
Abstract
The market for the application of probiotics as a livestock health improvement supplement has increased in recent years. However, most of the available products are quality-controlled using low-resolution techniques and un-curated databases, resulting in misidentification and incorrect product labels. In this work, we deployed two workflows and compared results obtained by full-length 16S rRNA genes (16S) and metagenomic (Meta) data to investigate their reliability for the microbial composition of both liquid and solid forms of animal probiotic products using Oxford Nanopore long-read-only (without short-read). Our result revealed that 16S amplicon data permits to detect the bacterial microbiota even with the low abundance in the samples. Moreover, the 16S approach has the potential to provide species-level resolution for prokaryotes but not for assessing yeast communities. Whereas, Meta data has more power to recover of high-quality metagenome-assembled genomes that enables detailed exploration of both bacterial and yeast populations, as well as antimicrobial resistance genes, and functional genes in the population. Our findings clearly demonstrate that implementing these workflows with long-read-only monitoring could be applied to assessing the quality and safety of probiotic products for animals and evaluating the quality of probiotic products on the market. This would benefit the sustained growth of the livestock probiotic industry.
Similar content being viewed by others


Introduction
Probiotics are live microorganisms that confer a health benefit on the host by restoring the gut microbial balance and improving the immune system and are mostly intended to serve human health1. Besides human health, the use of probiotics such as Bacillus, Lactobacillus, Streptococcus, and Saccharomyces, to promote animal health and growth performance has gained increasing attention since it can alleviate the need for antibiotic use in animal husbandry2,3. The guidance on the characterisation of microorganisms used as feed additives or as production organisms requires specific information on probiotics including taxonomic information, antimicrobial susceptibility and production, and the toxigenicity and pathogenicity of the strain should be identified4. A statement of quantity, benefit, and use-by date also needs to be provided in the probiotic product label5. Since microbial safety and benefits are known to be strain-specific, accurate identification and labelling are critical for ensuring the safety and efficacy of the probiotic product and gaining consumer trust.
Labelling discrepancies in the probiotic product due to either the absence of listed taxon or the presence of non-listed taxon have been broadly demonstrated. A study of 55 European probiotic products showed as many as 40–47% of the probiotic products were mislabelled6. In addition, many probiotic products were shown to contain unlisted, possibly pathogenic bacteria that may undermine the safety and quality of the product7,8. Inappropriate labelling such as misspelling, obsolete, or non-existing nomenclature, failure to indicate the number of live cells, and in some instances failure to yield any living culture was commonly reported8,9,10. Hence, the European Society for Pediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Working Group for Probiotics and Prebiotics conducted a systematic review on quality assessment of the commercial probiotic products available worldwide. The working group identified the misidentification at the genus/species/strain level and contamination as worrisome issues among other quality problems i.e., incongruent number of viable cells and decreased functional properties. Majority of the probiotic products tested, regardless of the country of origin, yielded unsatisfactory results with more than one labelling inconsistency. A call for improved quality control of probiotic products including precise identification of microorganisms to the strain level has been issued11.
The majority of these studies have used conventional methods i.e. selective culture techniques, physiology, biochemistry, PCR identification, or used an out of date database for bacterial identification, which is time-consuming, limited to specific bacteria in a multispecies mixed product, and leads to misidentification. Recently, high-throughput next-generation sequencing is an emerging trend that attempts to accurately assess the microbial composition and any possibly pathogenic bacteria detected in the product. For example, whole-genome sequencing, microbiome, and metagenomic analysis using short-read sequencing was used to verify the microbial composition, and possible contamination of commercial probiotic products sold in the Canada, China, and United States marketplaces12,13,14. Given such approaches do not enable immediate species-level identification in a mixed microbial community, a gold standard PCR technique using strain-specific primers is required to verify the presence of certain organisms. Recently, the short-read metagenomic-based technique has been noticed to analyse probiotic supplements through both partial 16S rRNA-targeted sequencing (V3-V4)15 and shotgun metagenomic sequencing14,16. Though short-read sequences are typically classified at the family- or genus-level, greater taxonomic resolution to strain-level is of more interest in the quality assessment of probiotic products17. The MinION™ sequencer from Oxford Nanopore Technologies (ONT) is one of the single-molecule based sequencers that can be conducted in real-time with little equipment and is portable, enhancing its potential as a field tool for remote sites18,19. Since very long reads with no limitation of read length can be generated, ONT nanopore sequencer can help to resolve complex structure variants and repetitive regions in the genomes and can provide location of antimicrobial resistance (AMR) genes on mobile element or plasmid20 and nowadays, an alternative base-calling approach was improved by reaching the base accuracy up to > 99% (https://nanoporetech.com/accuracy). Therefore, long-read data could increase accuracy of microbial classification for both entire 16S rRNA gene and metagenome samples with bacterial mixtures which previously demonstrated in a gut microbiota study19,21.
