
Abstract
Selective elimination of tumors has always been the mainstay of oncology research. The on-going research underlying the cellular apoptotic mechanisms reveal caspases activation, especially the key effector caspase-3, as a personalized tumor-selective therapeutic strategy. Our continued research protocol has exploited new optimized Passerini α-acyloxy carboxamides as efficient apoptotic inducers via caspase-3/7 dependent mechanism with highly selective anticancer profiles. The adopted design rationale relied on excluding structural alerts of previous leads, while merging various pharmacophoric motifs of natural and synthetic caspase activators via optimized one-pot Passerini reaction conditions. The prepared compounds resulting from Passerini reaction were screened for their cytotoxic activities against colorectal Caco-2 and liver HepG-2 cancer cells compared to normal fibroblasts utilizing MTT assay. Notably, all compounds exhibited promising low-range submicromolar IC50 against the studied cancer cell lines, with outstanding tumor selectivity (SI values up to 266). Hence, they were superior to 5-fluorouracil. Notably, 7a, 7g, and 7j conferred the highest potencies against Caco-2 and HepG-2 cells and were selected for further mechanistic studies. Caspas-3/7 activation assay of the hit compounds and flow cytometric analysis of the treated apoptotic cancer cells demonstrated their significant caspase activation potential (up to 4.2 folds) and apoptotic induction capacities (up to 58.7%). Further assessment of Bcl2 expression was performed being a physiological caspase-3 substrate. Herein, the three studied Passerini adducts were able to downregulate Bcl2 in the treated Caco-2 cells. Importantly, the mechanistic studies results of the three hits echoed their preliminary MTT antiproliferative potencies data highlighting their caspase-3 dependent apoptotic induction. Finally, the in silico predicted physicochemical and pharmacokinetic profiles, as well as ligand efficiency metrics were drug-like.
Similar content being viewed by others

Introduction
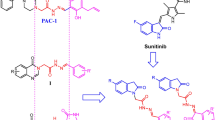
Evading apoptosis, the normal programmed cell death process that maintains tissue homeostasis1, is a cancer hallmark2,3. Apoptosis is mediated through a cascade of intrinsic and extrinsic signaling pathways that mainly converge on caspase-dependent proteolysis of numerous vital proteins, DNA cleavage, and membrane blebbing4. Caspases comprise a characteristic family of cysteinyl-aspartate-specific proteases that are capable of cleaving an aspartate amino acid residue from their specific substrates5,6 inducing irreversible cell death. Caspases are classified into initiator (caspase-10, -9, -8 and -2) and effector (caspase-7, -6 and -3) groups7. Caspase-3 and -7 are considered the key effector caspases executing apoptosis8,9,10. These findings thus directed cancer research programs to set the caspases family, especially the effector members, as attractive anticancer targets for inducing apoptosis induction in cancer cells11. Moreover, the cellular levels of procaspases, the inactive precursors of effector caspases, are usually elevated in cancer relative to normal tissue, introducing an ideal opportunity for tumor-targeting to selectively generate cytotoxic caspases in cancer through weaponizing procaspases overexpression. Accordingly, caspase-mediated apoptotic induction is considered a highly tumor-selective anticancer strategy12. Since mature caspases are inhibited using the cellular inhibitor of apoptosis proteins (IAPs) that are essentially inactivated by the secondary mitochondria-derived activator of caspases (SMAC/Diablo)7, early drug discovery trials have focused on SMAC that triggers caspase-dependent apoptosis13,14. However, its direct clinical application was hampered by the drawbacks of its peptide nature. A tetrapeptide of SMAC was then envisioned as the principal motif for designing peptidomimetics that can function as the endogenous protein. Pioneer studies introduced series of efficient apoptotic inducers designed as capped tripeptides to reduce their peptide character (Fig. 1; I and II)16. This active research was mirrored by several trials to optimize novel small-molecule caspase-dependent apoptotic inducers. Early studies employing high throughput screening caspase-based assays introduced a new series of potent caspase activators derived from nicotinamides III (Fig. 1)16. Further fruitful work led to various preclinical-stage caspase activators have been introduced17,18. Optimized fragment-based design studies identified nonpeptidic clinical-stage apoptotic inducers that are based on an essential amide core (Fig. 1, IV) that is crucial for activity and should not be changed or removed19. Along this line, our research group have utilized the multicomponent Passerini-3CR as a facial synthetic strategy to synthesize libraries of amide-based caspase activators inducers V (Fig. 1)20 incorporating the thematic structural features of lead compounds (Fig. 1). Among the evaluated series, the hit derivatives exerted profound anticancer potency against various human cancers20.

Lead Small-molecules caspase activators.
Rationale design
Herein, we continued our optimization protocol utilizing the lead Passerini scaffold (Fig. 2). Firstly, the newly synthesized α-acyloxy carboxamides were designed to incorporate an ester to the amide counterpart as a substitute to the nitro group being a structural alert and a possible toxicophore21,22 aiming to enhancing the molecule’s safety profile and anticancer selectivity. Secondly, the acyloxy counterpart was rationally selected inspired by pharmacophoric motifs of natural caspase activators, where indole and quinoline rings were installed to the skeleton being the main cores of marine bisindole alkaloids23,24 from the 3,3′-diindolylmethane (DIM) family25 and quinoline alkaloids23. Finally, to possibly limit “the molecular obesity” for better pharmacokinetics as well as to enrich the deduced SAR in this study, structure simplification strategy was adopted, where the heterocyclic acyloxy counterparts were simplified to diverse aromatic (benzoate, phenylacetate, and cinnamate) and aliphatic (thioglycolate) moieties. As part of the followed mimicry design approach, various substituents that conferred caspase-dependent antitumor activities to reported lead molecules were selected including halogens20, and thiol26 group.

Design rationale of the target Passerini adducts.
All the target Passerini adducts were initially screened via the MTT assay27 against human fibroblasts Wi-38 for their cytotoxic potential against human fibroblasts (Wi-38) and two cancer cell lines namely HepG-2 (liver) and Caco-2 (colon) being among the most common cancers worldwide according to the updated WHO list28. The selectivity against the cancer versus normal cells were evaluated. Activation assay of caspase-3/7 and flow cytometric analysis of apoptosis were performed to the most promising compounds. Further apoptosis studies were performed to quantify the expression of Bcl2 that is a physiological caspase-3 substrate29. Thereafter, the hit compounds were subjected to computational studies to predict their physicochemical properties, ADME parameters and efficiency metrics.
Results and discussion
Chemistry
Multicomponent reactions (MCRs) have become excellent processes in organic studies due to their bond formation efficiency since three or more components react together in one-pot synthesis to achieve higher yields of pure products, enhanced reaction rate, better selectivity, improved ease of manipulation, rapid optimization of reaction, ability to effect, chemo-, regio-, and stereoselective transformations, economical and eco-friendly processes. Isocyanide based multicomponent reactions (IMCRs) are one of the most important fields in the last decade. IMCRs represent as interesting close of multicomponent reactions due to their versatile biological properties. Moreover, they were useful for the formation of novel, complex and biological active compounds30,31.
Mario Passerini performed the first IMCR by combining three components (an isocyanide, carboxylic acid, and oxo-compound) to form α-acyloxy carboxamides. Passerini products are scaffolds that have been used for synthesizing large libraries of bioactive drugs32,33.
Novel synthesized α-acyloxy carboxamides have shown cytotoxic activities with different human cancer cell lines. Three-component Passerini reaction was reported by using p-nitrophenyl isocyanide previously, in which the nitro group as an electron withdrawing group with cyclohexanone and different carboxylic acids. Also unexpected products obtained by using trifluoro- and trichloro-acetic acids forming hydroxy and spiro compounds, respectively. In this respect, we report herein the Passerini reaction between ethyl-4-isocyanobenzoate 4 with carboxylic acid derivatives 5a–k and cyclohexanone 6 forming biologically active products20.
