-
Telomere maintenance mechanisms
References
Nakasu, S. & Nakasu, Y. Malignant progression of diffuse low-grade gliomas: A systematic review and meta-analysis on incidence and related factors. Neurol. Med. Chir. https://doi.org/10.2176/jns-nmc.2021-0313 (2022).
Cancer Genome Atlas Research, N et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372, 2481–2498. https://doi.org/10.1056/NEJMoa1402121 (2015).
Ceccarelli, M. et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563. https://doi.org/10.1016/j.cell.2015.12.028 (2016).
Louis, D. N. et al. International Society Of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 24, 429–435. https://doi.org/10.1111/bpa.12171 (2014).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 23, 1231–1251. https://doi.org/10.1093/neuonc/noab106 (2021).
Brat, D. J. et al. cIMPACT-NOW update 5: Recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 139, 603–608. https://doi.org/10.1007/s00401-020-02127-9 (2020).
Chan, A. K. et al. Loss of CIC and FUBP1 expressions are potential markers of shorter time to recurrence in oligodendroglial tumors. Mod. Pathol. 27, 332–342. https://doi.org/10.1038/modpathol.2013.165 (2014).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 111011–111033 (2013).
Park, C. et al. Clinical and genomic characteristics of adult diffuse midline glioma. Cancer Res Treat 53, 389–398. https://doi.org/10.4143/crt.2020.694 (2021).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. https://doi.org/10.1093/bioinformatics/btw313 (2016).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474. https://doi.org/10.1038/nature26000 (2018).
Maksimovic, J., Phipson, B. & Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Res 5, 1281. https://doi.org/10.12688/f1000research.8839.3 (2016).
Shirahata, M. et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 136, 153–166. https://doi.org/10.1007/s00401-018-1849-4 (2018).
Reis, G. F. et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J. Neuropathol. Exp. Neurol. 74, 442–452. https://doi.org/10.1097/Nen.0000000000000188 (2015).
Kocakavuk, E. et al. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat. Genet. 53, 1088. https://doi.org/10.1038/s41588-021-00874-3 (2021).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. https://doi.org/10.1016/j.ccr.2010.12.014 (2011).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483. https://doi.org/10.1038/nature10866 (2012).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. https://doi.org/10.1038/nature10860 (2012).
Habiba, U. et al. Loss of H3K27 trimethylation is frequent in IDH1-R132H but not in non-canonical IDH1/2 mutated and 1p/19q codeleted oligodendroglioma: A Japanese cohort study. Acta Neuropathol. Commun. 9, 95. https://doi.org/10.1186/s40478-021-01194-7 (2021).
Filipski, K. et al. Lack of H3K27 trimethylation is associated with 1p/19q codeletion in diffuse gliomas. Acta Neuropathol. 138, 331–334. https://doi.org/10.1007/s00401-019-02025-9 (2019).
Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. https://doi.org/10.1038/nature16490 (2016).
Barthel, F. P., Wesseling, P. & Verhaak, R. G. W. Reconstructing the molecular life history of gliomas. Acta Neuropathol. 135, 649–670. https://doi.org/10.1007/s00401-018-1842-y (2018).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011. https://doi.org/10.1016/j.cell.2017.05.015 (2017).
Weissmann, S. et al. The tumor suppressor CIC directly regulates MAPK pathway genes via histone deacetylation. Cancer Res. 78, 4114–4125. https://doi.org/10.1158/0008-5472.CAN-18-0342 (2018).
Baumgarten, P. et al. Loss of FUBP1 expression in gliomas predicts FUBP1 mutation and is associated with oligodendroglial differentiation, IDH1 mutation and 1p/19q loss of heterozygosity. Neuropathol. Appl. Neurobiol. 40, 205–216. https://doi.org/10.1111/nan.12088 (2014).
Zaytseva, O. & Quinn, L. M. Controlling the master: Chromatin dynamics at the MYC promoter integrate developmental signaling. Genes 8, 118. https://doi.org/10.3390/genes8040118 (2017).
Gartel, A. L. et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 98, 4510–4515. https://doi.org/10.1073/pnas.081074898 (2001).
Elman, J. S. et al. Identification of FUBP1 as a long tail cancer driver and widespread regulator of tumor suppressor and oncogene alternative splicing. Cell Rep. 28, 3435–3449. https://doi.org/10.1016/j.celrep.2019.08.060 (2019).
Bell, R. J. et al. Understanding TERT promoter mutations: A common path to immortality. Mol. Cancer Res. 14, 315–323. https://doi.org/10.1158/1541-7786.MCR-16-0003 (2016).