Metagenomics facilitates to retrieve metagenome-assembled genomes (MAGs) with high contiguity and completeness from metagenomic data by assembling sequencing reads into contigs and grouped into single-taxon bins, can then be used for further high-accuracy taxonomic identification and gene annotation22. MAGs data also enables the prediction of AMR genes, virulence elements and biosynthetic gene clusters which can determine the presence of any microbial contaminant and further AMR-associated microorganism monitoring in the environment23,24,25. Long-read based MAGs have been noted to elucidate the microbial profiling in complex ecosystems, such as canine feces26, chicken gut27 and activated sludge28. Nevertheless, there is limited information using long-read data in probiotic products especially animal feed supplements. Given this, here, we compared two quality-checking techniques to investigate the list of microorganisms declared on the labels and any possible harm detected in animal probiotic feed-additive products available in Thailand. Two workflows, full-length 16S RNA gene and long-read metagenomics, were presented and compared. Using metagenomic data generated by ONT, we also demonstrated useful computational tools for MAGs taxonomy classification. AMR, virulence factors (VF), biosynthetic gene clusters (BGCs), and bacteriocins in probiotics products for animal were also characterised from MAGs.
Results
Animal probiotic product investigation workflows
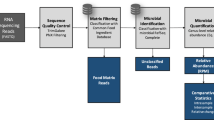
To investigate animal probiotic products available in the Thai market, both liquid and solid forms of commercial feed-additive products; designated as Product A and B, were collected and analysed. We applied long-read nanopore sequencing technology to assess detailed microbial compositional of the products as described in Fig. 1. Both full-length 16S rRNA amplicons and long-read metagenomic DNA libraries were prepared and subjected to nanopore sequencing. To display the relative abundance of each taxon in the sample, full-length 16S amplicon (16S) and metagenomic (Meta) data were taxonomically separated at the species-level using both NanoCLUST (NC) and Kraken2 (KK) tools. Probiotic genomes were reconstructed using metagenome-assembled genomes (MAGs) approach, taxonomically classification was achieved using the GTDB-Tk and NCBI database. Antimicrobial resistance (AMR) genes, virulence factor (VF), secondary metabolites biosynthetic gene cluster (BGC) and bacteriocin genes were predicted from either metagenome or MAGs data.

Schematic represents full-length 16S rRNA gene and metagenomic sequencing (A) and bioinformatic pipelines (B) for labelled strains investigation of animal probiotic products. Firstly, metagenomic DNA and 16S amplicon of animal probiotic products A and B were prepared and sequenced using Nanopore sequencer. Then, the nanopore reads were qualified and filtered by adapter trimming using porechop. After that, the 16S amplicon data were taxonomically identified by NanoCLUST and Kraken2 database while the metagenomic data were the taxonomically assigned using Kraken2 and assemble to the genome using the metagenome assembled-genomes (MAGs) approach. Genome taxonomy was determined using the GTDB-Tk database and only unidentified MAGs were identified by BLAST against the NCBI database. Finally, antimicrobial resistance (AMR) and function genes were predicted from metagenomic and MAGs data.
Taxonomic determination using nanopore reads
In this study, we initially performed long-read sequencing on animal probiotic Products A and B (Table 1) to investigate microbial profiling through full-length 16S amplicon, metagenomic, and MAGs profiling. Approximately 210 Mbp and 1.1 Gbp (Product A) and 253 Mbp and 469 Mbp (Product B) of 16S amplicon and metagenomic sequence data were respectively generated. The statistics of sequencing reads in animal probiotic products are summarised in Table S1 and Fig. 2. Overall, even at low abundance (< 1%), the composition of probiotic Products A and B were found to be in near perfect agreement with what was labelled by the producer for 16S data, especially Bacillus licheniformis which was only found in 16S data against Kraken2 (16S-KK, Fig. 2A). Furthermore, classification of 16S data against the NCBI database using NanoCLUST (16S-NC) gave the higher number in total percentage of relative abundance in both probiotic Products A and B than Kraken2 classification (16S-KK). On the other hand, only strains with high percentage of relative abundance, such as Lactobacillus plantarum (20.8%) and Lactobacillus paracasei (37.9%) in Product A, for example, were rescued by metagenomic data (Fig. 2A). As compared to an analysis of metagenomic data against Kraken2 (Meta-KK), the assembly of metagenomic data (MAGs) yields the highest taxonomic identity (Meta-KK) (Fig. 2).

Percentage of relative abundance of labelled and additional identified strains of animal probiotic Product A (A) and Product B (B). Strains are retrieved from either 16S amplicon data against Kraken2 (16S-KK) and NanoCLUST (16S-NC) or metagenomic data against Kraken2 (Meta-KK) and metagenomic-assembled genomes (MAGs). The additional identified strains exhibit representative strains from at least three techniques. The total relative abundance values are indicated as a total of classified bacteria in both labelled and additional identified strains by each classification approach. Missing data indicate strains that could not be identified from metagenomic sequenced data.
For Product A, all tools identified L. pararcasei (recently reclassified as Lacticaseibacillus pararcasei) as the dominant taxon, followed by L. plantarum (recently reclassified as Lactiplantibacillus plantarum) whereas L. acidophilus, listed by the manufacturers, was not detected in the sample. Among these, other labelled bacterial species, Lactobacillus fermentum (recently reclassified as Limosilactobacillus fermentum) and Bacillus subtilis was recovered from 16S-KK, 16S-NC, and Meta-KK at the low relative abundant (< 1% relative abundance). In addition, Bacillus licheniformis was merely recovered by 16S-KK (0.1%) (Fig. 2A). For Product B, the 16S amplicon sequencing allowed to identify the bacteria in the Bacillus genera such as B. subtilis and B. coagulans (recently reclassified as Weizmannia coagulans), which is listed on the product label, but the yeast Saccharomyces cerevisiae was not detected. For complete identification of both bacteria and yeast, the metagenome sequencing yielded more complete information (Fig. 2B). Similar to Product A, several unlisted species were identified such as Bacillus velezensis, B. licheniformis and B. thermoamylovorans (recently reclassified as Caldibacillus thermoamylovorans) as significant components by all sequencing-identification tools. Interestingly, an unlisted bacteria B. velezensis was identified as comprising up to 18% in Product A, and 40% in Product B, depending on the identification tools used (Fig. 2). The reason for this high prevalence either from misidentification or contamination during production process requires further investigation.