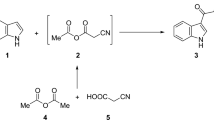
Herein, our strategy to carry out the Passerini reaction utilizing the isocyanide 4, which was synthesized by performing of esterification reaction of 4-amino benzoic acid (1) by reflux in absolute ethanol to afford the corresponding ester 4-amino ethyl benzoate (2) which known as benzocaine drug that used as pain reliever34. Consequently, formylation of the latter utilizing formic acid and toluene to give 4-formamido ethyl benzoate (3)35 followed by dehydration using PPh3/Et3N/I2 protocol where the reaction was proceeded for 1 h to afford the corresponding isonitrile ethyl-4-isocyanobenzoate (4) in good yield36 (Fig. 3). The formed isocyanide is unstable, and it is preferable to store it at a low temperature (5–10 °C). The structure of 4 was confirmed by IR that showed strong band at 2124 and 1717 cm−1 that characterizes the isocyano and carbonyl of the ester groups respectively.

Synthesis of ethyl-4-isocyanobenzoate 4.
A novel series of the α-acyloxy carboxamide derivatives 7a–k were prepared via Passerini reaction by using 4 in excess cyclohexanone as a ketone and solvent with miscellaneous carboxylic acids namely indole-3-acetic acid, quinaldic acid, benzoic acid, p-chlorobenzoic acid, o-chlorobenzoic acid, o-iodobenzoic acid, phenylacetic acid, (E)-3-(4-(trifluoromethyl)cinnamic acid, N-Boc-4-aminohippuric acid, (phenoxycarbonyl)-l-phenylalanine, and 2-mercaptoacetic acid 5a–k, respectively; at room temperature (Fig. 4). The obtained Passerini derivatives were analyzed in detail by FT-IR and NMR spectroscopy. 1H-NMR spectrum of 7a–k showed signals for –NH in amido groups resonating from δH 10.9–8.9 ppm. The ten Protons of c-Hexylidene group also appeared at range from δH 3.1–1.1 ppm. The proton of –NH indole group 7a showed singlet signal at δH 10.94 ppm. Also, two protons of –CH2 group (CO-CH2-Ar) for compound 7g appeared as singlet signal at δH 3.78 ppm. Proton spectrum for compound 7h recorded two signals corresponding to olefinic hydrogens at δH 7.71 and 6.86 ppm. Three amide protons for product 7i showed three signals at δH 9.74, 9.62 and 8.90 ppm. For product 7j, three protons of l-phenylalanine moiety recorded signals at δH 4.95 ppm for –CH2 and from δH 4.44–4.39 ppm for –CH. Proton of Mercapto group –SH in 7k showed signals resonating from δH 0.85–0.80 ppm. 13C-NMR spectrum recorded signals resonated from δc 171.8–161.2 ppm corresponding to amido and ester CO groups, respectively. Carbons of c-Hexylidene (O–C–CO) resonating at range from δc 84.0–81.1 ppm. Also, Cyclohexyl carbons recorded signals resonating from δc 36.0–20.6 ppm. For products 7f and 7k, carbons of C-I and CH2-SH showed signals recorded at δc 94.7 and 40.7 ppm, respectively.

Synthesis of α-acyloxy carboxamide derivatives 7a–k via Passerini reaction.
(P-3CR) utilizing trihaloacetic acids via Passerini reaction showed unexpected derivatives. Where, trifluoroacetic acid (TFA) afforded Ethyl 4-(1-hydroxycyclohexanecarboxamido)benzoate 9 in a good yield after hydrolyzing the formed Passerini product ethyl 4-(1-(2,2,2-trifluoroacetoxy)cyclohexanecarboxamido)benzoate 8 (Fig. 5). 1H-NMR spectrum confirmed the structure of 9, where two protons correspond to amide –NH and hydroxyl group –OH appeared clearly as two singlet signals at δH 9.91 and 5.49 ppm, respectively. Also, 13C-NMR spectrum showed signals for carbonyl of the amide group and carbons of tertiary alcohol at δc 177.1 and 74.5 ppm, respectively.

Synthesis of 9 and 11 via Passerini reaction using trihaloacetic acids.
Alternatively, P-3CR using trichloroacetic acid (TCA) afforded the unexpected spiro compound; Ethyl 4-(2,4-dioxo-1-oxa-3-azaspiro[4.5]decan-3-yl)benzoate (11), where Passerini product; ethyl 4-(1-(2,2,2-trichloroacetoxy)cyclohexanecarboxamido)benzoate 10 was formed first, then releasing of –CCl3 group through intramolecular nucleophilic nitrogen attack on trichloro acetyl group, followed by cyclization to form 11 (Fig. 5). 1H-NMR approved the structure of 11, where protons of aromatic moiety recorded two obvious doublets signals at δH 8.06 and 7.62 ppm. Moreover, c-Hexylidene protons signals appeared at δH 2.04 and 1.30 ppm. 13C-NMR confirmed the spiro carbon signal at δc 85.0 ppm, Also, CO groups of oxazolinedione ring recorded two signals at δc 174.0 and 152.8 ppm.
Regarding the stability of the currently studied Passerini adducts, literature review revealed a recent valuable study demonstrating that the stability of the ester in the Passerini skeleton could be influenced by other substructures present based on experiments carried on a library of more than Passerini adducts37. Being concerned with the medicinal chemistry applications of Passerini reaction, the study focused on investigating the metabolic stability of the scaffold towards esterases. Accordingly, when the group directly attached to the ester moiety (R3; Fig. 6) is an ortho-substituted or ortho, ortho′-disubstituted aromatic ring, the Passerini α-acyloxy carboxamides are metabolically stable to esterases37.

The general structure of Passerini α-acyloxy carboxamides.
This is in accordance with previous studies that generally showed that steric hindrance can reduce or suppress the hydrolytic activity of esterases, while the presence of electron-withdrawing groups on the acyl side can facilitate the enzymatic cleavage38. The skeleton ester is also stable toward hydrolysis when the R1 group is a bulky substituent regardless of the nature of the R3 substituent. The study was also pointed to the stability of the terminal aromatic ester as exemplified by experimental observation of the different hydrolytic stabilities of methyl benzoate and methyl 2,6-dimethylbenzoate toward esterases, where the former is readily hydrolyzed, while the latter is completely resistant to esterases37.
Biology
Cytotoxicity screening
The newly synthesized Passerini adducts 7a–k were preliminarily screened utilizing the microculture MTT assay39 to evaluate their potential antiproliferative activities against normal human fibroblasts (Wi-38), colorectal (Caco-2) and hepatocellular (HepG-2) cancer cells compared to the standard chemotherapy 5-fluorouracil that promote caspase-dependent apoptosis in various cancers40,41 (Table 1). Obviously, all derivatives were superior to 5-fluorouracil against the screened cell lines concerning potency and selectivity. 7g was ranked as the most potent (IC50 = 0.06 μM) and selective (SI = 262–266) Passerini adduct against the screened cell lines, followed by 7j and 7a (IC50 = 0.07–0.12 μM, SI = 97–243). 7e, 7f, 7i and 9 exhibited slightly lower potency and recorded comparable low range submicromolar IC50 values. These derivatives were notably tumor-selective with SI ranging from 69 to 180. The remainder derivatives were relatively less active, but still promising regarding potency and selectivity.
