
Abstract
Arabidopsis SENSITIVITY TO RED LIGHT REDUCED 1 (SRR1) delays the transition from vegetative to reproductive development in noninductive conditions. A second-site suppressor screen for novel genes that overcome early flowering of srr1-1 identified a range of suppressor of srr1-1 mutants flowering later than srr1-1 in short photoperiods. Here, we focus on mutants flowering with leaf numbers intermediate between srr1-1 and Col. Ssm67 overcomes srr1-1 early flowering independently of day-length and ambient temperature. Full-genome sequencing and linkage mapping identified a causative SNP in a gene encoding a Haloacid dehalogenase superfamily protein, named HAD-FAMILY REGULATOR OF DEVELOPMENT AND FLOWERING 1 (HDF1). Both, ssm67 and hdf1-1 show increased levels of FLC, indicating that HDF1 is a novel regulator of this floral repressor. HDF1 regulates flowering largely independent of SRR1, as the effect is visible in srr1-1 and in Col, but full activity on FLC may require SRR1. Furthermore, srr1-1 has a delayed leaf initiation rate that is dependent on HDF1, suggesting that SRR1 and HDF1 act together in leaf initiation. Another mutant flowering intermediate between srr1-1 and wt, ssm15, was identified as a new allele of ARABIDOPSIS SUMO PROTEASE 1, previously implicated in the regulation of FLC stability.
Similar content being viewed by others

Introduction
Higher plants carefully time the transition to flowering, allowing for maximal acquisition of biomass during the growth season to enhance reproductive success. Environmental factors such as day length and temperature are integrated with endogenous developmental processes, creating a network of signaling pathways to find the optimal time for flowering1,2.
In the facultative long day (LD) plant Arabidopsis thaliana, a long daily light period promotes flowering via the photoperiodic pathway3,4. Day length is sensed in the leaves by photoreceptors and integrated into the flowering time network via the circadian clock. The key regulator of the photoperiodic pathway is CONSTANS (CO) and the oscillatory pattern of the CO transcript is in large parts shaped by the repressive action of CYCLING DOF FACTORS (CDFs) which prevent CO accumulation in the morning5,6. Instead, CO protein accumulates in the evening and activates the floral integrator FLOWERING LOCUS T (FT). The FT protein moves from leaves to the shoot apical meristem (SAM) to induce flowering7,8,9. Important antagonists of CO are TEMPRANILLO (TEM) transcription factors which repress expression of FT, thereby counteracting the effect of CO and balancing the floral response to LDs10.
The phytohormone gibberellin (GA) has a well-established function in the control of flowering11. GA is required for flowering in short days (SDs)12 as GA accumulation in the SAM activates LEAFY and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) which enables flowering without a photoperiodic stimulus13,14. In addition, GA has spatially separated functions in LDs, by promoting expression of FT in the vascular tissue and activation of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) transcription factors in the meristem15.
Vernalization, the exposure to an extended period of cold, promotes flowering by down-regulating the MADS-box transcription factor FLOWERING LOCUS C (FLC)16,17. FLC is also down-regulated by endogenous regulators, collectively referred to as the autonomous pathway (AP) which comprises a suite of RNA binding proteins and chromatin modifiers18,19,20. FLC negatively regulates a set of floral activators, among them FT21. It has evolved as a central regulatory hub for flowering time in the Brassicaceae and the mechanisms of FLC transcriptional regulation and epigenetic silencing are extensively studied (reviewed in22). Additionally, FLC expression might also be regulated indirectly by phosphorylation of AP components through Casein Kinase II and either upregulated or repressed through Protein Phosphatase 2A dephosphorylation, depending on the subunit involved23,24.
Apart from extended periods of low temperature, ambient temperature also impacts flowering, with increasing temperature accelerating floral transition. Mutants deficient in the photoreceptor PhyB flower earlier than wild type (wt) plants at 22 °C but not at 16 °C, indicating that PhyB is involved in temperature perception25.
We have previously characterized the role of SENSITIVITY TO RED LIGHT REDUCED (SRR1) in flowering time control. SRR1 was originally identified as a component of phyB signaling due to the reduced sensitivity to red light in srr1-126. Srr1-1 mutant plants also have a reduced ability to sense the length of the photoperiod, leading to particularly early flowering in SDs27. By promoting the expression of direct repressors of FT including CDF1 and the TEM transcription factors, SRR1 represses flowering in non-inductive conditions27. However, SRR1 function is not limited to the photoperiodic pathway. Levels of the key floral repressor FLC are also repressed in the srr1-1 mutant. In other Brassicaceae, Brassica rapa BrFLC2 was identified as a candidate for a major flowering QTL co-regulating a cis-QTL containing BrSRR1, thus associating BrSRR1 with flowering time control28. Moreover, Brassica napus harbours five SRR1 homologs which show subfunctionalization29. Recently, SNPs in the SRR1 orthologue of Brassica juncea were shown to be associated with flowering time also in this species30.
SRR1 is a pioneer protein with hitherto uncharacterized biochemical properties. To begin to understand the molecular underpinnings of its action in flowering time control we embarked on a second-site suppressor screen in Arabidopsis. A screen of an ethylmethanesulfonate (EMS) mutagenized srr1-1 population revealed several mutants that partially rescued the early flowering of srr1-1.
Here, we identify two of the suppressor of srr1-1 mutants (ssm), ssm15 and ssm67, which suppress srr1-1 early flowering independent of temperature and day-length. Full genome resequencing combined with a classical mapping approach identified the causal mutation in ssm15 as a G to A single nucleotide polymorphism (SNP) in exon 6 of ARABIDOPSIS SUMO PROTEASE 1 (ASP1), a protein already known to regulate FLC protein stability31. Ssm67 revealed a C to T transition in the At5g10460 gene encoding a Haloacid dehalogenase (HAD) superfamily protein. HADs represent an evolutionary ancient group of enzymes with diverse functions32 but have not been implicated in flowering time regulation so far. Besides the name-giving haloacid dehalogenases that cleave carbon-halogen bonds and are mostly present in prokaryotes, the superfamily comprises ATPases, phosphoesterases, phosphonatases and sugar phosphomutases acting on a wide variety of substrates. Their core catalytic domain consists of a Rossmannoid-fold, a distinct structure of alternating β-strands and α-helices which encompasses the active sites characterized by four diagnostic motifs. Motif I contains two Asp residues (DxD), catalysing a nucleophilic attack on the substrate. Motif II and III contain a conserved Thr, Ser or Lys residue and motif IV is comprised of the sequences DD, GDxxxD, or GDxxxxD. Many HADs also contain capping domains with a high degree of sequence variation that shield the catalytic site. Based on their size and their structural properties, they are defined as either C0, C1, or C2 caps. Together with the catalytic motifs, they determine the substrate specificity of the enzyme33. Although there are HADs that act on proteins as their substrates, the vast majority targets small molecules whose identities cannot be deduced from the sequence of the catalytic sites but have to be determined experimentally32,33,34. In Arabidopsis, about 200 genes encoding HAD enzymes were identified, but to date only few have been characterized35,36,37. Our investigation revealed that the mutation in the HAD responsible for the ssm67 phenotype acts to reduce levels of the floral repressor FLC. This is the first time that HADs could be assigned a role in flowering time control.
