
Abstract
Sargassum is one of the most important brown algal genera that can be used as food and raw material for medicinal purpose, and has various beneficial effects. As the classification of Sargassum species is currently based on their morphological characteristics, organellar genome sequences of Sargassum would provide important information for accurate identification of species and developing species-specific markers. We sequenced the complete organellar genomes of six Sargassum species, including the first complete chloroplast genome sequences of S. fulvellum, S. serratifolium, S. macrocarpum, and S. siliquastrum, and the first complete mitochondrial genome sequences of S. fulvellum, S. serratifolium, and S. macrocarpum. The chloroplast genomes of the 6 Sargassum species contained 139 protein-coding genes (PCGs), and the mitochondrial genomes possessed 37 PCGs. A comparative study was performed between the newly sequenced organellar genomes and 44 other species belonging to class Phaeophyceae. Phylogenetic relationships using PCGs shared by Phaeophyceae species were constructed with IQ-TREE 2 using the maximum likelihood method. In addition, we developed real-time PCR markers based on SNPs to distinguish the 6 Sargassum species. Our results provide useful information for establishing phylogenetic relationships between brown algae.
Similar content being viewed by others

Introduction
Sargassum is a genus of brown algae (Phaeophyceae) that is distributed along the coastline of the lower intertidal zones, especially in northeast Asia. It forms marine ecosystems that provide food and habitat for diverse organisms in the sea1. Some species of Sargassum, such as S. fusiforme and S. horneri can be consumed and are raw materials for the development of medicine2,3, particularly in Asian countries. Recent studies have shown that, seaweed, including Sargassum have been known to have various beneficial compounds4,5,6; consequently, the seaweed market is constantly expanding, globally.
Sargassum species are distributed worldwide within intertidal and subtidal regions, forming dense submarine forests. Some species such as S. horneri can form harmful blooms known as “golden tides” and be spread on the ocean surface7,8.
The classification of brown algae, especially Sargassum, is based on the morphological characteristics of the thallus, stem, branches, and life-cycle types9,10. Unfortunately Sargassum species have limited morphological differences7,11 that can vary according to the environment, growth conditions, or developmental stages. Therefore, it is difficult to classify these species based on morphological characteristics alone.
Plastids and mitochondria are organelles that have evolved by endosymbiotic interactions with cyanobacteria and proteobacteria, respectively12. These organelles possess independent genomes; thus, the genetic information of these organelles is widely used as molecular markers to infer evolutionary relationships13,14. As next-generation sequencing (NGS) technologies have become cheaper and faster, a greater number of complete organellar genome sequences have been published. The Sargassum genus contains over 360 species14, but only a few organellar genomes of the genus have been completely sequenced despite the development of sequencing technologies.
In this study, we examined 6 Sargassum species (S. confusum, S. fulvellum, S. horneri, S. macrocarpum, S. serratifolium, and S. siliquastrum). These 6 species are used as raw materials for food, medicine, and therapeutic products in Korea and their economic value is growing15. Unfortunately, raw and processed materials of different brown algae species are typically mixed during the manufacturing process, due to the difficulties in morphological identification, which can lead to deterioration in product quality. Various Sargassum species have been taxonomically misclassified before (i.e., S. macrocarpum and S. serratifolium were thought to be the same species in Korea and Japan in the 1980s)16. In addition, many brown macroalgae within Sargassum are invasive, and there is a need for accurate identification to determine the risk. Since the classification of Sargassum species has recently improved with the use of NGS platforms for sequencing organellar genomes, the precise identification of the species is possible. This will prevent mixing and contamination by other cheap brown algae, thus improving consumers’ health and safeguarding their rights.
Here, we sequenced, assembled, and annotated the organellar genomes of 6 Sargassum species, including the first complete genome sequence of both the chloroplast and mitochondria of S. fulvellum, S. macrocarpum, and S. serratifolium, and the chloroplast of S. siliquastrum using the Illumina sequencing platform. Based on the genome structures and gene information of these organellar genomes, we conducted comparative studies and reconstructed their phylogenetic relationships with other Phaeophyceae species. Furthermore, we developed real-time PCR markers based on SNP that can distinguish each of the 6 species to prevent contamination of Sargassum in processed foods with other brown algae. As the demand of Sargassum for human consumption and as a raw material for medicine is growing17,18, it is important to distinguish each species using quick and easy methods. We demonstrated that sequence variations in the chloroplast and mitochondrial genomes of Sargassum species can distinguish between many target species. The obtained results provide valuable information for promoting the phylogenetics of brown algae and for developing molecular markers.