It is worth mentioning that the observed cytotoxicity pattern of the investigated Passerini esters highlighted their promising tumor selectivity profile. This could be safely deduced when compared with the cytotoxicity profiles of the previously studied Passerini leads with nitro group substituents20 as exemplified by the MTT assay data of the structurally related derivatives illustrated in Fig. 7. It is obvious that simple replacement of the nitro group by an ester moiety enhanced the tumor selectivity profile of the Passerini scaffold by approximately 85 folds reflecting their higher safety on normal cells.

The effect of substituting nitro group by ester on the cytotoxicity profile of Passerini scaffold.
Interestingly, this observation justified our design strategy and optimization protocol adopting replacement of the nitro group being considered as structural alert by the biocompatible ester group.
Apoptosis studies
Caspase-dependent apoptosis
Caspase-3/7 activation assay20,42 was performed to the most active anticancer derivatives to record the relative caspase-3/7 activity fold increments in HepG-2 and Caco-2 cells after treatment with IC50 doses (Table 1) of the studied compounds. Results (Table 2) showed that the three compounds activated caspase-3/7 and induced cancer cell apoptosis. Slightly higher activation potentials (3.08–4.18 folds) were detected in Caco-2 cells compared to HepG-2 (2.94–4.11 folds). The most potent antiproliferative Passerini adduct 7g displayed the highest caspase-3/7 relative fold increments (up to 4.17) in the studied cancer cells compared to the other derivatives 7j, 7a and the reference drug, respectively. These results echoed the MTT assay results (Table 1). Thus, it could be postulated that caspase-3/7 mediated apoptotic induction may be the possible antiproliferative mechanism of the studied compounds.
Bcl2 expression
Bcl2 protein inhibits apoptosis and is normally cleaved at Asp-34 during apoptosis by caspases or in vitro by recombinant caspase-329. Furthermore, several studies demonstrated that Bcl2 expression is downregulated in various tissues as a result of caspase-3 activation43 and vice versa44 In this study, RT-qPCR of Bcl2 results showed that the studied compounds could repress Bcl2 expression by more than 10 folds relative to 5-FU-treated Caco-2 cells (Table 3). 7g revealed the highest potential for downregulating Bcl2 by 11.5 folds, followed by 7j and 7a.
Morphological examination of the induced apoptosis
Caco-2 and HepG-2 cells treated with the hit derivatives 7a, 7g and 7j were microscopically examined, after 72 h, compared to untreated and 5-FU-treated cells (Fig. 8). As shown, the hit compounds-treated cells appeared with severe shrinkage and morphological alterations indicating antiproliferative activities and high apoptotic induction capacities of the studied adducts.

Morphological alterations of the hit compounds 7a, 7g and 7j-treated cancer cell lines after 72 h, untreated cancer cells and 5-fluororuracil-treated cells.
Flow cytometric analysis of induced apoptosis
Flow cytometric analysis was performed on the annexin-stained apoptotic Caco-2 cells treated with IC50 doses of the hit derivative 7a, 7g and 7j for 72 h. Caco-2 cells were selected for the study being more sensitive to the caspase-dependent apoptotic potential of the studied compounds compared to HepG-2 cells. Herein, results showed that our efficient caspase activators induced cancer cells apoptosis by ≥ 51%. The most active caspase activator 7g displayed the highest apoptotic induction potential as indicated by 58.7% total apoptotic cell population in the treated cells, followed by 7j (52.6%) and 7a (50.9%), respectively, compared to the untreated control (1.8%) as shown in Table 4 and Fig. 9. Interestingly, the detected potencies could be correlated to the results of MTT and caspase activation assays.

Flow cytometric analysis of apoptosis induction in the most active compounds-treated Caco-2 compared to untreated control cells, after staining with annexin V/propidium iodide.
Computational prediction of the hits’ ligand efficiency metrics, physicochemical properties, ADMET, and drug-likeness
As useful ligand identification tool, computational studies are usually performed to predict the most promising compounds ligand efficiency metrics, physicochemical and pharmacokinetic parameters, as well as drug-likeness. In the current study, the ligand efficiency (LE) and lipophilic ligand efficiency (LLE) metrics of the most promising caspase activators 7a, 7g and 7j were computed (Table 5) as reported20,45,46. LE represents molecular size to potency balance47,48,49, whereas LLE illustrates how efficiently the active compound exploits its lipophilic character to exert potency50, accordingly, the hit compounds could be more reliably prioritized corrected for their molecular sizes and lipophilicity.
Results (Table 5) displayed promising LE and LLE metrics of the studied compounds. 7g exceeded the acceptable LE limit against the screened cell lines. 7a was slightly beyond the lower LE limit, whereas 7j displayed the lowest predicted LE value among the group. Interestingly, the three compounds recorded lead-like LLE values ranging from 3.36 to 4.02.
Importantly, the physicochemical properties that formulate Lipinski's rule of five51, Veber and Egan`s rules52,53 bioavailability parameters were computed utilizing SwissADME web tool54 to assess the predicted drug-likeness of the studied compounds. Results (Table 6) showed that the three compounds were in full accordance with Egan’s parameters. 7a and 7g fulfilled Lipinski’s and Veber’s parameters, unlike 7j that violated Lipinski’s molecular weight limit and Veber’s number of rotors. Topological polar surface area (TPSA) and aqueous solubility were also predicted being considered as useful descriptors of bioavailability55. The studied compounds recorded drug-like TPSA (150 < A2)56,57, acceptable solubility requirement, with 7g at the top of the list.
Pre-ADMET58 and ProTox-II59 online servers were employed to predict the ADMET parameters for the current study hits. The three compounds recorded high predicted intestinal absorption (> 92.6%), medium Caco-2 (PCaco2 = 25.18–32.72 nm/s) and low MDCK (PMDCK = 0.07–6.74 nm/s) models permeabilities, strong plasma proteins binding (90.83–92.69%) and medium CNS absorption (BBB = 0.10–1.89). The compounds were predicted to be devoid of CYP2D6 inhibition and were classified as class IV chemicals according to the Globally Harmonized System for Classification and Labeling of Classified Chemicals (GHS), with high predicted median lethal oral dose (LD50 = 530–1739 mg/kg) in rodents. Additional predicted toxicity assessment showed that the three studied compounds were not expected to be hepatotoxic, carcinogens nor mutagens, thus they could be predicted to be safe druggable leads.
The references values were considered as previously reported for TPSA56,57, HIA60, PPB58, BBB61, Caco262,63, MDCK64, LD5059, Lipinski rule51, Veber’s rule52 and Egan’s rule53.
Structure–Activity relationship
The observed activity revealed that the currently modified Passerini scaffold generally conserved the intrinsic potency of the leads (Fig. 1), with enhanced tumor cell selectivity. However, both potency and selectivity were functions of the acyloxy counterpart nature and size. The bioinspired 2-(1H-indol-3-yl)acetate ester (7a) conferred excellent potency and selectivity to the main scaffold against screened cancer cell lines. Substituting 2-(1H-indol-3-yl)acetate motif was substituted with quinoline-2-carboxylate (7b) lowered the molecule’s potency and selectivity by nearly 2 folds. Structure simplification approach of the acyloxy counterpart of (7a) to benzoate (7c) attenuated the antiproliferative potency by 3 folds against Caco-2 cells and, by 1.5 folds against HepG-2 cells. This was mirrored by lowered selectivity to Caco-2 (5 folds) and HepG-2 (2 folds). Further substitution of the benzoate group with o-chloro (7e) or o-iodo (7f) led to increase in potency (2 folds) and selectivity (6 folds) against Caco-2 cells and supplemental enhancement in activity against HepG-2 with 2–3 folds more selectivity compared to the unsubstituted derivative (7c). Shifting the chloro substituent in (7e) to the para- position (7d) didn’t favor both potency and selectivity against the studied cell lines. The scaffold’s potency and selectivity profiles were optimized by modifying the benzoate group to phenylacetate (7g), or phenoxycarbonyl phenylalaninate (7j) that afforded the highest recorded potency and selectivity with the evaluated series. Other versatile substituents namely trifluoromethylcinnamate (7h), 4-(boc)aminobenzoylglycinate (7i) or thioglycolate (7k) exhibited relatively moderate activities being superior to the benzoate derivative (7c) against Caco-2 and comparable to it against HepG-2. Importantly, the hit antiproliferative derivatives recorded similar caspase-3/7 activation potential pattern highlighting the possibility that caspase-3/7 mediated apoptotic induction may be their main antiproliferative mechanism.