Results
A second site suppressor screen of the srr1-1 mutant
Arabidopsis thaliana SRR1 delays the transition to flowering by indirectly repressing the expression of the floral integrator FT under noninductive growth conditions27. In a search for novel genes important for the regulation of flowering by SRR1, a second site suppressor screen of the srr1-1 loss-of-function mutant was performed. The srr1-1 mutant flowers particularly early in SDs. As loss of SRR1 and PhyB additively accelerate flowering time at ambient temperatures, the screen was performed at 16 °C, where PhyB has no influence on flowering time25. Srr1-1 seeds were EMS-mutagenized and for M2 plants the number of leaves at bolting was recorded.
In the initial screen, we identified several ssm candidates which bolted significantly later than srr1-1 and produced viable seeds. Here, we focus on candidate suppressors with a flowering phenotype intermediate between srr1-1 and Col-7 wt, defined by a significant difference from srr1-1 as well as from wt (Fig. 1a). As the initial screen was performed at 16 °C, the five ssm mutants ssm5, ssm15, ssm67, ssm209, and ssm242 were rescreened at 20 °C in SDs to determine whether the flowering phenotype was temperature-dependent. The mutants again partially rescued early flowering of the srr1-1 mutant, similarly as at 16 °C. The only exception was ssm209 which did not flower significantly later than srr1-1 and therefore showed a temperature-dependent suppression of the srr1-1 flowering phenotype (Fig. 1b).

Flowering time of suppressor of srr1-1 (ssm) candidate mutants. Candidate ssm mutants with an intermediary phenotype between Col-7 wt and srr1-1 selected in an initial screen were rescreened in SDs at 16 °C (a), in SDs at 20 °C (b), and LDs at 20 °C (c). The number of leaves was counted for 10–12 plants each. Data are means ± s.d. To determine the statistical significance, an ANOVA followed by a post-hoc Dunnett-test was performed (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n.s. not significant).
Similarly, to account for any photoperiodic effects, flowering time of the ssm mutants was also tested in 20 °C LD conditions. All plant lines flowered earlier than in SDs, which shows that recognition of day length via the photoperiodic pathway was not compromised. The five ssm mutants still were flowering intermediate between srr1-1 and wt, therefore partially rescuing the early flowering of srr1-1 independently of day length (Fig. 1c).
Here, we focus on the candidate ssm67 which reproducibly produced the most robust phenotypes and flowered with a significant difference from srr1-1 as well as from wt under all conditions tested (Fig. 1a–c). Additionally, as ssm15 showed a strong intermediate phenotype especially at 16 °C SDs (Fig. 1a), we also identified the causal mutation of this mutant. Ssm5 also showed robust phenotypes under all tested conditions, but the plants were extremely difficult to propagate and produced little viable seeds, therefore we had to exclude this mutant from further analysis.
Identification of the ssm15 mutation
To identify the causal single nucleotide polymorphism (SNP) of the ssm15 mutant, a bulk segregant analysis using full genome sequencing was performed, as outlined in Suppl. Fig. S138. Therefore, ssm15 was back-crossed to srr1-1. F1 plants were selfed and the resulting segregating BC1F2 population was grown and scored for flowering time in SDs at 16 °C. Plants that displayed the suppressor phenotype were identified and material from each of these plants was pooled. The resulting DNA pool was submitted to full genome re-sequencing, with a coverage of ca. 60 ×. A pool of srr1-1 plants was sequenced as a reference. The sequencing revealed 5845 SNPs different between the TAIR10 reference genome and the srr1-1 or ssm15 mutants, respectively. Of these, 771 SNPs were unique between srr1-1 and ssm15. A SNP index was calculated to score the identified SNPs, leading to a number of candidate genes which were, however, distributed on chromosomes I, III, and IV (Suppl. Fig. S2a).
To asses which SNP was associated with the suppressor phenotype, a mapping population was created by crossing ssm15 with the Landsberg erecta wt. Mapping with SSLP and dCAPS markers (Suppl. Table S1) revealed the lowest recombination frequency with the marker F21M12 on the upper arm of chromosome I (Suppl. Fig. S2b). For candidates on chromosome I in the full genome re-sequencing, we ultimately found a G to A transition in exon 6 of ARABIDOPSIS SUMO PROTEASE 1 (ASP1), leading to an Ala to Thr exchange (Suppl. Fig. S2c). ASP1 was recently shown to positively regulate the transition to flowering at least partly by repressing FLC protein stability31, confirming a role in flowering time control also revealed here in our suppressor screen. As ASP1 has already been characterized with respect to flowering, we decided to focus on ssm67.
Identification of the ssm67 mutation
To identify the causal SNP of the ssm67 mutant, a bulk segregant analysis using full genome sequencing was performed, as outlined before (Suppl. Fig. S1). F1 plants of the backcross of ssm67 to srr1-1 were selfed and the resulting segregating BC1F2 population was scored for flowering time in SDs at 16 °C. Sequencing of pooled plants with the suppressor phenotype revealed 5500 SNPs different between the TAIR10 reference genome and the srr1-1 or ssm67 mutants, respectively, with 739 SNPs unique between srr1-1 and ssm67. Calculation of the SNP index revealed a number of candidate loci on each of the chromosomes II, III, and V (Fig. 2a).

Identification of the causal mutation in ssm67. (a) Distribution of all identified SNPs in the ssm67 genome and their SNP index compared to the srr1-1 genome. A ΔSNP index closer to − 1.0 indicates SNPs unique for ssm67. The red dots denote SNPs ≤ − 0.7, all SNPs below this value were evaluated as potential causal SNPs. (b) Linkage mapping of the causal mutation in a ssm67 × L. erecta mapping population using SSLP and CAPS markers. (c) Scheme of the At5g10460 Haloacid dehalogenase-like hydrolase (HAD). The position of the Ala to Val exchange is indicated. The consensus sequences of the HAD signature motifs I-IV are given below the boxes, the sequences of HDF1 are given above the boxes. (d) Flowering time of the segregating BC1F2 ssm67 population. Leaf number was scored in LDs for 10–12 plants each. −/− homozygous wt, +/− heterozygous for causal mutation, +/+ homozygous for causal mutation. Data are means ± s.d. A t test was employed to determine statistical significance (***P ≤ 0.001, n.s. not significant). (e) Complementation of the ssm67 late flowering phenotype by genomic HDF1. The leaf number of two independent lines homozygous for the genomic HDF1 construct in the ssm67 background, Col-7 wt plants, and the srr1-1 and ssm67 mutants was determined in SDs. For all lines, 15–20 plants were counted. Data are means ± s.d. A t test was employed to determine statistical significance. (*P ≤ 0.05, n.s. not significant).
To map the ssm67 locus, a linkage mapping population was created by crossing the ssm67 mutant with L. erecta and screened for plants with the late-flowering suppressor phenotype. A rough mapping localized the ssm67 locus to the top of chromosome V (Fig. 2b). This was confirmed by fine-scale mapping with the markers F15M7 and T31P16.
In the corresponding region on the top of chromosome V, three SNP candidates were located that caused nonsynonymous amino acid changes (Suppl. Table S2). To distinguish between these mutant candidates, dCAPS markers for the SNP in each candidate gene were designed to genotype individuals from a srr1-1 × ssm67 BC1F2 population with a stable ssm67 phenotype (Suppl. Table S1). Only one of the SNPs was present in all lines with a suppressor phenotype, namely a C to T transition in position 683 of the At5g10460 locus. This mutation was further confirmed by Sanger sequencing. The gene consists of seven exons and encodes a HAD superfamily protein of unknown function with a length of 306 amino acids. The mutation leads to an Alanine to Valine change in amino acid 228 (Fig. 2c).