MAGs-based taxonomic classification
Next, we sought to reconstruct genomes from the metagenome sequencing to classify organisms to the species-level from these samples. The MAG approach enabled the recovery of seven and four genomes of bacterial species in products A and B, respectively, with the quality of MAG by showing above 80% genome completeness and less than 10% contamination (Fig. 3, Table S4). The completeness of the genomes retrieved from MAGs ranged from 17% to 99.4% (average, 80.7%) for Product A and from 3.4% to 99% (average, 67.4%) for Product B. While genome contamination was less than 3.6% and 0.2% for all isolate genomes for Product A and B, with the exception of bin 4 of the Product B (Table S4). Furthermore, most of MAGs had a high percentage of average nucleotide identity (ANI) in the range of 97–99% in both products A and B, indicating that the MAGs obtained belonged to the same population as the isolates (Table S4).

Metagenome-assembled genomes (MAGs) from animal probiotic products (A,B). Circos plot representation of features on the circular genome. Labelled and additional identified strains are shown in teal and sea green. The outer circle represents the complete genome of the closest strains. The distribution of MAG contigs all over the closest genome is located in the first inner circle. The whole-genome average nucleotide identity (ANI) percentage is calculated by FastANI and shown in the second inner circle. The percentage of completeness of the reconstructed genomes is indicated in the third inner circle.
Abundance of AMR, VF, BGC, and bacteriocin genes in probiotic feed supplements
To evaluate the existence of antimicrobial resistance (AMR) and virulence factor (VF) genes in bacterial composition in the animal probiotic products, we used the AMR and VF screening tool, ABRicate, to annotate from either metagenomic or MAGs data against NCBI AMRFinderPlus, RestFinder, and VFDB databases. AMR analysis of metagenomic data from products A and B revealed that two and nine AMR genes (Fig. S1) respectively were distributed in animal probiotic products with ranges from 80–98.4% identity and 82–99.7% coverage (Table S5). Among these, chloramphenicol resistance gene, cfr(B), was commonly found both in products A and B. Overall, there was a higher number of AMR genes in Product B than those in Product A which were identified against antimicrobial drugs in class aminoglycosides [ant(4')-Ib and aadK], chloramphenicol (cat4), macrolides [erm(34), erm(D), and mphK] and quinolone (qnrD1). Whereas, the tetracycline efflux associated gene [tet(L)], was only found in Product A (Fig. S1, Table S5). Correlation of AMR analysis in metagenomics data and MAGs confirmed the present of genes involved in two (chloramphenicol and tetracycline) and three (aminoglycosides, chloramphenicol, and macrolides) classes of antimicrobial drugs with ranges from 87.3 to 99.8% identity and 98.7–100% coverage (Table 2, Fig. S1) in Product A and B, respectively. With no observation of any plasmids from labelled reconstructed genomes (Table S4), indicating that all of annotated AMR genes may locate on the chromosome. No VFs were identified in the MAGs.
Biosynthetic gene cluster (BGC) encoding for secondary metabolites and bacteriocins of the MAGs were identified using antiSMASH and BAGEL4. In this work, we identified seven and ten putative BGCs in the recovered MAGs from Product A and B (Fig. S2, Table S6). Of these, three BGCs (Non-Ribosomal Peptide Synthetase or NRPS, post-translationally modified peptides or RiPPs, and trans-AT polyketide synthases or transAT-PKS) were found in both products. In the MAGs of Product A found that transAT-PKS gene clusters were the most prevalent BGCs (n = 3), while NRPS was dominantly found in the Product B (n = 8). Among these, three BGCs (arylpolyene, lanthipeptide-class-iv, and PKS) and six BGCs (batalactone, epipeptide, ladderane, lanthipeptide-class-ii, sactipeptide, and siderophore) were exclusively annotated in the either Product A or B (Fig. S2, Table S6). Furthermore, BAGEL4 revealed eight and eleven classes of bacteriocins and RiPPs from MAGs of the Product A and B. Of these, three classes (amylocyclicin, LCI, and UviB) were found in both samples. The carnocin CP52 was particularly abundant in the Product A, whereas, four (amylocyclicin, ComX2, lassopeptide and UviB) of eleven were the most abundant in the Product B (Fig. S3, Table S7).