Conclusion
This work portrays structure optimization of a promising Passerini-derived caspase activator scaffold towards enhanced tumor selectivity via excluding a possible toxicophore, introducing bioinspired substituents, and structure-simplification design approaches. Initial screening revealed excellent selectivity against colorectal Caco-2 and hepatocellular HepG-2 cancer cells rather than normal ones (SI up to 266.8). All derivatives were superior to 5-fluorouracil with sub-micromolar IC50 values. 7a, 7g and 7j were the study hits. The three derivatives activated caspase up to 4.2 folds in the treated cancer cells, downregulated Bcl2 and induced apoptosis (up to 58.7%). Computational studies predicted their acceptable efficiency metrics and drug-like pharmacokinetic parameters.
Experimental
Chemistry
The Materials and equipment were described in the supporting information.
Synthesis of 4-amino ethyl benzoate (2)
A solution of 4-amino benzoic acid 1 (20 g, 0.14 mol) in anhydrous EtOH (150 ml) and sulphuric acid (17 ml) was heated until the consumption of the acid was complete (typically 1–2 h). After cooling, the mixture was neutralized by using solution of washing soda. The formed desired product was filtered, and washed using distilled water to yield a white solid of 2 (22 g, 92%); m.p = 74–76 °C.
Synthesis of 4-Formamido ethyl benzoate 335.
To a solution of ethyl 4-aminobenzoate 2 (11 g, 0.06 mol) in toluene (100 ml), formic acid (3.98 g, 0.08 mol) was added dropwise, followed by heating to 85–90 °C for 2 h. After cooling at r.t, the product was formed, filtered and washed using distilled water to yield a white solid of 3 (10.2 g, 79%); m.p = 138–140 °C.
Synthesis of ethyl-4-isocyanobenzoate 436
To a mixture of formamide 3 (5.0 g, 0.026 mol), I2 (9.87 g, 0.039 mol) and Ph3P (10 g, 0.038 mol) in DCM (75 ml), Et3N (7.79 g, 10.7 ml, 0.07 mol) was added dropwise. The reaction mixture was stirred at r.t for 1 h. The reaction mixture was diluted by adding CHCl3 (50 ml) and then washed using an ice-cold saturated solution of Na2S2O3. The aqueous phase was extracted using DCM and the organic phase was washed with brine, dried over anhydrous Na2SO4, and filtered. The combined organic phase was evaporated and purified using column chromatography (1:2, EtOAc–n-hexane) to give the corresponding brown solid of isocyanide 4 (2 g, 55%); m.p = 93–96 °C, lit.36 101–105 °C.
General method for Passerini reactions20
A mixture of ethyl-4-isocyanobenzoate 4 (50 mg, 0.286 mmol, 0.5 eq), carboxylic acids derivatives namely indole-3-acetic acid, quinaldic acid, benzoic acid, 4-chlorobenzoic acid, 2-chlorobenzoic acid, 2-iodobenzoic acid, phenylacetic acid, (E)-3-(4-(trifluoromethyl)cinnamic acid, N-Boc-4-aminohippuric acid, (phenoxycarbonyl)-l-phenylalanine, and 2-mercaptoacetic acid 5a–k, respectively (0.57 mmol, 1 eq) and excess cyclohexanone 6 (2.86 mmol, 5 eq) was stirred at r.t for 24 h and was monitored by TLC. Then, the reaction was diluted using 5 ml of DCM and neutralized using saturated sodium bicarbonate solution. The organic layer was collected, dried using anhydrous Na2SO4 and purified using flash column chromatography (1:3, EtOAc–n-hexane).
Ethyl-4-(1-(2-(1H-indol-3-yl)acetoxy)cyclohexanecarboxamido)benzoate 7a
Orange powder; yield 50%; m.p = 79–81 °C; Rf 0.25 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3372 (OCNH), 1697 (br, OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.94 (s, 1H, NH-CH), 9.88 (s, 1H, OC-NH), 7.88 (d, J = 9.0 Hz, 2H, Ar–H), 7.77 (d, J = 9.0 Hz, 2H, Ar–H), 7.50 (d, J = 8.0 Hz, 1H, Ar–H), 7.33 (d, J = 8.5 Hz, 1H, Ar–H), 7.25 (d, J = 2.0 Hz, 1H, Ar–H), 7.04 (t, J = 7.5 Hz, 1H, Ar–H), 6.91 (t, J = 7.0 Hz, 1H, Ar–H), 4.27 (q, J = 7.5 Hz, 2H, OCH2–CH3), 3.87 (s, 2H, OC–CH2), 2.13 (d, J = 13.5 Hz, 2H, c-Hex-H), 1.77–1.71 (td, J = 14.5, 4.0 Hz, 2H, c-Hex-H), 1.59–1.50 (m, 3H, c-Hex-H), 1.41–1.34 (m, 2H, c-Hex-H), 1.30 (t, J = 7.5 Hz, 3H, OCH2–CH3), 1.25–1.20 (m, 1H, c-Hex-H); 13C-NMR (125 MHz, DMSO-d6) δC: 171.6 (OCNH), 170.6 (C–O–CO), 165.4 (EtO–CO), 143.4, 136.1, 129.9, 127.1, 124.3, 124.2, 121.1, 119.4, 118.5, 118.4, 111.4, 106.9 (Ar–C), 81.2 (O–C–CO), 60.5 (OCH2), 31.3, 31.0, 24.6, 20.8 (c-Hex-C), 14.3 (CH3). Anal. calcd. for C26H28N2O5 (448.51): C, 69.63; H, 6.29; N, 6.25; found C, 69.42; H, 6.18; N, 6.43.
1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl quinoline-2-carboxylate 7b
Orange powder; yield 56%; m.p = 103–105 °C; Rf 0.32 (1:2, EtOAc–n-hexane); IR: vmax/cm–1 3398 (OCNH), 1717, 1680 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.07 (s, 1H, –NH), 8.58 (d, J = 9.0 Hz, 1H, Ar–H), 8.17 (d, J = 8.5 Hz, 1H, Ar–H), 8.13 (d, J = 8.5 Hz, 1H, Ar–H), 8.08 (d, J = 8.0 Hz, 1H, Ar–H), 7.87–7.83 (m, 3H, Ar–H), 7.76–7.71 (m, 3H, Ar–H), 4.23 (q, J = 6.5 Hz, 2H, OCH2–CH3), 2.33 (d, J = 14.0 Hz, 2H, c-Hex-H), 2.04 (s, 1H, c-Hex-H), 1.95–1.89 (m, 2H, c-Hex-H), 1.66 (s, 5H, c-Hex-H), 1.25 (t, J = 7.5 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 171.1 (OCNH), 165.4 (EtO–CO), 163.2 (C–O–CO), 147.8, 146.9, 143.4, 137.8, 130.8, 129.9, 129.0, 128.9, 128.1, 124.4, 121.2, 119.6 (Ar–C), 82.7 (O–C–CO), 60.5 (OCH2), 31.5, 30.7, 24.6, 21.1 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C26H26N2O5 (446.50): C, 69.94; H, 5.87; N, 6.27; found C, 70.01; H, 5.97; N, 6.32.