Genotyping of the segregating BC1F2 ssm67 population scored for its flowering phenotype revealed that plants either heterozygous or homozygous for the causal mutation flowered significantly later than srr1-1, confirming a dominant segregation of the ssm67 mutation and explaining the SNP distribution pattern observed in the sequencing analysis (Fig. 2d).
To further test whether At5g10460 is the causal gene for the ssm67 phenotype, a construct consisting of ca. 1.5 kb of the endogenous promoter and the genomic sequence of At5g10460 was introduced into ssm67 using Agrobacterium-mediated transformation. The construct was able to revert the intermediate flowering phenotype of ssm67 back to the early flowering of srr1-1, indicating that At5g10460 indeed is responsible for the ssm67 late flowering phenotype (Fig. 2e). Based on the flowering phenotype and the overall appearance of the plants, we named this protein HAD-FAMILY REGULATOR OF DEVELOPMENT AND FLOWERING 1 (HDF1) and the identified allele hdf1-1. HADs represent an evolutionary ancient group of enzymes with diverse functions32. The active sites are characterized by four diagnostic motifs. Motif I contains two Asp residues (DxD), catalysing a nucleophilic attack on the substrate. Motif II and III contain a conserved Thr, Ser or Lys residue and motif IV is comprised of the sequences DD, GDxxxD, or GDxxxxD.
We performed a whole sequence alignment of 93 HAD superfamily proteins from a dataset compiled of previously identified HADs35,37 and our own search in the Araport 11 database. We found all four diagnostic motifs to be well conserved. However, HDF1 contains only the first of the two conserved Asp in motif I which in HAD phosphatases serves as a nucleophile attacking the phosphoryl group of the substrate. The second Asp is positioned two residues C-terminal of the first Asp and protonates the leaving group in the first part of the reaction and deprotonates a water molecule in the second part of the reaction34. In this position, HDF1 instead contains a Tyr. The absence of the second Asp in HDF1 raises the possibility that is does not act as a phosphatase. Instead, the presence of a Tyr in the Asp + 2 position is characteristic for HADs with dehalogenase function in bacteria33. Interestingly, we found six candidates in Arabidopsis also missing the Asp in the + 2 position, but none of them displays a Tyr.
Based on the alignment, we constructed a phylogenetic tree of all 93 HADs we previously identified (Suppl. Fig. S3a). From this tree, we further investigated the branch with the closest neighboring HDF1 paralogs (Fig. 3). Due to the lack of other proteins with a Tyr in the + 2 position, HDF1 groups together with the uncharacterized HADs At1g14310 displaying a Gly in the + 2 position of motif I and At2g41250 displaying a Val residue in the + 2 position. In close vicinity we find the CoA phosphohydrolase NUDX1139 and the thiamin monophosphate phosphatase TH240 (Suppl. Fig. S3b). Although HDF1 was the only HAD family protein with a Tyr in the + 2 position that we have found in the Col reference genome, it seems to be well conserved in other plant species, especially the Brassicaceae, with one copy in Arabidopsis lyrata, three copies in Camelina sativa, one copy each in Brassica rapa and Brassica oleracea, and two copies in Brassica napus (Suppl. Fig. S4). This points towards a very distinctive role of HDF1, as it apparently emerged early in the evolution of the Brassicaceae but has retained a very high degree of sequence conservation.

Phylogenetic analysis of HDF1. Phylogenetic tree of HAD superfamily proteins from Arabidopsis neighboring HDF1. Alignment of the predicted sequences was made using MAFFT version 7. The phylogenetic tree was constructed using the PHYML 3.0 web server. The complete tree can be seen in Suppl. Fig. S3.
HADs are divided into several subclasses, based on the location of a so-called “capping domain”41,42. HDF1 belongs to the Class 2A, where the capping domain is found between the second and third motifs. The presence of the larger C2 cap shielding the active site suggests that the enzyme acts on a small molecule rather than on a protein. So far, the substrate remains elusive. HDF1 was identified in a search for sequences related to AtFHy1, a flavin mononucleotide (FMN) hydrolase involved in turnover of FMN that we also find distantly related to HDF135. Subsequently, it was shown that recombinant AtFHy1 in vitro dephosphorylates 5-amino-6-ribitylamino-2,4(1H,3H) pyrimidinedione 5′-phosphate (ARPP), an intermediate of the riboflavin biosynthetic pathway, while recombinant HDF1 does not, excluding a role in riboflavin biosynthesis43.
Impact of the ssm67 mutant on development
Many mutants in genes regulating flowering time display additional pleiotropic phenotypes that affect development during the vegetative growth stages44. We previously found phenotypical differences between srr1-1 and wt that we attributed mainly to a misregulation of PhyB-mediated red light signaling, showing that the function of SRR1 was not limited to control flowering time26. Therefore, we assayed how vegetative development was affected in srr1-1 and ssm67.
A measure of plant development is the rate of emergence of new leaves. To examine whether the higher number of leaves at bolting in ssm67 compared to srr1-1 (Fig. 1) is due to changes in the leaf initiation rate, leaf numbers were counted every third day on plants grown in SDs. This revealed that srr1-1 itself has a slower leaf initiation than wt, contributing to its lower leaf number at flowering (Fig. 4a). Thus, SRR1 in addition to its role in flowering likely has a role in vegetative development. The ssm67 mutant had a statistically significant (p < 0.05) increase in leaf initiation rate compared to srr1-1, therefore partially rescuing also this phenotype of srr1-1 (Fig. 4a). The average number of days to flowering was not significantly different between srr1-1 and ssm67, but ssm67 produced more leaves in the same time frame than srr1-1 (Fig. 4b). Thus, HDF1 may act in the same molecular pathway as SRR1 to regulate leaf initiation.

The ssm67 mutation affects plant development. (a) Leaf initiation rate of ssm67 compared to Col-7 wt and srr1-1. (b) Flowering time of ssm67 compared to Col-7 wt and srr1-1 in SDs, measured in days to bolting (left) and leaves to bolting (right) from n = 15–20 plants. Data are means ± s.d. To determine statistical significance, a Kruskal–Wallis test was performed (*P ≤ 0.05, ***P ≤ 0.001, n.s. not significant). (c) Rosette size of ssm67 compared to Col-7 wt and srr1-1 in SDs. (d,e) Trichome distribution. Col-7 wt, srr1-1, and ssm67 mutant plants were grown in SDs at 20 °C. The leaf with the first abaxial trichome (d) and the number of days until first abaxial trichome (e) were observed (n = 15–20 plants, data are means ± s.d). To determine statistical significance, a Kruskal–Wallis test was performed (n.s. not significant).
To quantify general growth, rosette size was tracked in SD-grown plants from 30 days after germination until plants started to flower. This revealed that srr1-1 plants themselves have a smaller rosette compared to wt, but also that the hdf1-1 mutation in ssm67 leads to a further decrease in size (Fig. 4c). This points towards an SRR1 independent effect of HDF1 on general plant growth, in addition to the changed leaf initiation rate.
To determine whether in addition to the adult-to-reproductive transition the juvenile-to-adult developmental phase shift is altered, abaxial trichome development was monitored in plants growing in SDs. The number of leaves at appearance of the first abaxial trichome was recorded as well as the days until emergence of the first trichome. This showed that there was no difference in juvenile-to-adult shift between any of the genotypes (Fig. 4d). Both ssm67 and srr1-1 reached the adult phase a little later than wt, but the differences were not significant (Fig. 4e). This possibly reflects the slower leaf initiation rate in these genotypes compared to the wt. Thus, ssm67 does not affect the juvenile-to-adult phase transition.