Materials and methods
Sargassum samples collection
Algal thalli were collected of S. confusum from Chujado island, Jeju, Republic of Korea (33°57′ N, 126°17′ E), S. fulvellum and S. horneri from Jeopdo island, Jeollanamdo, Republic of Korea (34°22′ N, 126°18′ E), S. macrocarpum from Geumodo island, Jeollanamdo, Republic of Korea (34°31′ N, 127°46′ E), S. serratifolium from Seongsan, Jeju, Republic of Korea (33°27′ N, 126°55′ E), and S. siliquastrum from Sikdo, Jeollabukdo, Republic of Korea (35°37′ N, 126°17′ E). All the species samples were collected and identified according to the morphological characteristics (Fig. S1) by professional experts (Sung-Je Choi & Tae-Ho Seo) in Korean algae. Permission to collect samples was granted by the Ministry of Oceans and Fisheries, Republic of Korea. Our studies were complied with local and national regulations and following Kangwon National University (Chuncheon, Republic of Korea) and the Ministry of Oceans and Fisheries (Sejong, Republic of Korea) regulations.
DNA isolation and organellar genome sequencing, assembly, and annotation
The total Sargassum genomic DNA was extracted from the blades using the Exgene™ Plant SV Kit (GeneAll®, Seoul, Korea) according to the manufacturer’s instructions. Paired-end libraries of the samples were constructed and subsequently sequenced on the Illumina NovaSeq 6000 platform according to the manufacturer’s instructions. Raw reads were quality- and adapter-trimmed for GetOrganelle19 and only adapter-trimmed for NOVOPlasty20, for de novo assembly using Trimmomatic21. The organellar genomes were reconstructed and manually corrected using assembled sequences, by comparing the two assemblers. Annotation of the organellar genome was performed using GeSeq22 and Geneious Prime software23 with a custom reference GenBank database of 44 Phaeophyceae species (Table S1). The open reading frames of protein-coding genes (PCGs) were identified by manually comparing reference GenBank data and ‘Find ORFs’ function in Geneious software. tRNA genes were predicted using tRNAscan-SE24 v2.0.7 annotator in GeSeq with default parameters. Circular organellar genome maps of Sargassum were drawn using OGDRAW software25.
Referential genome modification
We selected 44 Phaeophyceae species (two accessions for Lessonia spicata organellar genomes) (Table S1) for which chloroplast and mitochondrial genome data were available. All chloroplast and mitochondrial genomes were modified to start with the ycf37 gene and rnl rRNA gene, respectively, for convenience in comparative analysis. Additionally, as some reference genomes possessed old gene names in their annotation, we provisionally re-annotated them using the 6 newly constructed Sargassum organellar genomes in this study to standardize gene names manually from old to new, (e.g., ycf3 to pafI, ycf40 to thiS) for comparative analysis and identification of shared genes among Phaeophyceae species.
Repeat sequences analysis
Simple sequence repeats (SSRs) were detected using MISA26 with the parameters set at 10 > for mono-, > 5 for di- and tri-, > 3 for tetra-, penta-, and hexa-nucleotide SSRs. Dispersed repeats were identified using REPuter27 with parameters of Hamming distance 3, sequence identity ≥ 90%, and minimum repeat size ≥ 30 bp.
Comparative analysis
The progressiveMauve28 alignment tool was used to analyze gene arrangement. The mVISTA software29 was used to analyze the divergences among the assembled Sargassum chloroplast and mitochondrial genomes, and the selected reference Phaeophyceae species in the Shuffle-LAGAN mode.
Codon usage analysis
The codon usage bias was analyzed for all protein-coding genes (PCGs) using the CodonW program (http://codonw.sourceforge.net/), and the RSCU (relative synonymous codon usage) was visualized with the R script provided in Phylosuite30.