Ethyl-4-(1-(benzoyloxy)cyclohexanecarboxamido)benzoate 7c
Off-white powder; yield 55%; m.p = 140 – 142 °C; Rf 0.48 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3342 (OCNH), 1718, 1679 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.96 (s, 1H, -NH), 8.04 (d, J = 7.5 Hz, 2H, Ar–H), 7.87 (d, J = 9.0 Hz, 2H, Ar–H), 7.76 (d, J = 9.0 Hz, 2H, Ar–H), 7.69 (t, J = 7.0 Hz, 1H, Ar–H), 7.56 (t, J = 7.5 Hz, 2H, Ar–H), 4.26 (q, J = 7.0 Hz, 2H, OCH2–CH3), 2.31 (d, J = 13.5 Hz, 2H, c-Hex-H), 1.88 (td, J = 14.0, 2.5 Hz, 2H, c-Hex-H), 1.67 (d, J = 10.5 Hz, 3H, c-Hex-H), 1.60–1.51 (m, 2H, c-Hex-H), 1.33–1.31 (m, 1H, c-Hex-H), 1.28 (t, J = 7.5 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 171.3 (OCNH), 165.4 (EtO–CO), 164.4 (C–O–CO), 143.4, 133.6, 129.9, 129.6, 128.8, 124.4, 119.6 (Ar–C), 81.7 (O–C–CO), 60.5 (OCH2), 31.4, 24.6, 21.1 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C23H25NO5 (395.45): C, 69.86; H, 6.37; N, 3.54; found C, 69.93; H, 6.42; N, 3.61.
1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl 4-chlorobenzoate 7d
Grey powder; yield 56%; m.p = 193–195 °C; Rf 0.54 (1:2, EtOAc–n-hexane); IR: vmax/cm–1 3277 (OCNH), 1717, 1684 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.92 (s, 1H, –NH), 8.00 (d, J = 9.0 Hz, 2H, Ar–H), 7.84 (d, J = 8.5 Hz, 2H, Ar–H), 7.71 (d, J = 8.5 Hz, 2H, Ar–H), 7.60 (d, J = 8.5 Hz, 2H, Ar–H), 4.23 (q, J = 7.5 Hz, 2H, OCH2–CH3), 2.26 (d, J = 13.5 Hz, 2H, c-Hex-H), 1.85 (td, J = 13.5, 2.5 Hz, 2H, c-Hex-H), 1.63 (d, J = 9.5 Hz, 3H, c-Hex-H), 1.55–1.48 (m, 2H, c-Hex-H), 1.32–1.28 (m, 1H, c-Hex-H), 1.25 (t, J = 7.0 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 171.1 (OCNH), 165.3 (EtO–CO), 163.6 (C–O–CO), 143.3, 138.5, 131.4, 130.0, 129.0, 128.7, 124.4, 119.6 (Ar–C), 82.1 (O–C–CO), 60.5 (OCH2), 31.4, 30.7, 24.5, 21.1 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C23H24ClNO5 (429.89): C, 64.26; H, 5.63; N, 3.26; found C, 64.19; H, 5.53; N, 3.39.
1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl 2-chlorobenzoate 7e
White powder; yield 51%; m.p = 130–132 °C; Rf 0.42 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3341 (OCNH), 1729, 1692 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.02 (s, 1H, −NH), 7.94–7.92 (m, 1H, Ar–H), 7.89 (d, J = 8.5 Hz, 2H, Ar–H), 7.78 (d, J = 9.0 Hz, 2H, Ar–H), 7.63–7.59 (m, 2H, Ar–H), 7.53–7.50 (m, 1H, Ar–H), 4.27 (q, J = 14.0, 7.0 Hz, 2H, OCH2–CH3), 2.31 (d, J = 13.5 Hz, 2H, c-Hex-H), 1.94–1.90 (td, J = 13.5, 3.0 Hz, 2H, c-Hex-H), 1.68–1.61 (m, 5H, c-Hex-H), 1.34–1.32 (m, 1H, c-Hex-H), 1.29 (t, J = 7.0 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 170.9 (OCNH), 165.4 (EtO–CO), 163.8 (C–O–CO), 143.4, 133.5, 132.0, 131.4, 130.9, 130.2, 130.0, 127.5, 124.5, 119.6 (Ar–C), 83.1 (O–C–CO), 60.5 (OCH2), 31.3, 24.5, 21.1 (c-Hex-C), 14.3 (CH3). Anal. calcd. for C23H24ClNO5 (429.89): C, 64.26; H, 5.63; N, 3.26; found C, 64.33; H, 5.71; N, 3.42.
1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl 2-iodobenzoate 7f
Off-white powder; yield 76%; m.p = 148–150 °C; Rf 0.45 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3328 (OCNH), 1730, 1693 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.00 (s, 1H, –NH), 8.02 (d, J = 7.0 Hz, 1H, Ar–H), 7.93 (dd, J = 7.5, 2.0 Hz, 1H, Ar–H), 7.88 (d, J = 8.5 Hz, 2H, Ar–H), 7.79 (d, J = 9.0 Hz, 2H, Ar–H), 7.57 (t, J = 6.5 Hz, 1H, Ar–H), 7.30 (td, J = 8.0, 1.5 Hz, 1H, Ar–H), 4.26 (q, J = 7.5 Hz, 2H, OCH2–CH3), 2.31 (d, J = 14.0 Hz, 2H, c-Hex-H), 1.91 (td, J = 13.5, 3.0 Hz, 2H, c-Hex-H), 1.68–1.61 (m, 5H, c-Hex-H), 1.38–1.32 (m, 1H, c-Hex-H), 1.29 (t, J = 7.5 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 170.9 (OCNH), 165.4 (EtO–CO), 165.2 (C–O–CO), 143.4, 140.9, 135.4, 133.3, 130.8, 130.0, 128.3, 124.4, 119.7 (Ar–C), 94.7 (C-I), 82.9 (O–C–CO), 60.5 (OCH2), 31.3, 24.5, 21.1 (c-Hex-C), 14.3 (CH3). Anal. calcd. for C23H24INO5 (521.34): C, 52.99; H, 4.64; N, 2.69; found C, 53.11; H, 4.88; N, 2.91.
Ethyl -4-(1-(2-phenylacetoxy)cyclohexanecarboxamido)benzoate 7g
Off-white powder; yield 72%; m.p = 90 – 93 °C; Rf 0.48 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3386 (OCNH), 1737, 1708, 1681 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.87 (s, 1H, –NH), 7.89 (d, J = 9.0 Hz, 2H, Ar–H), 7.77 (d, J = 9.0 Hz, 2H, Ar–H), 7.33–7.28 (m, 4H, Ar–H), 7.26–7.23 (m, 1H, Ar–H), 4.27 (q, J = 7.0 Hz, 2H, OCH2–CH3), 3.78 (s, 2H, CO–CH2–Ar), 2.12 (d, J = 13.5 Hz, 2H, c-Hex-H), 1.74 (td, J = 14.0, 3.5 Hz, 2H, c-Hex-H), 1.53–1.50 (m, 3H, c-Hex-H), 1.35–1.32 (m, 2H, c-Hex-H), 1.29 (t, J = 7.5 Hz, 3H, OCH2–CH3), 1.24–1.19 (m, 1H, c-Hex-H); 13C-NMR (125 MHz, DMSO-d6) δC: 171.3 (OCNH), 170.1 (C–O–CO), 165.4 (EtO–CO), 143.3, 134.3, 129.9, 129.5, 128.4, 128.3, 126.9, 124.4, 119.5 (Ar–C), 81.4 (O–C–CO), 60.5 (OCH2), 40.9 (CO–CH2), 31.3, 24.5, 20.7 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C24H27NO5 (409.47): C, 70.40; H, 6.65; N, 3.42; found C, 70.66; H, 6.81; N, 3.70.