Discussion
There are several commercially available probiotic products available on the market, including animal probiotic products, but very few have been formally evaluated and demonstrated to contain the bacterial strains on the label by either conventional 16S rRNA gene amplification or short-read sequencing data analysis14,15,16. The emergence of long-read sequencing technology such as PacBio and Nanopore, allows the sequence of either full-length the 16S rRNA gene or long-read whole metagenome analysis which can be used to compare the sequences in public databases for microbial identification29,30. Although long-read sequencing has been used to monitor probiotic products for humans such as cottage cheese and paocai brine31,32, remarkably it has been used far less in animal feed additive products. Furthermore, recent developments in state-of-the-art software packages using metagenome data such as ATLAS, for instance, allowed to recovery of genomes from metagenome data33. However, most bioinformatic tools are optimised for either short-read or hybrid (short- and long-read) and a few lees implemented for Nanopore-long-read-only application22. Here, we modified and deployed the workflow for animal probiotics product investigation using a Nanopore long-read-only (Fig. 1). Applying Oxford Nanopore long-read sequencing and developing a bioinformatics pipeline for metagenomics study using only Nanopore long-read was firstly validated using ZymoBIOMICS™ Microbial Community DNA Standard as a control. Metagenomic profiling demonstrated the expected microbial species at anticipated abundances (Table S8 and Fig. S4), suggesting that the developed workflow is suitable for analysing long-read metagenomics data. In addition, our result found that this workflow is successful in applying both targeted 16S rRNA gene (16S) and metagenomic (Meta) data incorporated with bioinformatic tools and a suitable public database for taxonomically microbial classification. This approach also allowed metagenome-assembled genomes (MAGs) of probiotic strains to be reconstructed and used for taxonomic assignments which are consequently useful and a guideline for further application such as quality and consistency control of strains.
This work employed both 16S amplicon and metagenomic data to investigate the composition of probiotic strains in the products and demonstrates that 16S results in consistent patterns of microbial taxonomic identification when compared to metagenomics. However, using targeted 16S rRNA gene sequencing through the amplification step may introduce PCR biases in bacterial quantifying taxa resulting in underestimation of the abundance of bacterial species34,35,36. Moreover, despite the fact that targeted 16S rRNA gene sequencing is considered the gold standard for microbial species identification, only bacterial and archaea communities are detected, while fungi and viruses are excluded, with no specific ONT kits currently available for fungal community classification37. Conversely, even though metagenomics missed to detect the microbial species in the low abundance sample, however, MAGs enable the identification of the yeast, Saccharomyces cerevisiae in Product B (Fig. 2), which is overlooked using targeted 16S rRNA gene sequencing. Of note, the probiotic products used in this work (Products A and B) were listed to contain two bacterial genera (Bacillus and Lactobacillus) and only one yeast strain (S. cerevisiae in the Product B; Table 1). Of these, only two of the six strains listed on the label of Product A were detected using all methods, with some strains being found in low abundance within the sample. Remarkably, one strain listed as a component of the product, L. acidophilus, was not detected from the targeted 16S rRNA gene or metagenomic workflows, indicating that it was not present in Product A (Fig. 2). This work also demonstrated that additional strains were also present in the probiotic products. Levilactobacillus brevis, Lacticaseibacillus rhamnosus, Bacillus velezensis and Pediococcus acidilactici were all identified by at least three of the investigating methods in the Product A, while B. velezensis, B. licheniformis and Caldibacillus thermoamylovoran were identified in the Product B (Fig. 2). Among these, B. velezensis was a dominant taxon in both products which were detected in all methods used in this work. Our results confirmed the presence of this bacteria and other species by showing a high percentage of ANI value and showed a high distribution of our assembled contigs to the closest reference genome (Fig. 3, Table S4). B. velezensis is a member of B. subtilis group complex and was later reclassified as a new species based on genomic and secondary metabolites diversity38,39 and is considered safe and implicated as a probiotic for the animal feed-additive products for poultry40,41,42. Therefore, the lack of an updated database for safety evaluation by authorities and the highly similar prevalence of characters and sequences among this group may easily lead to misidentification and labelling discrepancies.
In this work, we used three databases, Kraken2, NCBI Refseq, and GTDB-Tk, for microbial taxonomic classification. Overall, classifying using blastn and the NCBI Refseq database in the NanoCLUST tool using 16S data (16S-NC) gave the percentage of total microbial relative abundance than Kraken2 (16S-KK) in both animal probiotic products. Generally, NanoCLUST pipeline is classified using blastn of the polished consensus through the NCBI Refseq database43 while Kraken2 builds its own database not only from the 16S database but also from Greengenes, SILVA, and RDP by using k-mer base assignments. Even though Kraken2 allowed a greater amount of reference genomic data for microbial classification, however, it was noted that Kraken2 would not identify a large proportion of reads correctly at the species level (8.93% mean absolute percentage error)44, particularly when genomes from various species or genera have a high level of genome similarity, as is the case in several taxonomic groups such as Bacillus45 highlighted for B. velezensis in this work. Compared with conventional 16S amplicon, both Kraken2 and GTDB-Tk respectively provided better consistency and a higher relative abundance of taxonomic annotation for metagenomic and MAGs of the Product A. Even two MAGs of Product A were not attributed to existing species in the GTDB-Tk database, however, the result was a consistent proportion of bacterial classification annotated from Kraken2 (Meta-KK) (Fig. 2), suggesting that both Kraken2 and GTDB-Tk are suitable to be used as reference database for bacterial identification.