Ferreira, M. S. V. et al. Alternative lengthening of telomeres is the major telomere maintenance mechanism in astrocytoma with isocitrate dehydrogenase 1 mutation. J. Neurooncol. 147, 1–14. https://doi.org/10.1007/s11060-020-03394-y (2020).
Dratwa, M., Wysoczanska, B., Lacina, P., Kubik, T. & Bogunia-Kubik, K. TERT-regulation and roles in cancer formation. Front. Immunol. 11, 589929. https://doi.org/10.3389/fimmu.2020.589929 (2020).
Hayashi, Y. et al. Diagnostic potential of TERT promoter and FGFR3 mutations in urinary cell-free DNA in upper tract urothelial carcinoma. Cancer Sci. 110, 1771–1779. https://doi.org/10.1111/cas.14000 (2019).
Labussiere, M. et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br. J. Cancer 111, 2024–2032. https://doi.org/10.1038/bjc.2014.538 (2014).
Killela, P. J. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 110, 6021–6026. https://doi.org/10.1073/pnas.1303607110 (2013).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. https://doi.org/10.1038/npp.2012.112 (2013).
Bouyahya, A. et al. The role of epigenetic modifications in human cancers and the use of natural compounds as epidrugs: Mechanistic pathways and pharmacodynamic actions. Biomolecules 12, 367. https://doi.org/10.3390/biom12030367 (2022).
Priesterbach-Ackley, L. P. et al. Brain tumour diagnostics using a DNA methylation-based classifier as a diagnostic support tool. Neuropathol. Appl. Neurobiol. 46, 478–492. https://doi.org/10.1111/nan.12610 (2020).
Galbraith, K. & Snuderl, M. DNA methylation as a diagnostic tool. Acta Neuropathol. Commun. 10, 71. https://doi.org/10.1186/s40478-022-01371-2 (2022).
Pegg, A. E. Repair of O(6)-alkylguanine by alkyltransferases. Mutat. Res. 462, 83–100. https://doi.org/10.1016/s1383-5742(00)00017-x (2000).
Gorlia, T. et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: Prognostic factor analysis of EORTC and NCIC trial 26981–22981/CE.3. Lancet Oncol. 9, 29–38. https://doi.org/10.1016/S1470-2045(07)70384-4 (2008).
van den Bent, M. J. et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic Oligodendrogliomas and Oligoastrocytomas. A report from EORTC study 26951. Clin. Cancer Res. 19, 5513–5522. https://doi.org/10.1158/1078-0432.CCR-13-1157 (2013).
Horbinski, C., McCortney, K. & Stupp, R. MGMT promoter methylation is associated with patient age and 1p/19q status in IDH-mutant gliomas. Neuro Oncol. 23, 858–860. https://doi.org/10.1093/neuonc/noab039 (2021).
Ferrer-Luna, R. et al. Whole-genomic survey of oligodendroglial tumors: correlation between allelic imbalances and gene expression profiles. J. Neurooncol. 103, 71–85. https://doi.org/10.1007/s11060-010-0369-4 (2011).
Acknowledgements
This study was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant number: HI14C1277) and the Institute for Information & Communications Technology Promotion (IITP) grant funded by the Korean government (MSIP) (No.2019-0567).
Author information
Authors and Affiliations
Contributions
S.-H.P. designed and supervised the study and wrote the entire manuscript. K.L. analyzed the NGS data and wrote the manuscript. S.-H.P., S.-I.K., and E.E.K. reviewed histopathology slides and anonymized molecular data for qualitative analysis. J.-K.W. partially contributed to the pathological diagnosis. C.-K.P. operated and treated the patients, and provided clinical data. Seung Hong Choi analyzed radiological image data. H.Y., Y.-M.S. and H.L. analyzed the NGS data derived from the NGS studies using a customized brain tumor targeted gene panel. All authors have reviewed and edited the final manuscript. All materials had been obtained for the electronic medical record of the patients, which were anonymized and retrospectively reviewed. No extra-human materials were obtained from the patients for this study. Under the Korean Bioethics and Safety Act, additional consent to publish was waived.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, K., Kim, SI., Kim, E.E. et al. Genomic profiles of IDH-mutant gliomas: MYCN-amplified IDH-mutant astrocytoma had the worst prognosis. Sci Rep 13, 6761 (2023). https://doi.org/10.1038/s41598-023-32153-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32153-y
- Springer Nature Limited