Ethyl-4-(1-((3-(4-(trifluoromethyl)phenyl)acryloyl)oxy)cyclohexanecarboxamido)benzoate 7 h
Orange crystal; yield 57%; m.p = 145–147 °C; Rf 0.80 (1:2, EtOAc–n-hexane); IR: vmax/cm–1 3343 (OCNH), 1715, 1639 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.93 (s, 1H, –NH), 7.95 (d, J = 8.0 Hz, 2H, Ar–H), 7.84 (d, J = 9.0 Hz, 2H, Ar–H), 7.76–7.73 (m, 4H, Ar–H), 7.71 (s, 1H, Ar–CH=CH), 6.86 (d, J = 16.0 Hz, 1H, Ar–CH=CH), 4.23 (q, J = 7.0 Hz, 2H, OCH2–CH3), 2.20 (d, J = 14.0 Hz, 2H, c-Hex-H), 1.81 (td, J = 16.0 ,4.0 Hz, 2H, c-Hex-H), 1.62–1.45 (m, 6H, c-Hex-H), 1.26 (t, J = 7.5 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 171.8 (OCNH), 165.9 (EtO–CO), 165.1 (C–O–CO), 143.9 (CH=CH–Ar), 143.6, 138.5, 130.4, 129.6, 126.3, 124.8 (CF3), 121.7 (Ar–C), 120.0 (CH=CH–Ar), 82.0 (O–C–CO), 61.0 (OCH2), 31.9, 25.1, 21.5 (c-Hex-C), 14.7 (CH3). Anal. calcd. for C26H26F3NO5 (489.48): C, 63.80; H, 5.35; N, 2.86; found C, 63.99; H, 5.52; N, 3.08.
Ethyl-4-(1-(2-(4-((tert-butoxycarbonyl)amino)benzamido)acetoxy)cyclohexane carboxamido) benzoate 7i
Reddish brown powder; yield 62%; m.p = 115–117 °C; Rf 0.26 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3334 (br, NH's), 1713, 1644 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.74, 9.62 (2 s, 2H, 2NH's), 8.90 (t, J = 5.5 Hz, 1H, CH2–NH), 7.88–7.71 (m, 6H, Ar–H), 7.48 (d, J = 9.0 Hz, 2H, Ar–H), 4.26–4.22 (m, 3H, CH2–NH, OCH2–CH3), 4.08 (d, J = 5.5 Hz, 1H, CH2–NH), 2.11 (d, J = 12.5 Hz, 2H, c-Hex-H), 2.04 (s, 2H, c-Hex-H), 1.75 (td, J = 14.5, 4.5 Hz, 2H, c-Hex-H), 1.55 (bs, 4H, c-Hex-H), 1.44 (s, 9H, C(CH3)3), 1.26 (t, J = 7.0 Hz, 3H, OCH2–CH3). 13C-NMR (125 MHz, DMSO-d6) δC: 171.2 (OCNH), 169.0 (EtO–CO), 166.7 (CH2–NH–CO), 165.4 (C–O–CO), 152.6 (NH–CO–O), 143.4, 143.2, 142.7, 130.0, 129.9, 128.2, 126.7, 124.4, 119.5, 117.2 (Ar–C), 81.9 (O–C–CO), 79.6 (C(CH3)3), 60.5 (OCH2), 41.8 (CH2-NH), 31.5, 30.7, 28.1 (C(CH3)3), 20.7 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C30H37N3O8 (567.63): C, 63.48; H, 6.57; N, 7.40; found C, 63.66; H, 6.77; N, 7.88.
Ethyl-4-(1-((2-((phenoxycarbonyl)amino)-3-phenylpropanoyl)oxy) cyclohexanecarboxamido) benzoate 7j
Reddish brown crystal; yield 71%; m.p = 103–105 °C; Rf 0.48 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3336 (br, NH's), 1709 (br, OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.53 (s, 1H, –NH), 7.90 (d, J = 8.0 Hz, 1H, –NH), 7.86 (d, J = 8.5 Hz, 2H, Ar–H), 7.73 (d, J = 9.0 Hz, 2H, Ar–H), 7.28–7.17 (m, 10H, Ar–H), 4.95 (q, J = 12.5 Hz, 2H, Ar–CH2), 4.44–4.39 (m, 1H, CH), 4.24 (q, J = 7.0 Hz, 2H, OCH2–CH3), 3.15 (dd, J = 14.5, 5.0 Hz, 1H, c-Hex-H), 2.86 (dd, J = 13.5, 4.0 Hz, 1H, c-Hex-H), 2.08 (d, J = 11.0 Hz, 2H, c-Hex-H), 1.75–1.70 (m, 2H, c-Hex-H), 1.51 (d, J = 8.5 Hz, 3H, c-Hex-H), 1.43–1.34 (m, 1H, c-Hex-H), 1.26 (t, J = 7.5 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 171.0 (O–CO–CH), 170.6 (NH–CO–C), 165.4 (EtO–CO), 156.4 (NH–O–O), 143.1, 137.5, 136.8, 130.0, 129.1, 128.3, 128.2, 127.9, 127.6, 126.5, 124.5, 119.4 (Ar–C), 82.1 (O–C–CO), 65.6 (OCH2), 60.5 (CH–CH2), 55.6 (CH–CH2), 36.0, 31.4, 31.1, 24.5, 20.7 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C32H34N2O7 (558.62): C, 68.80; H, 6.13; N, 5.01; found C, 69.11; H, 6.40; N, 5.33.
Ethyl-4-(1-(2-mercaptoacetoxy)cyclohexane-1-carboxamido)benzoate 7k
Yellow crystal; yield 50%; m.p = 108–110 °C; Rf 0.35 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3318 (OCNH), 2570 (SH), 1741, 1705, 1679 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.84 (s, 1H, –NH), 7.87 (d, J = 8.5 Hz, 2H, Ar–H), 7.75 (d, J = 8.0 Hz, 2H, Ar–H), 4.26 (q, J = 7.0 Hz, 2H, OCH2–CH3), 4.03–3.80 (m, 2H, CH2-SH), 2.15 (d, J = 12.5 Hz, 2H, c-Hex-H), 1.79 (t, J = 12.0 Hz, 2H, c-Hex-H), 1.60–1.38 (m, 5H, c-Hex-H), 1.29 (t, J = 7.0 Hz, 3H, OCH2–CH3), 1.25–1.21 (m, 1H, c-Hex-H), 0.85–0.80 (m, 1H, –SH). 13C-NMR (125 MHz, DMSO-d6) δC: 171.0 (OCNH), 168.1 (EtO–CO), 165.3 (O–CO–CH2), 143.2, 129.9, 124.5, 119.6, 118.6 (Ar–C), 82.4 (O–C–CO), 60.5 (OCH2), 40.7 (CH2-SH), 31.3, 24.5, 20.8 (c-Hex-C), 14.2 (CH3). Anal. calcd. for C18H23NO5S (365.44): C, 59.16; H, 6.34; N, 3.83; found C, 59.31; H, 6.51; N, 3.97.