Hdf1-1 single mutants flower late
To isolate the novel hdf1-1 allele from the ssm mutant background, ssm67 was crossed to the Col-7 wt. Subsequently, plants without the srr1-1 mutation were selected based on the loss of the Basta resistance of the srr1-1 T-DNA26 and confirmed by PCR to carry the causal hdf1-1 SNP.
The single hdf1-1 mutant plants were further characterized to examine whether the ssm67 flowering time phenotype was dependent on SRR1. Hdf1-1 flowers later than wt plants in LDs (Fig. 5a) and particularly in SDs (Fig. 5b). The delay of flowering in ssm67 relative to srr1-1 is similar to the delay of flowering in hdf1-1 relative to Col-7, suggesting that the hdf1-1 flowering phenotype is mostly independent of SRR1.

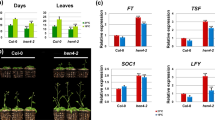
The role of HDF1 in flowering time control. Col-7 wt plants, srr1-1, ssm67 and hdf1-1 single mutants selected from a backcross of ssm67 with Col-7 were grown in LDs (a) and SDs (b). The number of leaves was counted when the bolt was 0.5 cm in height (data are means ± s.d from n = 20–25 plants). A t test was employed to determine statistical significance (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). (c) Expression levels of HDF1. Col-7 wt, srr1-1, ssm67 and hdf1-1 single mutants were grown in SDs and harvested at 4-h intervals over the course of on day. HDF1 transcript levels were monitored by RT-qPCR and normalized to PP2A. Data are means ± s.d. of three biological replicates. (d) Flowering time in response to GA. Plants were grown in SDs and sprayed with 100 µM GA3 or 0.1% DMSO as control (data are means ± s.d from n = 20–25 plants). A t test was employed to determine statistical significance (***P ≤ 0.001, n.s. not significant). (e) Expression levels of FLC, SOC1 and FT. Plants were grown in SDs for three weeks and harvested at zt4. Transcript levels were monitored by RT-qPCR and normalized to PP2A. Data are means ± s.d. of three biological replicates. (f) Proposed working model for the regulation of flowering time and vegetative development through HDF1 and SRR1. Flowering is inhibited by FLC while vegetative development could be influenced indirectly by additional unknown factors.
The HDF1 expression profile was tested in a diurnal SD cycle in wt, srr1-1, ssm67, and hdf1-1 background. This revealed that expression of HDF1 is somewhat elevated during the dark period (Fig. 5c). There was no difference in expression between srr1-1 and wt, but expression of HDF1 was reduced in both ssm67 and hdf1-1. This suggests a possible feedback regulation of the HDF1 protein on its own expression. Furthermore, HDF1 is strongly expressed in all plant tissues according to the Arabidopsis eFP browser (Suppl. Fig. S5). This suggests that its role is not limited to floral transition.
Impact of HDF1 on the flowering time network
In the initial screen, ssm67 flowered much later in SDs than in LDs (Fig. 1c). Similarly, flowering of the hdf1-1 single mutant was delayed in SDs compared to LDs (Fig. 5a,b), indicating that HDF1 is not involved in sensing day length. Therefore, we tested the effect of hdf1-1 on flowering pathways other than the photoperiodic pathway.
To determine whether the flowering phenotype of hdf1-1 is dependent on GA, ssm67 and hdf1-1 plants were tested for flowering in response to exogenous GA application. Plants grown in SDs were treated with GA every three days from 2 weeks after sowing by spraying 100 µM of GA3 or 0.1% DMSO as control. All plant lines reacted to the GA treatment with early flowering. The ratio of leaves with and without GA was 0.57 for wt and 0.65 for srr1-1 indicating that either srr1-1 reacts weaker to GA or that leaf numbers could not be reduced below a certain threshold with the applied amount of GA. Ssm67 flowered with the same number of leaves as srr1-1 after GA application and a ratio of 0.59, likely due to the higher initial leaf number in untreated plants. The hdf1-1 mutant still flowered with significantly more leaves than wt after GA treatment and the ratio was 0.56, indicating that hdf1-1 responds to GA in a similar fashion than wt (Fig. 5d). We concluded that HDF1 is not involved in GA dependent flowering, but given the weaker reaction of srr1-1, SRR1 might play a role in this regulatory pathway.
To examine whether the floral repressor FLC plays a role in the delayed flowering of hdf1-1 and ssm67 compared to srr1-1 and wt, FLC transcript levels were measured. Whereas FLC levels are reduced in srr1-1, we found a three-fold increase of FLC in hdf1-1 compared to Col-7 which could explain the delay in flowering. Interestingly, despite the strong effect in hdf1-1, the FLC level in ssm67 was only reverted back to wt suggesting the possibility that the effect of HDF1 on FLC is at least partly dependent on SRR1 (Fig. 5e). Although FLC is reverted back to wt level in ssm67, the plants still flower with less leaves than wt, which can be explained by the slower leaf initiation rate compared to Col-7 (Fig. 4a). In line with the observed FLC levels, we also measured a lower level of the floral activator SOC1 in hdf1-1 and a somewhat elevated level in srr1-1 compared to wt. The expression level of FT was not changed in hdf1-1, but was elevated in srr1-1 and ssm67, although with some variation (Fig. 5e). This could possibly be explained by the overall low expression of floral activators under SD conditions caused by a lack of photoperiodic induction.
We noticed that HDF1 is located in close proximity to the FLC locus on chromosome five and both genes likely co-segregate. As L.er was used as a parent to create the mapping population from which hdf1-1 was isolated and Col and L.er differ in the allelic state of FLC45 we checked whether the FLC allele from Col or L.er was present in ssm67 and hdf1-1. Both lines proved homozygous for the Col FLC allele, thus lacking the characteristic 1.2 kb insertion indicative of the weak L.er allele (Suppl. Fig. S6a,b). Therefore, we could rule out differences in the allelic state of FLC as a cause for the observed differences in FLC abundance. Taken together, our data suggest that FLC could be a major cause for the reverted flowering phenotype of ssm67. This leads us to a model where SRR1 impacts flowering time via HDF1 through the regulation of FLC and additionally affects plant development during the vegetative phase also through HDF1, but possibly also involving other factors. Whether there is a reciprocal interaction of SRR1 and HDF1 has yet to be determined (Fig. 5f).
Discussion
In a search for novel genes important for the regulation of flowering by SRR1, we embarked on a second-site suppressor screen for mutants that rescue the srr1-1 early-flowering phenotype under noninductive SD conditions. As the loss of SRR1 and PhyB additively accelerate flowering time at higher temperatures, the screen was performed at 16 °C, where PhyB has no influence on flowering time25. Candidate mutants were then rescreened at 20 °C. This screening scheme allowed for identification of flowering suppressors in an early-flowering background reducing the risk of overseeing mutant candidates whose increase in leaf number would be difficult to quantify in wt background due to leaf senescence of earlier developed leaves. The screen identified a range of mutant candidates with a flowering phenotype intermediate between srr1-1 and Col-7 (Fig. 1a).