Probiotics are becoming increasingly popular and recognized as generally recognized as safe (GRAS) strains for humans and animals, mostly including species of Bacillus, Bifidobacterium, Lactobacillus, Streptococcus, and the yeast Saccharomyces boulardii46,47. Even though it is classified as a GRAS strain, it is not exempt from acquiring antimicrobial resistance (AMR) genes following the FDA regulation48. Recently, a putative aadk, cat, erm(D), lsa(B), and tet(L) genes were characterized from five commercial Bacillus used as probiotic feed additives49. Moreover, cfr(B) gene, an RNA methyltransferase, was reported as a multidrug-resistant phenotype and conferred resistance to some macrolide antibiotics such as phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics found in Bacillus isolates from swine feces, suggesting that the possible transmission of AMR gene to host cells50,51,52. Consistent with the result of this study, cfr(B) gene was reconstructed in the genome of B. velezensis and B. subtilis of Products A and B, respectively. Because no plasmids were found in the MAGs of labelled probiotics in this study, the putative AMR-related genes may be intrinsic in the chromosomes of these related Bacillus species, indicating that such genes are less able to transmit to other bacterial species and may be safe for the host. However, further investigation on the distribution of AMR genes is needed to support this conclusion.
The rise of antibiotic-resistant bacteria due to overuse of antibiotics, both in humans and animals, has led to concern regarding the impact of AMR on global public health53. To decrease the use of antibiotics in animal production, the application of bacteriocins-producing probiotics for livestock has been strongly encouraged due to their immunomodulatory effect and maintenance of balance in the gastrointestinal microbiota54, potentially functioning as an alternative to antibiotic growth promoter55. For instance, various species of Lactobacillus including L. acidilactici, L. johnsonii L. mucosae, and L. plantarum isolated from the gastrointestinal tract of piglets showed significant antibacterial activities against Escherichia coli and Enterobacteria56,57. With the de novo assembly, it was possible to determine genes encoding bacteriocins and biosynthetic gene clusters (BGC) for secondary metabolites present within the assemblies, potentially increasing the chances of discovering novel antimicrobials using function-based metagenomic analysis as previously described58,59. Thus, the application of metagenomics approaches to animal probiotic products not only allows a deep understanding of microbial profiling but is also useful for identification of BGCs encoding bacteriocins which could have beneficial effects within probiotics in livestock production.
Conclusion
In this work, we highlight a useful workflow for ensuring the safety and quality of commercial probiotic feed supplements using only Nanopore long-read. Implementation of two workflows using either 16S amplicon (16S) or metagenome (Meta) data enables animal probiotic product quality and safety assurance. The analysis of two animal probiotic products, one in liquid form and another in solid form found that both 16S and Meta data showed inconsistency in the product labels and was of special concern of the presence of antimicrobial resistance genes. Moreover, the metagenome data can be used for more in-depth analysis of the product quality/consistency/efficacy such as the production of bacteriocins and other secondary metabolites. These findings provide a guide for selecting an appropriate method for the safety assessment of animal probiotic products. In addition, using Nanopore long-read-only incorporated with our developed workflows are promising technique, and can be used as an efficient tool to monitor and ensure probiotic product quality and safety, both for the producer and regulatory body.
Materials and methods
Animal probiotic product samples
Two commercial animal probiotic products available in Thailand were collected on January 2022. The sample, designated as Product A and B, were used directly for metagenomic DNA extraction.
Metagenomic DNA extraction
Metagenomic DNA was extracted from both samples of animal probiotic products. Either bacterial pellet collected from 15 mL of Product A or 10 g of Product B were extracted using the ZymoBIOMICS™ DNA miniprep Kit (D4300; Zymo Research, USA) following the modified protocol by changing from 20 to 3 min for bead-beating step to avoid DNA shearing. Next, the purity of the extracted DNA was checked by using a Nanodrop Spectrophotometer (Thermo Fisher Scientific, USA) and was quantified by a Qubit® 4.0 Fluorometer (Invitrogen, USA).
Library preparation and nanopore sequencing
The 20 ng of metagenomic DNA was used as a template for amplifying 16S rRNA genes using the 16S Barcoding Kit (SQK-RAB204; Oxford Nanopore Technologies, UK) containing the 27F/1492R primer set. PCR amplification was performed using LongAmp™ Taq 2X Master Mix (New England Biolabs, UK) with the following conditions: initial denaturation at 95 °C for 1 min, 25 cycles of 95 °C for 20 s, 55 °C for 30 s, and 65 °C for 2 min, followed by a final extension at 65 °C for 5 min. The PCR product was cleaned up using AMPure XP (Beckman Coulter, USA) and a total of 100 ng DNA barcoded libraries was used for rapid adapter attachment. For the metagenome library preparation was performed using the Rapid Barcoding Sequencing Kit (SQK-RBK004; Oxford Nanopore Technologies, UK). Briefly, a total of 150 ng of metagenomic DNA was used for library preparation by cleaved with transposase enzyme to produce chemically modified ends and a barcode was added to each DNA sample, finally ligated with an adapter. The library was loaded into the R9.4.1 flow cell (FLO-MIN106; Oxford Nanopore Technologies, UK) and sequenced using MinION (Mk1C) with the default setting. ZymoBIOMICS™ Microbial Community DNA Standard (D6305; Zymo Research Corp, CA, USA) was used as a control.
Sequence processing and taxonomic classification
Base-calling and demultiplexing were performed using Guppy v6.0.1 in the SUP (super accuracy) mode60. Read quality was assessed with Nanoplot v1.20.060. Adapters and barcodes were removed from the reads using Porechop v0.2.4 (https://github.com/rrwick/ Porechop). The reads were filtered using NanoFilt v2.8.060 with a mean quality score of > 10 with at least 1000-bp read length for 16S amplicon and a mean quality score of > 9 with at least 200-bp read length for metagenomic data. The taxonomy of 16S amplicon was assigned against both RefSeq databases (PlusPFP-8; 5/17/2021) using Kraken2 v2.1.244 and NCBI databases using NanoCLUST version eb6a2c82 (committed on Dec 20, 2021)43. Whereas, metagenomics data were only classified to species-level against the RefSeq databases (PlusPFP-8; 5/17/2021) using Kraken2 pipeline44. Percentage of relative species abundance is calculated by dividing the number of species from one group by the total number of species from all groups.