Ethyl 4-(1-hydroxycyclohexanecarboxamido)benzoate 9
Reddish brown crystal; yield 50%; m.p = 160–163 °C; Rf 0.32 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 3326 (OH), 3264 (OCNH), 1718, 1656 (OC, NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.91 (s, 1H, –NH), 7.85 (q, J = 12.0, 8.5 Hz, 4H, Ar–H), 5.49 (s, 1H, OH), 4.23 (q, J = 6.5 Hz, 2H, OCH2–CH3), 1.70 (td, J = 13.0, 4.0 Hz, 2H, c-Hex-H), 1.56 (d, J = 12.0 Hz, 5H, c-Hex-H), 1.52–1.47 (m, 2H, c-Hex-H), 1.26 (t, J = 7.0 Hz, 3H, OCH2–CH3), 1.19–1.14 (m, 1H, c-Hex-H); 13C-NMR (125 MHz, DMSO-d6) δC: 177.1 (OCNH), 165.9 (EtO–CO), 143.7, 130.5, 124.8, 119.5 (Ar–C), 74.5 (O–C–CO), 60.9 (OCH2), 34.2, 31.2, 25.5, 21.3 (c-Hex-C), 14.7 (CH3). Anal. calcd. for C16H21NO4 (291.34): C, 65.96; H, 7.27; N, 4.81; found C, 66.12; H, 7.51; N, 4.90.
Ethyl-4-(2,4-dioxo-1-oxa-3-azaspiro[4.5]decan-3-yl)benzoate 11
Orange crystal; yield 60%; m.p = 143–145 °C; Rf 0.77 (1:2, EtOAc–n-hexane); IR: vmax/cm−1 1815 (CO–N–CO), 1736 (EtO–CO); 1H-NMR (500 MHz, DMSO-d6) δH: 8.06 (d, J = 9.0 Hz, 2H, Ar–H), 7.62 (d, J = 8.5 Hz, 2H, Ar–H), 4.30 (q, J = 7.0 Hz, 2H, OCH2–CH3), 2.04 (s, 2H, c-Hex-H), 1.80 (td, J = 12.0, 4.0 Hz, 2H, c-Hex-H), 1.72–1.68 (m, 2H, c-Hex-H), 1.62–1.48 (m, 4H, c-Hex-H), 1.29 (t, J = 7.5 Hz, 3H, OCH2–CH3); 13C-NMR (125 MHz, DMSO-d6) δC: 174.0 (N–CO), 165.0 (EtO–CO), 152.8 (N–CO–O), 135.4, 129.8, 129.7, 126.6 (Ar–C), 85.0 (O–C–CO), 61.1 (OCH2), 31.2, 30.7, 23.8, 20.8 (c-Hex-C), 14.1 (CH3). Anal. calcd. for C17H19NO5 (317.34): C, 64.34; H, 6.03; N, 4.41; found C, 64.64; H, 6.32; N, 4.52.
Biological evaluation
Cytotoxicity screening
Wi-38, Caco-2 and HepG-2 cells were cultured in DMEM medium-contained 10% FBS, seeded as 5 × 103 cells per well in 96-well cell culture plate and incubated at 37 °C in 5% CO2 incubator. After 24 h, serial concentrations of the test compounds were incubated with cells for 72 h. Cell viability was assayed via MTT protocol27 (Supplementary data). IC50 values of the synthesized compounds were measured via the Graphpad Instat software. Morphological changes were examined using phase contrast inverted microscope with a digital camera (Olympus, Japan).
Caspase-3/7 activation assay
ApoONE® Caspase-3/7 kit was used following the manufacturer’s instructions (Supplementary data).
Real-time quantitative PCR Analysis of Bcl2
Colon cancer cell line (Caco-2) was incubated with the most effective anticancer compounds, at 0.06 µM, for 72 h in 5% CO2 incubator. RNAs of untreated and treated cancer cells were extracted using Gene JET RNA purification kit (Thermo Scientific, USA). Then cDNAs were synthesized using cDNA Synthesis Kit (Thermo Scientific, USA). Real time PCR was performed using SYBR green master mix and specific primers (Forward/Reverse) as detailed in the supplementary data.
Flow cytometric analysis of apoptosis
The hit compounds were assayed for their proapoptotic effects by incubation MDA-MB 231 cells for 72 h, at their minimum IC50 doses. After trypsinization, the treated and untreated cancer cells were stained with fluorescein isothiocyanate (FITC)-annexin V/ propidium iodide (PI) then subjected to flow cytometric analysis of apoptosis as previously reported20 (Supplementary data).
Data analysis and statistics
Statistical analysis is described in the supplementary data.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Reed, J. C. & Tomaselli, K. J. Drug discovery opportunities from apoptosis research. Curr. Opin. Biotechnol. 11(6), 586–592 (2000).
Reed, J. C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 17(9), 2941–2941 (1999).
Wong, R. S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 30(1), 1–14 (2011).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17(7), 395–417 (2020).
Leung, D., Abbenante, G. & Fairlie, D. P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 43(3), 305–341 (2000).
Nicholson, D. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 6(11), 1028–1042 (1999).
Olsson, M. & Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 18(9), 1441–1449 (2011).
Thornberry, N. A. Caspases: Key mediators of apoptosis. Chem. Biol. 5(5), R97–R103 (1998).
Porter, A. G. & Jänicke, R. U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6(2), 99–104 (1999).
Clark, A. & MacKenzie, S. H. Targeting cell death in tumors by activating caspases. Curr. Cancer Drug Targets 8(2), 98–109 (2008).
Jiang, X. et al. Distinctive roles of PHAP proteins and prothymosin-α in a death regulatory pathway. Science 299(5604), 223–226 (2003).
Boudreau, M. W., Peh, J. & Hergenrother, P. J. Procaspase-3 overexpression in cancer: A paradoxical observation with therapeutic potential. ACS Chem. Biol. 14(11), 2335–2348 (2019).
Du, C., Fang, M., Li, Y., Li, L. & Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102(1), 33–42 (2000).
Chai, J. et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406(6798), 855–862 (2000).
Oost, T. K. et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem. 47(18), 4417–4426 (2004).
Cai, S. X. et al. Discovery of substituted N-phenyl nicotinamides as potent inducers of apoptosis using a cell-and caspase-based high throughput screening assay. J. Med. Chem. 46(12), 2474–2481 (2003).
Bai, L., Smith, D. C. & Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 144(1), 82–95 (2014).
Sun, H. et al. Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc. Chem. Res. 41(10), 1264–1277 (2008).
Johnson, C. N. et al. A fragment-derived clinical candidate for antagonism of X-linked and cellular inhibitor of apoptosis proteins: 1-(6-[(4-Fluorophenyl) methyl]-5-(hydroxymethyl)-3, 3-dimethyl-1 H, 2 H, 3 H-pyrrolo [3, 2-b] pyridin-1-yl)-2-[(2 R, 5 R)-5-methyl-2-([(3R)-3-methylmorpholin-4-yl] methyl) piperazin-1-yl] ethan-1-one (ASTX660). J. Med. Chem. 61(16), 7314–7329 (2018).
Ayoup, M. S. et al. Design, synthesis and biological evaluation of novel α-acyloxy carboxamides via Passerini reaction as caspase 3/7 activators. Eur. J. Med. Chem. 168, 340–356 (2019).
Benigni, R. & Bossa, C. Structural alerts of mutagens and carcinogens. Curr. Comput. Aided Drug Des. 2(2), 169–176 (2006).
Alves, V. M. et al. Alarms about structural alerts. Green Chem. 18(16), 4348–4360 (2016).
Li, W. et al. Isolation and identification of aromatic compounds in Lion’s Mane Mushroom and their anticancer activities. Food Chem. 170, 336–342 (2015).
Salucci, S., Burattini, S., Buontempo, F., Orsini, E., Furiassi, L., Mari, M., Lucarini, S., Martelli, A. M., Falcieri, E. Marine bisindole alkaloid: A potential apoptotic inducer in human cancer cells. Eur. J. Histochem. EJH 62(2) (2018).