To identify the position of the causal mutation of ssm67, a combination of linkage mapping and full genome sequencing was used. The dominant segregation pattern of the mutation in ssm67 hindered the enrichment of homozygous candidates for the bulk-segregant analysis, leading to a distribution of potential SNP candidates in the sequencing data on three chromosomes, with no candidate SNP reaching a ΔSNP index over − 0.8 (Fig. 2a). The linkage mapping, however, revealed that a region on the top of chromosome V harbors the causative SNP (Fig. 2b), narrowing down the number of candidate loci to three (Suppl. Table S2). The SNP in position 683 of HDF1 could then be confirmed as the causal mutation by genotyping an independent segregation BC1F2 population, revealing that its presence was necessary for the late flowering phenotype (Fig. 2d). The complementation of ssm67 with the genomic copy of the HDF1 gene restored the srr1-1 flowering phenotype, further confirming that the mutation in HDF1 is responsible for the delay in flowering (Fig. 2e). Our results show the strength of combining a classical linkage mapping and state-of-the-art full genome resequencing when the mutated population does not display a purely Mendelian segregation pattern. In this way, we were able to identify a novel flowering time regulator from a dominantly segregating population, which masked its position in the sequencing data.
The novel flowering time regulator we identified turned out to be a HAD superfamily protein. These proteins are characterized by a core catalytic domain that is known as a Rossmannoid fold and is comprised of four diagnostic motifs33. The mutant allele we identified in our screen affects none of these motifs directly, but is instead located in the intervening region between motif III and IV (Fig. 3c). The Ala to Val exchange is considered a conservative replacement, as both are aliphatic non-polar amino acids. This raises the possibility that the observed effects in the ssm67 and hdf1-1 plants are rather mild. In the future, the CRISPR/Cas technology can be used to engineer a complete loss-of-function allele both in the wt background and the srr1-1 background. This may provide more insights into the degree of genetic interaction between HDF1 and SRR1 in flowering time control and development.
The HAD superfamily is by far dominated by ATPases and acid-phosphatases46, but information on substrates cannot be gained from sequence comparison alone but instead has to be determined experimentally. So far, there is only minor evidence on what as possible substrate could be. The presence of a large C2 cap between the conserved motifs II and III, restricting access to the active site, points to HDF1 acting on a small molecular rather than on a larger substrate. HDF1 was found previously to be related to AtFHy1 encoding an FMN hydrolase. AtFHy1 indeed was shown to dephosphorylate the riboflavin biosynthesis pathway intermediate 5-amino-6-ribitylamino-2,4(1H,3H) pyrimidinedione 5′-phosphate (ARPP) in vitro whereas HDF1 was not active on this substrate43.
The close relationship to HAD type phosphatases may suggest that HDF1 also acts as a phosphatase. The absence of the diagnostic second Asp in motif I, however, could also suggest that HDF1 may act as a haloacid dehalogenase, as found in bacteria. Additionally, the possibility that HDF1 acts on multiple substrates cannot be ruled out, as HADs with a C2 cap were found to be less substrate specific than HADs with a C1 or a C0 cap47.
It is likely that HDF1 plays a more general role in plant metabolism than solely being limited to floral transition. This may be reflected by the developmental phenotypes we found in ssm67. The rosette size of ssm67 mutants was strongly reduced and the leaf emergence rate was accelerated in ssm67 compared to srr1-1 (Fig. 4c–e). The widespread expression throughout development (Arabidopsis eFP browser) as well as the high level of conservation in the Brassicaceae also point in this direction (Suppl. Fig. S4). This is further strengthened by a recent study that identified 41 HAD genes in rice and 40 HAD genes in Arabidopsis. 17 of the HADs from rice were induced upon phosphate starvation and thus were predicted to be involved in phosphate recycling under Pi stress conditions37. As the search was limited to HADs that show a conserved DxD pattern in motif I, HDF1 was omitted from the search.
HDF1 expression shows a weak diurnal fluctuation, with an elevated level in the dark period (Fig. 5c). Notably, HDF1 expression was reduced in both ssm67 and hdf1-1, pointing to a positive feedback regulation that might be disturbed by the Ala to Val exchange. As such, it is likely that the mutation we identified creates a hypomorphic allele. HDF1 expression was previously found to be reduced in phyA and phyB mutants, placing red light as a putative upstream regulator of HDF148. However, as HDF1 expression was not strongly altered in srr1-1, the effect of PhyB may be independent of signaling through SRR1.
To define how HDF1 acts in the network of flowering time regulators, we tested the involvement in several known pathways. Ssm67 and hdf1-1 were late flowering in SDs as well as in LDs, indicating that the photoperiodic response is intact and that HDF1 is not involved in sensing day length (Fig. 5a,b). Furthermore, ssm67 flowered with more leaves than srr1-1 both at 16 °C and 20 °C, suggesting that HDF1 does not affect the temperature response (Fig. 1a,b).
When plants grown in SDs were treated with GA, the transition to flowering was accelerated in hdf1-1 and ssm67 similarly as in wt, ruling out also this prominent pathway of flowering time regulation. Notably, the response of srr1-1 to GA was somewhat reduced. This may indicate that either early flowering could not be accelerated further, or that srr1-1 reacts weaker to GA (Fig. 5d).
Ultimately, we monitored FLC levels to address a role in the autonomous regulation of flowering time. We found FLC to be strongly increased in hdf1-1. Interestingly, despite the strong effect in hdf1-1, the FLC level in ssm67 was only reverted back to wt. (Fig. 5e). The FLC allele from the Landsberg ecotype carries a 1.2 kb insertion of a Mutator-like transposon in intron 1 causing low FLC steady-state abundance45. To rule out that the differences in FLC levels of hdf1-1 and ssm67 are a consequence of segregating FLC alleles either from the Landsberg background used to generate the mapping population or the Col background used for backcrossing, we genotyped the FLC allele. We found that in srr1-1, hdf1-1 and ssm67 the FLC allele from Col was present in a homozygous state (Suppl. Fig. S6). This suggests the possibility that functional SRR1 is needed for proper HDF1 function on FLC and that the differences in FLC expression between ssm67 and hdf1-1 are not caused by FLC alleles from different genetic backgrounds. But, as hdf1-1 is late-flowering compared to Col-7 wt in both LD and SD conditions, it is obvious that besides this partial dependency on SRR1, HDF1 influences flowering also independently of SRR1. FLC is a strong repressor of flowering, but its roles are not limited to flowering time pathways. Instead, among its numerous targets are many other transcripts implicated in the regulation of developmental phase transition, i.e. hormonal response genes, floral meristem identity genes, or the transcription factor SPL15 involved in the juvenile to adult phase transition49,50. This could mean that changes in the FLC level might be responsible for other developmental phenotypes of srr1-1.
To our knowledge, this is the first time that a HAD superfamily protein has been implicated in flowering time control in Arabidopsis. Previously, a protein phosphatase from tomato, LePS2;1, has been shown to be induced upon phosphate starvation, and overexpression in transgenic plants led to increased anthocyanin accumulation, changes in morphology and delayed flowering51. LePS2;1 dephosphorylates synthetic phosphor-Serine/Threonine peptides in vitro. It differs from HDF1 in that overexpression rather than loss-of-function delays flowering.
In addition to the ssm67 mutation, we characterized the ssm15 mutation which also flowers intermediate between srr1-1 and wt. Full genome sequencing led to the identification of a G to A transition in exon 6 of ARABIDOPSIS SUMO PROTEASE 1 (ASP1), leading to an Ala to Thr exchange (Suppl. Fig. S2c). ASP1 was recently shown to positively regulate the transition to flowering at least partly by repressing FLC protein stability31. FLC displayed wt levels in ssm15 (data not shown), The fact that we identified two regulators of FLC with very distinct functions may be a hint that FLC could be a determinant of SRR1 mediated flowering and further emphasizes the role of this repressor as a major hub of flowering time control in general.