Metagenomic assembly, polishing and MAGs taxonomic identification
Metagenome-assembled genome (MAGs) (quality score of > 9 with at least 200-bp read length) was performed by assembling using metaFlye v2.961. Nanopore assembly polishing was accomplished using two rounds of Racon v1.3.3 (https://github.com/lbcb-sci/Racon) and followed by one round of medaka v0.6.5 (https://github.com/nanoporetech /medaka). After that, nanopore reads were mapped to the polished nanopore assembly using minimap2 v2.2462. Automatic binning was performed using MetaBAT2 v2.15 with default settings63 and then annotated taxonomically using GTDB-Tk v1.5.1 against the GTDB R202 database (2022–04-08)64,65. Unmapped MAGs were further taxonomically annotated using blastn against NCBI database. The dereplicated bins were then checked for completeness and contamination using CheckM v1.0.1866 and MOB-suite v3.0.3 was used for plasmid typing from MAGs67. FastANI v1.3 was used to calculate the average nucleotide identity (ANI) of orthologous gene pairs in the scaffold and reference genomes68. A circular graphical display of bacterial genomic properties was done by pyCircos v0.3.0 (https://github.com/ponnhide/pyCircos).
AMR, VF, BGC, and bacteriocin genes annotation
The genetics determinants conferring AMR and VF genes were searched by ABRicate v1.0.1 (https://github.com/tseemann/abricate)69 against publicly available databases; NCBI AMRFinderPlus (PRJNA313047)70, ResFinder (2022–05-24)71, and VFDB (http://www.mgc.ac.cn/VFs/)72. Putative biosynthetic gene clusters (BGCs) for secondary metabolites was annotated by antiSMASH v6.073 with minimum contig length at 1000-bp and the minimal detection option selected so that only BGCs are detected. Annotation ribosomally synthesized and post translationally modified peptides (RiPPs) and bacteriocins on contigs were done using BAGEL4 v.1.274.
Data availability
The original contributions presented in the study are included in the article or supplementary material, further inquiries can be directed to the corresponding authors. The raw sequencing data are available at the NCBI Sequence Read Archive (SRA) under BioProject PRJNA82350075,76 with accession numbers SRR18682825 and SRR18682826 (16S amplicon sequencing) SRR18682824 and SRR18682823 (metagenomic sequencing).
References
Morelli, L. & Capurso, L. FAO/WHO guidelines on probiotics: 10 years later. J. Clin. Gastroenterol. 46, S1–S2 (2012).
Arsène, M. M. J. et al. The use of probiotics in animal feeding for safe production and as potential alternatives to antibiotics. Vet World 14, 319–328 (2021).
Bhogoju, S. & Nahashon, S. Recent advances in probiotic application in animal health and nutrition: A review. Agriculture 12, 304 (2022).
EFSA et al. Guidance on the characterisation of microorganisms used as feed additives or as production organisms. EFSA J. 16, e05206 (2018).
Jackson, S. A. et al. Improving end-user trust in the quality of commercial probiotic products. Front. Microbiol. 10, 739 (2019).
Temmerman, R., Pot, B., Huys, G. & Swings, J. Identification and antibiotic susceptibility of bacterial isolates from probiotic products. Int. J. Food Microbiol. 81, 1–10 (2003).
Hamilton-Miller, J. M. T. & Shah, S. Deficiencies in microbiological quality and labelling of probiotic supplements. Int. J. Food Microbiol. 72, 175–176 (2002).
Chen, T. et al. Microbiological quality and characteristics of probiotic products in China. J. Sci. Food Agric. 94, 131–138 (2014).
Weese, J. S. Evaluation of deficiencies in labeling of commercial probiotics. Can. Vet. J. 44, 982–983 (2003).
Weese, J. S. & Martin, H. Assessment of commercial probiotic bacterial contents and label accuracy. Can. Vet. J. 52, 43–46 (2011).
Kolaček, S. et al. Commercial probiotic products. A call for improved quality control: A position paper by the ESPGHAN working group for probiotics and prebiotics. J. Pediatr. Gastroenterol. Nutr. 65, 117–124 (2017).
Ullah, M., Raza, A., Ye, L. & Yu, Z. Viability and composition validation of commercial probiotic products by selective culturing combined with next-generation sequencing. Microorganisms 7, 188 (2019).
Shehata, H. R. & Newmaster, S. G. Combined targeted and non-targeted PCR based methods reveal high levels of compliance in probiotic products sold as dietary supplements in United States and Canada. Front. Microbiol. 11, 1 (2020).
Patro, J. N. et al. Culture-independent metagenomic surveillance of commercially available probiotics with high-throughput next-generation sequencing. mSphere 1, e00057–00016 (2016).
Lugli, G. A. et al. Compositional assessment of bacterial communities in probiotic supplements by means of metagenomic techniques. Int. J. Food Microbiol. 294, 1–9 (2019).
Lugli, G. A. et al. The probiotic identity card: A novel, “probiogenomics” approach to investigate probiotic supplements. Front. Microbiol. 12, 1. https://doi.org/10.3389/fmicb.2021.790881 (2022).