El-Mesery, M., Seher, A., El-Shafey, M., El-Dosoky, M. & Badria, F. A. Repurposing of quinoline alkaloids identifies their ability to enhance doxorubicin-induced sub-G0/G1 phase cell cycle arrest and apoptosis in cervical and hepatocellular carcinoma cells. Biotechnol. Appl. Biochem. 68(4), 832–840 (2021).
Tartier, L., McCarey, Y. L., Biaglow, J. E., Kochevar, I. E. & Held, K. D. Apoptosis induced by dithiothreitol in HL-60 cells shows early activation of caspase 3 and is independent of mitochondria. Cell Death Differ. 7(10), 1002–1010 (2000).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 65(1–2), 55–63 (1983).
Cancer today. https://gco.iarc.fr/today/home.
Kirsch, D. G. et al. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 274(30), 21155–21161 (1999).
Wahby, Y., Abdel-Hamid, H. & Ayoup, M. S. Two decades of recent advances of Passerini reactions: Synthetic and potential pharmaceutical applications. New J. Chem. 46(4), 1445–1468 (2022).
Alkan, B. et al. One-pot cascade polycondensation and Passerini three-component reactions for the synthesis of functional polyesters. Polym. Chem. 13(2), 258–266 (2022).
Zeng, L., Xu, S., Cui, S. & Zhang, F. Three-component synthesis of β-aminoxy amides. Org. Chem. Front. 9(14), 3757–3762 (2022).
Ramírez-López, S. C., Gámez-Montaño, R. Synthesis of α-acyloxycarboxamides via Passerini reaction (2021).
França, A. D. S., Leão, R. A. & de Souza, R. O. Two step continuous-flow synthesis of benzocaine. J. Flow Chem. 10(3), 563–569 (2020).
Wang, B., Zhang, P. Z., Xia, J. M., Jia, A. Q. & Zhang, Q. F. Synthesis, photochemical characterization, and thermal stability of a series of substituted formamidines as ultraviolet light absorbers. J. Vinyl Addit. Technol. 25(s2), E108–E113 (2019).
Wang, X., Wang, Q.-G. & Luo, Q.-L. Synthesis of isonitriles from N-substituted formamides using triphenylphosphine and iodine. Synthesis 47(01), 49–54 (2015).
Brunelli, F., Ceresa, C., Fracchia, L., Tron, G. C. & Aprile, S. Expanding the chemical space of drug-like Passerini compounds: Can α-acyloxy carboxamides be considered hard drugs?. ACS Med. Chem. Lett. https://doi.org/10.1021/acsmedchemlett.2c00420 (2022).
Wang, D. et al. Human carboxylesterases: A comprehensive review. Acta Pharm. Sin. B. 8(5), 699–712 (2018).
Ayoup, M. S., Abu-Serie, M. M., Abdel-Hamid, H. & Teleb, M. Beyond direct Nrf2 activation; reinvestigating 1, 2, 4-oxadiazole scaffold as a master key unlocking the antioxidant cellular machinery for cancer therapy. Eur. J. Med. Chem. 220, 113475 (2021).
Ponce-Cusi, R. & Calaf, G. M. Apoptotic activity of 5-fluorouracil in breast cancer cells transformed by low doses of ionizing α-particle radiation. Int. J. Oncol. 48(2), 774–782 (2016).
Mhaidat, N. M., Bouklihacene, M. & Thorne, R. F. 5-Fluorouracil-induced apoptosis in colorectal cancer cells is caspase-9-dependent and mediated by activation of protein kinase C-δ. Oncol. Lett. 8(2), 699–704 (2014).
Ayoup, M. S. et al. Battle tactics against MMP-9; discovery of novel non-hydroxamate MMP-9 inhibitors endowed with PI3K/AKT signaling attenuation and caspase 3/7 activation via Ugi bis-amide synthesis. Eur. J. Med. Chem. 186, 111875 (2020).
Li, Y., Lin, L. & Wang, Q. Correlation of expression levels of caspase-3 and Bcl-2 in alveolar lavage fluid in neonatal respiratory distress syndrome and prognosis. Exp. Ther. Med. 15(3), 2891–2895 (2018).
Grünenfelder, J. R. et al. Upregulation of Bcl-2 through caspase-3 inhibition ameliorates ischemia/reperfusion injury in rat cardiac allografts. Circulation 104, 202–206 (2001).
Keserü, G. M. & Makara, G. M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 8(3), 203–212 (2009).
Arnott, J. A., Kumar, R. & Planey, S. L. Lipophilicity indices for drug development. J. Appl. Biopharm. Pharmacokinet. 1(1), 31–36 (2013).
Reynolds, C. H., Tounge, B. A. & Bembenek, S. D. Ligand binding efficiency: Trends, physical basis, and implications. J. Med. Chem. 51(8), 2432–2438 (2008).
Andrews, P. R., Craik, D. J. & Martin, J. L. Functional group contributions to drug-receptor interactions. J. Med. Chem. 27(12), 1648–1657 (1984).
Orita, M., Ohno, K., Warizaya, M., Amano, Y., Niimi, T. Lead generation and examples: Opinion regarding how to follow up hits. In Methods in Enzymology, 383–419 (Elsevier, 2011).
Jabeen, I., Pleban, K., Rinner, U., Chiba, P. & Ecker, G. F. Structure–activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J. Med. Chem. 55(7), 3261–3273 (2012).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23(1–3), 3–25 (1997).
Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45(12), 2615–2623 (2002).
Egan, W. J., Merz, K. M. & Baldwin, J. J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 43(21), 3867–3877 (2000).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7(1), 1–13 (2017).
Veber, D. F., Johnson, S. R., Cheng, H., Smith, B. R., Ward, K. W., Kopple, K. D. J. Med. Chem. 45, 2615–2623 (2002)
Polinsky, A. & Shaw, G. B. High-speed chemistry libraries: Assessment of drug-likeness. In Practice of Medicinal Chemistry 147–157 (2003).
Ertl, P., Rohde, B. & Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 43(20), 3714–3717 (2000).
Lee, S. K., Lee, I. H., Kim, H. J., Chang, G. S., Chung, J. E., No, K. T. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. In EuroQSAR 2002 Designing Drugs and Crop Protectants: processes, problems and solutions 2003 418–420 (2003).
Banerjee, P., Eckert, A. O., Schrey, A. K. & Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46(W1), W257–W263 (2018).
Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man—Fact or myth. Pharm. Res. 14(6), 763–766 (1997).
Ma, X.-L., Chen, C. & Yang, J. Predictive model of blood–brain barrier penetration of organic compounds. Acta Pharmacol. Sin. 26(4), 500–512 (2005).
Yamashita, S. et al. Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur. J. Pharm. Sci. 10(3), 195–204 (2000).
Yazdanian, M., Glynn, S. L., Wright, J. L. & Hawi, A. Correlating partitioning and Caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm. Res. 15(9), 1490 (1998).
Irvine, J. D. et al. MDCK (Madin–Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 88(1), 28–33 (1999).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
M. S.A. idea and supervision of the experimental work. Y.W. carried out the experimental work. H.A-H. supervision and she reviewed the writing. M. M. A. carried out the biological part. M.T. carried out the biological part and writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ayoup, M.S., Wahby, Y., Abdel-Hamid, H. et al. Structure optimization of new tumor-selective Passerini α-acyloxy carboxamides as Caspase-3/7 activators. Sci Rep 12, 22390 (2022). https://doi.org/10.1038/s41598-022-26469-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26469-4
- Springer Nature Limited