In conclusion, our large scale genetic screen identified candidates that suppress the early flowering phenotype of srr1-1. On closer inspection, we could not only find flowering related phenotypes, but were able to describe a potential role of SRR1 in vegetative development for the first time. The leaf emergence rate in srr1-1 was significantly lower than in wt and this was in large parts rescued in the ssm67 mutant. This indicates that the developmental phenotype is at least partly dependent of HDF1. In contrast, ssm67 partly rescued the early flowering phenotype of srr1-1, but hdf1-1 also delayed flowering in wt. This might indicate that the effect on flowering time is regulated independently of SRR1. FLC was identified as a major conversion point in the suppressor candidates ssm15 and ssm67, suggesting that ASP1 and HDF1 bypass the requirement for SRR1 to downregulate floral repressors in inductive conditions by the strong repression of FLC.
Materials and methods
Plant material, growth conditions and suppressor screen
All Arabidopsis plant lines used in this study are derived from common laboratory strains. No seeds or plant material was collected from the wild and all methods involving plants were carried out according to institutional, national, and international guidelines and legislation.
The T-DNA mutant srr1-1 in the Col-7 background has been described26,27. All seeds were stratified for 3 days at 4 °C before grown on soil. Seeds were surface sterilized and stratified for 3 days at 4 °C before plating on agar-solidified half-strength MS (Murashige and Skoog) medium (Duchefa) supplemented with 0.5% sucrose and 0.5 g MES. Plants were grown in AR66-L3 Percival incubators (CLF Laboratories) in 100 µmol m−2 s−1 light intensity, with the light–dark and temperature conditions as indicated.
For the second-site suppressor screen, ca 20.000 srr1-1 seeds were mutagenized overnight in sodium phosphate buffer (pH 7.5)52. Subsequently, 0.3% ethylmethanesulfonate (Sigma Aldrich) was added and the mixture was incubated for 15 h with rotation. Seeds were then washed 20 × with water and distributed on soil in 280 batches. After 3 days of stratification at 4 °C, the seeds were transferred to LD growth conditions. M1 seeds were harvested in batches and 100 M2 seeds each from 11 randomly selected batches were used for the initial screen.
Flowering time analysis
Seeds were germinated as described above and grown on soil in a random fashion. Flowering time was determined by counting the rosette leaves when the bolt was > 0.5 cm tall53. For GA treatment, plants growing on soil were sprayed with 100 µM GA3 four to six h after lights on twice a week starting at day 14 after stratification. Mock treatment was performed by spraying with 0.1% DMSO/0.02% Tween 20.
Determination of leaf initiation rate and rosette size
Leaf initiation rate was scored by counting the number of rosette leaves every 3 days starting 10 days after sowing and plotting the number of leaves against days of growth. Counting was done until the plant started to flower.
Rosette size of the plants was monitored by taking pictures at the indicated time points starting 28 days after sowing. The area covered by the rosette was determined using the software Rosette tracker54.
Abaxial trichome analysis
As a measure of the juvenile-to-adult developmental transition, plants were scored for the presence of abaxial trichomes every 3 days starting 10 days after sowing. When an abaxial trichome was identified, the leaf number was notified and the plant was considered as adult and no longer scored.
DNA extraction for sequencing
Leaf material was sampled from 35 individuals in segregating ssm67 × srr1-1 BC1F2 and ssm15 × srr1-1 BC1F2 population consisting of 115 plants each. Plants with at least 25% more leaves at bolting compared to srr1-1 control plants were selected, the material was frozen in liquid N2 and ground to a fine powder. Equal amounts of each plant sample were pooled and this pool was extracted using a DNeasy Plant Maxi Kit (Qiagen) according to the manufacturer’s instructions, with the following exceptions: 2 × the amount of lysis buffer was used, all centrifugation steps were done for 10 min, and the empty column was centrifuged for 15 min and heated at 37 °C for 10 min before elution to avoid ethanol contamination.
Sequencing
Four µg of DNA from the ssm15 and ssm67 sequencing populations was sent to Novogene (Hong Kong) for sequencing and mapping to the reference genome. After quality control, libraries were constructed using the Illumina TruSeq Library Construction Kit. Pair-end sequencing were performed on Illumina® HiSeq platform, with the read length of PE150 bp at each end. Base calling was done with the CASAVA software. Mapping was performed by Novogene using the BWA, SAMtools and Picard softwares, followed by SNP/Indel detection (SAMtools) and variation annotation (ANNOVAR). The same process was used to sequence a pool of srr1-1 plants. SNPs were determined relative to the TAIR10 reference as of May 23rd, 2017.
To create a SNP candidate index and avoid false detection of polymorphisms, all SNPs also present in srr1-1 were filtered out, as well as low-quality and multiple-hit reads55. The SNP index was calculated by comparing the total number of reads to the number of reads for a non-reference base and plotted according to position on the chromosome.
Linkage mapping
To narrow down the number of candidates, a linkage mapping population was created by crossing the ssm67 mutant with the Landsberg erecta wt. Rough mapping was performed in the resulting segregating population by testing two markers per chromosome using PCR56,57. The region where candidate loci from the whole-genome sequencing approach were located were examined in more detail using additional markers. PCR was used to amplify nucleotide sequences from simple sequence length polymorphisms (SSLP) or cleaved amplified polymorphic sequences (CAPS) markers. In the case of the CAPS markers, the PCR product was digested with restriction enzymes according to the manufacturer’s instructions. The products were subsequently separated on an agarose gel and analyzed.
dCAPS primer design
To determine the presence of SNPs in the mutagenized plants, primers were designed to amplify the genomic region surrounding the SNP of interest. This was followed by digestion of the PCR product by restriction enzymes that only digested either the mutated product or the wt product, according to the Derived Cleaved Amplified Polymorphic Sequences (dCAPS) method. Primers were designed using the dCAPS finder software, 1–2 mismatches were added to the primer to create specific restriction sites, allowing digestion of only mutant or only wt sequence58. The sequence of the primers are listed in Suppl. Table S1.
Complementation of the ssm67 mutant
1.5 kb of the promoter and the genomic sequence of At5g10460 was amplified with primers adding BamHI restriction sites and cloned into the pJET1.2 cloning vector (Thermo Scientific). The fragment was cut and introduced into the binary vector pCAMBIA3300 using BamHI and transformed into Agrobacterium tumefaciens GV3101. Plants were transformed by floral dip59.
Transcript analysis
Total RNA was extracted from plant material using Universal RNA Purification Kit (Roboklon) following the manufacturer’s instructions. For cDNA synthesis, 2 µg of total RNA was DNase-treated using RQ1 RNase-free DNase (Promega) and reverse transcribed using AMV Reverse Transcriptase (Roboklon) according to the manufacturer’s instructions. qPCR was performed with iTaq Sybr Green Supermix (Bio-Rad) according to manufacturer’s instructions. The normalized expression level was determined using the ΔCt method, with PP2a (At1g13320) as a reference gene as described60. To amplify FT in SD grown samples, a nested PCR was performed. The primer sequences can be found in Suppl. Table S1.
Construction of the phylogenetic tree
To construct the phylogenetic tree of HAD family proteins, a dataset was compiled comprised of 21 genes from (dataset 1)35, 55 genes from (dataset 2)37, and 58 proteins which were identified as haloacid dehalogenase by textmining Araport 11 genes (dataset 3). Overall, 93 unique genes from the three datasets were used for further analysis.
The protein sequences were downloaded via the thalemine web interface (https://bar.utoronto.ca/thalemine/begin.do (accessed August 2021)). These sequences were aligned using the MAFFT version 7 online Alignment server (https://mafft.cbrc.jp/alignment/server/) with the ‘G-INS-i’ parameter for sequences with global homology and the BLOSUM62 scoring matrix for amino acids61,62.