Jeong, J. et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci Rep 11, 1727 (2021).
Leggett, R. M. & Clark, M. D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 68, 5419–5429 (2017).
Leggett, R. M. et al. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat. Microbiol. 5, 430–442 (2020).
Berbers, B. et al. Combining short and long read sequencing to characterize antimicrobial resistance genes on plasmids applied to an unauthorized genetically modified Bacillus. Sci. Rep. 10, 4310 (2020).
Matsuo, Y. et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol. 21, 35 (2021).
Yang, C. et al. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput. Struct. Biotechnol. J. 19, 6301–6314 (2021).
Youngblut, N. D. et al. Large-scale metagenome assembly reveals novel animal-associated microbial genomes, biosynthetic gene clusters, and other genetic diversity. mSystems 5, e01045–01020 (2020).
Zhou, Y. et al. Extensive metagenomic analysis of the porcine gut resistome to identify indicators reflecting antimicrobial resistance. Microbiome 10, 39 (2022).
Moss, E. L., Maghini, D. G. & Bhatt, A. S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 38, 701–707 (2020).
Cuscó, A., Pérez, D., Viñes, J., Fàbregas, N. & Francino, O. Long-read metagenomics retrieves complete single-contig bacterial genomes from canine feces. BMC Genom. 22, 330 (2021).
Feng, Y. et al. Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun. Biol. 4, 1305 (2021).
Singleton, C. M. et al. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 12, 2009 (2021).
Zhang, Y.-Z. et al. Nanopore basecalling from a perspective of instance segmentation. BMC Bioinf. 21, 136 (2020).
Noakes, M. T. et al. Increasing the accuracy of nanopore DNA sequencing using a time-varying cross membrane voltage. Nat. Biotechnol. 37, 651–656 (2019).
Jin, H. et al. Using PacBio sequencing to investigate the bacterial microbiota of traditional Buryatian cottage cheese and comparison with Italian and Kazakhstan artisanal cheeses. J. Dairy Sci. 101, 6885–6896 (2018).
Cao, J. et al. Assessment of bacterial profiles in aged, home-made Sichuan paocai brine with varying titratable acidity by PacBio SMRT sequencing technology. Food Control 78, 14–23 (2017).
Kieser, S., Brown, J., Zdobnov, E. M., Trajkovski, M. & McCue, L. A. ATLAS: a Snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinform. 21, 257 (2020).
Silvia, G. A., Sarma-Rupavtarm, R., Klepac-Ceraj, V. & Polz, M. F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71, 8966–8969 (2005).
Peterson, D. et al. Comparative analysis of 16S rRNA gene and metagenome sequencing in Pediatric gut microbiomes. Front. Microbiol. 12, 1–13 (2021).
Bonk, F., Popp, D., Harms, H. & Centler, F. PCR-based quantification of taxa-specific abundances in microbial communities: Quantifying and avoiding common pitfalls. J. Microbiol. Methods 153, 139–147 (2018).
Ciuffreda, L., Rodríguez-Pérez, H. & Flores, C. Nanopore sequencing and its application to the study of microbial communities. Comput. Struct. Biotechnol. J. 19, 1497–1511 (2021).
Fritze, D. Taxonomy of the genus Bacillus and related genera: the aerobic endospore-forming bacteria. Phytopathology 94, 1245–1248 (2007).
Mullins, A. J. et al. Reclassification of the biocontrol agents Bacillus subtilis BY-2 and Tu-100 as Bacillus velezensis and insights into the genomic and specialized metabolite diversity of the species. Microbiology 166, 1121–1128 (2020).
EFSA, Panel o. A. et al. Safety and efficacy of a feed additive consisting of Bacillus velezensis PTA-6507, B. velezensis NRRL B-50013 and B. velezensis NRRL B-50104 (Enviva® PRO 202 GT) for turkeys for fattening (Danisco Animal Nutrition). EFSA Journal 19, e06535 (2021).
EFSA, Panel o. A. et al. Safety and efficacy of Bacillus subtilisPB6 (Bacillus velezensisATCC PTA-6737) as a feed additive for chickens for fattening, chickens reared for laying, minor poultry species (except for laying purposes), ornamental, sporting and game birds. EFSA Journal 18, e06280 (2020).
Khalid, F. et al. Potential of Bacillus velezensis as a probiotic in animal feed: A review. J. Microbiol. 59, 627–633 (2021).
Rodríguez-Pérez, H., Ciuffreda, L. & Flores, C. NanoCLUST: a species-level analysis of 16S rRNA nanopore sequencing data. Bioinformatics 37, 1600–1601 (2021).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Fan, B., Blom, J., Klenk, H.-P. & Borriss, R. Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis form an “operational group B. amyloliquefaciens” within the B. subtilis species complex. Front. Microbiol. 8, 1 (2017).
Muzaffar, K., Jan, R., Ahmad Bhat, N., Gani, A. & Ahmed Shagoo, M. in Advances in Probiotics (eds Dharumadurai Dhanasekaran & Alwarappan Sankaranarayanan) 417–435 (Academic Press, 2021).
Chokesajjawatee, N. et al. Safety assessment of a nham starter culture Lactobacillus plantarum BCC9546 via whole-genome analysis. Sci Rep 10, 10241 (2020).