The resulting full-length sequence alignment was translated into the PHYLIP format and uploaded to the PHYML 3.0 web server (http://www.atgc-montpellier.fr/phyml/). The detection of the optimal substitution model was set to automatic model selection SMS63 with the Akaike Information Criterion (AIC) parameter. The tree was constructed using the fast likelihood-based method ‘aLRT SH-like’. The SMS of the first tree determined ‘WAG + G + I + F’ to be the best fitting model. The resulting tree was visualized using the PHYML 3.0 online resource PRESTO (http://www.atgc-montpellier.fr/presto/).
For construction of the phylogenetic tree of the HDF1 orthologs in other Brassicaceae, protein sequences from selected organisms were obtained from the UniProt database (http://www.uniprot.org). The tree was constructed using the same tools as outlined above, with the exception that the automatic model selection SMS determined ‘JTT + G’ to be the best fitting model.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information file. Sequencing data generated for ssm15 are accessible at NCBI’s Sequence Read Archive (SRA) via accession number [SRR22318065]. Sequencing data generated for ssm67 are accessible at SRA via accession number [SRR22318066].
References
Jung, C., Pillen, K., Staiger, D., Coupland, G. & von Korff, M. Recent advances in flowering time control. Front. Plant Sci. 7, 2011. https://doi.org/10.3389/fpls.2016.02011 (2016).
Amasino, R. M. & Michaels, S. D. The timing of flowering. Plant Physiol. 154, 516–520. https://doi.org/10.1104/pp.110.161653 (2010).
Romera-Branchat, M., Andrés, F. & Coupland, G. Flowering responses to seasonal cues: What’s new?. Curr. Opin. Plant Biol. 21, 120–127. https://doi.org/10.1016/j.pbi.2014.07.006 (2014).
Johansson, M. & Staiger, D. Time to flower: Interplay between photoperiod and the circadian clock. J. Exp. Bot. 66, 719–730. https://doi.org/10.1093/jxb/eru441 (2015).
Suarez-Lopez, P. et al. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 410, 1116–1120 (2001).
Sawa, M., Nusinow, D. A., Kay, S. A. & Imaizumi, T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science 318, 261–265 (2007).
Corbesier, L. et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033 (2007).
Mathieu, J., Warthmann, N., Kuttner, F. & Schmid, M. Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Curr. Biol. 17, 1055–1060 (2007).
Jaeger, K. E. & Wigge, P. A. FT protein acts as a long-range signal in Arabidopsis. Curr. Biol. 17, 1050–1054 (2007).
Castillejo, C. & Pelaz, S. The balance between CONSTANS and TEMPRANILLO activities determines FT expression to trigger flowering. Curr. Biol. 18, 1338–1343 (2008).
Blazquez, M. A., Green, R., Nilsson, O., Sussman, M. R. & Weigel, D. Gibberellins promote flowering of arabidopsis by activating the LEAFY promoter. Plant Cell 10, 791–800 (1998).
Wilson, R. N., Heckman, J. W. & Somerville, C. R. Gibberellin is required for flowering in Arabidopsis thaliana under short days. Plant Physiol. 100, 403–408. https://doi.org/10.1104/pp.100.1.403 (1992).
Blazquez, M. A., Ahn, J. H. & Weigel, D. A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nat. Genet. 33, 168–171 (2003).
Moon, J., Lee, H., Kim, M. & Lee, I. Analysis of flowering pathway integrators in Arabidopsis. Plant Cell Physiol. 46, 292–299 (2005).
Porri, A., Torti, S., Romera-Branchat, M. & Coupland, G. Spatially distinct regulatory roles for gibberellins in the promotion of flowering of Arabidopsis under long photoperiods. Development 139, 2198–2209. https://doi.org/10.1242/dev.077164 (2012).
Bastow, R. et al. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427, 164–167 (2004).
Michaels, S. D. & Amasino, R. M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956 (1999).
Macknight, R. et al. FCA, a gene controlling flowering time in Arabidopsis, encodes a protein containing RNA-binding domains. Cell 89, 737–745 (1997).
Simpson, G. G., Dijkwel, P. P., Quesada, V., Henderson, I. & Dean, C. FY is an RNA 3′ end-processing factor that interacts with FCA to control the Arabidopsis floral transition. Cell 113, 777–787 (2003).
Steffen, A., Elgner, M. & Staiger, D. Regulation of flowering time by the RNA-binding proteins AtGRP7 and AtGRP8. Plant Cell Physiol. 60, 2040–2050. https://doi.org/10.1093/pcp/pcz124 (2019).
Lee, H. et al. The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev. 14, 2366–2376 (2000).
Whittaker, C. & Dean, C. The FLC locus: A platform for discoveries in epigenetics and adaptation. Annu. Rev. Cell Dev. Biol. 33, 555–575. https://doi.org/10.1146/annurev-cellbio-100616-060546 (2017).
Mulekar, J. J. & Huq, E. Does CK2 affect flowering time by modulating the autonomous pathway in Arabidopsis?. Plant Signal. Behav. 7, 292–294. https://doi.org/10.4161/psb.18883 (2012).
Heidari, B., Nemie-Feyissa, D., Kangasjärvi, S. & Lillo, C. Antagonistic regulation of flowering time through distinct regulatory subunits of protein phosphatase 2A. PLoS One 8, e67987. https://doi.org/10.1371/journal.pone.0067987 (2013).
Halliday, K. J., Salter, M. G., Thingnaes, E. & Whitelam, G. C. Phytochrome control of flowering is temperature sensitive and correlates with expression of the floral integrator FT. Plant J. 33, 875–885 (2003).
Staiger, D. et al. The Arabidopsis SRR1 gene mediates phyB signaling and is required for normal circadian clock function. Genes Dev. 17, 256–268. https://doi.org/10.1101/gad.244103 (2003).
Johansson, M. & Staiger, D. SRR1 is essential to repress flowering in non-inductive conditions in Arabidopsis thaliana. J. Exp. Bot. 65, 5811–5820. https://doi.org/10.1093/jxb/eru317 (2014).
Xiao, D. et al. The Brassica rapa FLC homologue FLC2 is a key regulator of flowering time, identified through transcriptional co-expression networks. J. Exp. Bot. 64, 4503–4516 (2013).
Schiessl, S., Williams, N., Specht, P., Staiger, D. & Johansson, M. Different copies of SENSITIVITY TO RED LIGHT REDUCED 1 show strong subfunctionalization in Brassica napus. BMC Plant Biol. 19, 372. https://doi.org/10.1186/s12870-019-1973-x (2019).
Kang, L. et al. Genomic insights into the origin, domestication and diversification of Brassica juncea. Nat. Genet. 53, 1392–1402. https://doi.org/10.1038/s41588-021-00922-y (2021).
Kong, X., Luo, X., Qu, G.-P., Liu, P. & Jin, J. B. Arabidopsis SUMO protease ASP1 positively regulates flowering time partially through regulating FLC stability. J. Integr. Plant Biol. 59, 15–29. https://doi.org/10.1111/jipb.12509 (2017).
Seifried, A., Schultz, J. & Gohla, A. Human HAD phosphatases: Structure, mechanism, and roles in health and disease. FEBS J. 280, 549–571. https://doi.org/10.1111/j.1742-4658.2012.08633.x (2013).
Burroughs, A. M., Allen, K. N., Dunaway-Mariano, D. & Aravind, L. Evolutionary genomics of the HAD superfamily: Understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034. https://doi.org/10.1016/j.jmb.2006.06.049 (2006).
Gohla, A. Do metabolic HAD phosphatases moonlight as protein phosphatases?. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1866, 153–166. https://doi.org/10.1016/j.bbamcr.2018.07.007 (2019).
Rawat, R., Sandoval, F. J., Wei, Z., Winkler, R. & Roje, S. An FMN hydrolase of the haloacid dehalogenase superfamily is active in plant chloroplasts. J. Biol. Chem. 286, 42091–42098. https://doi.org/10.1074/jbc.M111.260885 (2011).
Caparrós-Martín, J. A., McCarthy-Suárez, I. & Culiáñez-Macià, F. A. HAD hydrolase function unveiled by substrate screening: Enzymatic characterization of Arabidopsis thaliana subclass I phosphosugar phosphatase AtSgpp. Planta 237, 943–954. https://doi.org/10.1007/s00425-012-1809-5 (2013).
Du, Z., Deng, S., Wu, Z. & Wang, C. Genome-wide analysis of haloacid dehalogenase genes reveals their function in phosphate starvation responses in rice. PLoS One 16, e0245600. https://doi.org/10.1371/journal.pone.0245600 (2021).
Schneeberger, K. et al. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods 6, 550–551 (2009).
Ito, D. et al. A comparative analysis of the molecular characteristics of the Arabidopsis CoA pyrophosphohydrolases AtNUDX11, 15, and 15a. Biosci. Biotechnol. Biochem. 76, 139–147. https://doi.org/10.1271/bbb.110636 (2012).
Mimura, M. et al. Arabidopsis TH2 encodes the orphan enzyme thiamin monophosphate phosphatase. Plant Cell 28, 2683–2696. https://doi.org/10.1105/tpc.16.00600 (2016).
Koonin, E. V. et al. Eukaryotic translation elongation factor 1 gamma contains a glutathione transferase domain—Study of a diverse, ancient protein superfamily using motif search and structural modeling. Protein Sci. 3, 2045–2054. https://doi.org/10.1002/pro.5560031117 (1994).
Morais, M. C. et al. The crystal structure of bacillus cereus phosphonoacetaldehyde hydrolase: Insight into catalysis of phosphorus bond cleavage and catalytic diversification within the HAD enzyme superfamily. Biochemistry 39, 10385–10396. https://doi.org/10.1021/bi001171j (2000).
Sa, N., Rawat, R., Thornburg, C., Walker, K. D. & Roje, S. Identification and characterization of the missing phosphatase on the riboflavin biosynthesis pathway in Arabidopsis thaliana. Plant J. 88, 705–716. https://doi.org/10.1111/tpj.13291 (2016).
Auge, G. A., Penfield, S. & Donohue, K. Pleiotropy in developmental regulation by flowering-pathway genes: Is it an evolutionary constraint?. New Phytol. 224, 55–70. https://doi.org/10.1111/nph.15901 (2019).
Gazzani, S., Gendall, A. R., Lister, C. & Dean, C. Analysis of the molecular basis of flowering time variation in Arabidopsis accessions. Plant Physiol. 132, 1107–1114. https://doi.org/10.1104/pp.103.021212 (2003).
Allen, K. N. & Dunaway-Mariano, D. Markers of fitness in a successful enzyme superfamily. Curr. Opin. Struct. Biol. 19, 658–665. https://doi.org/10.1016/j.sbi.2009.09.008 (2009).
Huang, H. et al. Panoramic view of a superfamily of phosphatases through substrate profiling. Proc. Natl. Acad. Sci. 112, E1974–E1983. https://doi.org/10.1073/pnas.1423570112 (2015).
Mazzella, M. A. et al. Phytochrome control of the Arabidopsis transcriptome anticipates seedling exposure to light. Plant Cell 17, 2507–2516 (2005).
Deng, W. et al. FLOWERING LOCUS C (FLC) regulates development pathways throughout the life cycle of Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 108, 6680–6685 (2011).
Mateos, J. et al. Combinatorial activities of SHORT VEGETATIVE PHASE and FLOWERING LOCUS C define distinct modes of flowering regulation in Arabidopsis. Genome Biol. 16, 31 (2015).
Baldwin, J. C., Karthikeyan, A. S., Cao, A. & Raghothama, K. G. Biochemical and molecular analysis of LePS2;1: A phosphate starvation induced protein phosphatase gene from tomato. Planta 228, 273. https://doi.org/10.1007/s00425-008-0736-y (2008).
Kim, Y., Schumaker, K. S. & Zhu, J. K. EMS mutagenesis of Arabidopsis. Methods Mol. Biol. 323, 101–103. https://doi.org/10.1385/1-59745-003-0:101 (2006).
Steffen, A., Fischer, A. & Staiger, D. Determination of photoperiodic flowering time control in Arabidopsis and barley. Methods Mol. Biol. 1158, 285–295 (2014).
De Vylder, J., Vandenbussche, F., Hu, Y., Philips, W. & Van Der Straeten, D. Rosette tracker: An open source image analysis tool for automatic quantification of genotype effects. Plant Physiol. 160, 1149–1159. https://doi.org/10.1104/pp.112.202762 (2012).
Uchida, N., Sakamoto, T., Tasaka, M. & Kurata, T. Identification of EMS-induced causal mutations in Arabidopsis thaliana by next-generation sequencing. Methods Mol. Biol. 1062, 259–270. https://doi.org/10.1007/978-1-62703-580-4_14 (2014).
Konieczny, A. & Ausubel, F. M. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 4, 403–410 (1993).
Koornneef, M., Alonso-Blanco, C. & Stam, P. Genetic analysis. Methods Mol. Biol. 323, 65–77. https://doi.org/10.1385/1-59745-003-0:65 (2006).
Neff, M. M., Turk, E. & Kalishman, M. Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 18, 613–615. https://doi.org/10.1016/s0168-9525(02)02820-2 (2002).
Clough, S. J. & Bent, A. F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. https://doi.org/10.1046/j.1365-313x.1998.00343.x (1998).
Streitner, C., Hennig, L., Korneli, C. & Staiger, D. Global transcript profiling of transgenic plants constitutively overexpressing the RNA-binding protein AtGRP7. BMC Plant Biol. 10, 221. https://doi.org/10.1186/1471-2229-10-221 (2010).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. https://doi.org/10.1093/bib/bbx108 (2017).
Kuraku, S., Zmasek, C. M., Nishimura, O. & Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 41, W22–W28. https://doi.org/10.1093/nar/gkt389 (2013).
Lefort, V., Longueville, J.-E. & Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. https://doi.org/10.1093/molbev/msx149 (2017).
Acknowledgements
We would like to thank Elisabeth Klemme and Kristina Neudorf for expert technical support, Prof. Sanja Roje, Washington State University, and Prof. C. Wang, Wuhan, for useful discussions on HAD family proteins and sequence information. We are grateful to Dr. Julieta Mateos for critical comments on the manuscript. This work was supported by DFG SPP1530 (grants JO1252/2-1 to MJ and STA653/5-2 to DS).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.J., D.S.: conceptualization of the project; M.J., D.S.: funding acquisition; M.J., A.S., N.K.: investigation; M.L.: phylogenetic tree; D.S., A.S., and M.J.: writing of the manuscript. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johansson, M., Steffen, A., Lewinski, M. et al. HDF1, a novel flowering time regulator identified in a mutant suppressing sensitivity to red light reduced 1 early flowering. Sci Rep 13, 1404 (2023). https://doi.org/10.1038/s41598-023-28049-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28049-6
- Springer Nature Limited