Mathur, S. & Singh, R. Antibiotic resistance in food lactic acid bacteria—A review. Int. J. Food Microbiol. 105, 281–295 (2005).
Agersø, Y. et al. Antimicrobial susceptibility testing and tentative epidemiological cutoff values for five Bacillus species relevant for use as animal feed additives or for plant protection. Appl. Environ. Microbiol. 84, e01108-01118 (2018).
Dai, L. et al. First report of the multidrug resistance gene cfr and the phenicol resistance gene fexA in a Bacillus strain from swine feces. Antimicrob. Agents Chemother. 54, 3953–3955 (2010).
Long, K. S., Poehlsgaard, J., Kehrenberg, C., Schwarz, S. & Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 50, 2500–2505 (2006).
Hansen, L. H., Planellas, M. H., Long, K. S. & Vester, B. The order Bacillales hosts functional homologs of the worrisome cfr antibiotic resistance gene. Antimicrob. Agents Chemother. 56, 3563–3567 (2012).
Talebi Bezmin Abadi, A., Rizvanov, A. A., Haertlé, T. & Blatt, N. L. World Health Organization report: Current crisis of antibiotic resistance. BioNanoSci. 9, 778–788 (2019).
Hernández-González, J. C., Martínez-Tapia, A., Lazcano-Hernández, G., García-Pérez, B. E. & Castrejón-Jiménez, N. S. Bacteriocins from lactic acid bacteria. A powerful alternative as antimicrobials, probiotics, and immunomodulators in veterinary edicine. Animals 11, 979 (2021).
Jha, R., Das, R., Oak, S. & Mishra, P. Probiotics (direct-fed microbials) in poultry nutrition and their effects on nutrient utilization, growth and laying performance, and gut health: A systematic review. Animals 10, 1863 (2020).
Huang, C., Qiao, S., Li, D., Piao, X. & Ren, J. Effects of Lactobacilli on the performance, diarrhea incidence, VFA concentration and gastrointestinal microbial flora of weaning pigs. Anim. Biosci. 17, 401–409 (2004).
Chiang, M.-L. et al. Optimizing production of two potential probiotic Lactobacilli strains isolated from piglet feces as feed additives for weaned piglets. Anim. Biosci 28, 1163–1170 (2015).
de Abreu, V. A. C., Perdigão, J. & Almeida, S. Metagenomic approaches to analyze antimicrobial resistance: An overview. Front. Genet. 11, 575592 (2021).
de Castro, A. P., Fernandes, G. d. R. & Franco, O. L. Insights into novel antimicrobial compounds and antibiotic resistance genes from soil metagenomes. Front. Microbiol. 5, 00489 (2014).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Kolmogorov, M. et al. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat Methods 17, 1103–1110 (2020).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Kang, D. D. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359–e7359 (2019).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Parks, D. H. et al. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucl. Acids Res. 50, D785–D794 (2021).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Robertson, J. & Nash, J. H. E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4, 1. https://doi.org/10.1099/mgen.1090.000206 (2018).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Seemann, T. ABRicate: mass screening of contigs for antiobiotic resistance genes. https://github.com/tseemann/abricate (2016).
Feldgarden, M. et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63, e00483-e1419 (2019).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644 (2012).
Chen, L., Zheng, D., Liu, B., Yang, J. & Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucl. Acids Res. 44, D694–D697 (2015).
Blin, K. et al. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucl. Acids Res. 49, W29–W35 (2021).
van Heel, A. J. et al. BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucl. Acids Res. 46, W278–W281 (2018).
Khongchatee, A. et al. Full-Length 16S rRNA Gene Amplicon and Metagenome Taxonomic Profiling of Beneficial Microbes in Poultry and Swine Probiotic Product. Microbiol Resour Announc 11, e0069022 (2022).
Sudjai, A. et al. Long-Read 16S rRNA Amplicon and Metagenomic Data of Swine Feed-Additive Probiotics Product. Microbiol Resour Announc. 11, e0039722 (2022).
Acknowledgements
We special thanks to Prof. Dr. Paul Hoskisson from Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK for comments and proofreading of the manuscript. We also thanks to Ms. Kantgamon Suriye from the Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University; Ms. Aunthikarn Sudjai, and Mr. Adison Khongchatee from the Division of Bioinformatics and Data Management for Research, Faculty of Medicine Siriraj Hospital, Mahidol University for helping in animal probiotic sample preparation, gDNA extraction and some part of bioinformatics analysis.
Funding
This research project is supported by Mahidol University (Basic Research Fund: fiscal year 2023, R016641012) and Thailand Science Research and Innovation (TSRI).
Author information
Authors and Affiliations
Contributions
T.W. and S.F. guided and completed the whole experimental design. W.K., P.J., T.A., and T.W. involved in the data collection. W.K. and T.A. were responsible for the arrangement of data. W.K., P.J., T.A., S.F. and T.W. were responsible for analysing the data and participated in the interpretation of the results. P.L., N.C. and S.F. provided A.M.R. information and valuable comments on probiotic safety assessments. W.K., P.J., N.P. and T.W. wrote the initial draft with all authors providing critical feedback and edits to subsequent revisions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kruasuwan, W., Jenjaroenpun, P., Arigul, T. et al. Nanopore Sequencing Discloses Compositional Quality of Commercial Probiotic Feed Supplements. Sci Rep 13, 4540 (2023). https://doi.org/10.1038/s41598-023-31626-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31626-4
- Springer Nature Limited