Phylogenetic analysis
PCG sequences shared by 44 Phaeophyceae organellar genomes and six newly constructed Sargassum organellar genomes were used for phylogenetic analysis. Each of the 132 and 33 shared genes in the chloroplasts and mitochondria, respectively, were aligned using MAFFT31, and the ambiguously aligned regions in each alignment were deleted by trimAI32. The trimmed alignments of the chloroplast and mitochondrial genes were concatenated. We performed a maximum likelihood (ML) analysis using IQ-TREE 233 under the GTR + F + I + G4 model, which was the best-fit model chosen according to Bayesian Information Criterion (BIC) by the implemented program ModelFinder in IQ-TREE 2, with 1,000 bootstrap and 1,000 replicates of the SH-aLRT test. The procedures from alignment to ML analysis were performed using the implemented and plugin functions of Phylosuite.
Marker development for the identification of each of the 6 Sargassum species
The PCG sequences of the organellar genomes of the 6 Sargassum species were aligned using ClustalW. Primer pairs were designed with aligned PCG sequences based on SNPs using Beacon Designer™ (PRIMER Biosoft, Palo Alto, CA, USA). Quantitative real-time PCR for amplification was performed at 55–62 °C (depending on primer pairs) using the AccuPower® 2X GreenStar qPCR Master Mix (Bioneer, Daejeon, Korea) in a Quantstudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The size of the species-specific amplified PCR products was confirmed using 1.5% agarose gel electrophoresis (Fig. 7).
Results
Characterization of organellar genomes of Sargassum species
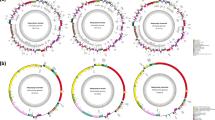
The DNA libraries were sequenced on Illumina platform, and we obtained average 25.3 Gb (from 24.2 Gb of S. horneri to 29.2 Gb of S. macrocarpum) of paired-end sequencing data, comprising from 67,195,726 (S. serratifolium) to 81,345,526 (S. macrocarpum) raw reads. Read coverage of each assembly varied from 466 to 1861X in chloroplast genomes and from 67 to 4540X in mitochondrial genomes. Among the 6 Sargassum species in this study, the organellar genomes of S. fulvellum, S. serratifolium, and S. macrocarpum were the first to be completely sequenced, assembled, and annotated organellar genomes in both chloroplast and mitochondria. Here we provided the organellar genome maps of S. fulvellum as Fig. 1 according to the alphabetical order of species name among the three species, and the organellar genome maps of the other 5 species were provided as Fig. S2 (chloroplast) and Fig. S3 (mitochondria). The chloroplast genomes of the 6 Sargassum species were assembled into a typical quadripartite circular structure containing large single copy (LSC) and small single copy (SSC) regions separated by 2 copies of an inverted repeat (IR) region, with sizes ranging from 124,175 to 124,517 bp (Fig. 1a, Fig. S2, and Table 1). The length of the LSC ranged from 73,403 bp (S. horneri) to 73,606 bp (S. serratifolium), while those of SSC and IRs were approximately 40,000 bp (39,900–40,175 bp) and 5400 bp (5373–5446 bp), respectively, with the exception of S. fulvellum (43,992 bp for SSC and 3413 bp for IRs). The GC content ranged from 30.3% to 30.6%. All 6 Sargassum species’ chloroplast genomes encoded 139 unique PCGs and 2 copies of each 3 rRNA genes. The chloroplast genomes encoded 28 tRNA genes (excluding S. horneri which had 29 tRNA genes), including 23 or 24 unique tRNA genes (Table 1). The IR regions and SC (single copy) region boundaries were analyzed, and high similarity was found among the six Sargassum species, with the exception of the SSC–IRa boundary of S. fulvellum (Fig. 2). The comparative location of the gene rpl21 (overlapped on the SSC–IRa boundary of the other 5 species) was almost the same in all six Sargassum species. However, the SSC of S. fulvellum was approximately 4 kbp longer than that of other species, and consequently, the rpl21 gene was located in the SSC region of S. fulvellum. Total chloroplast genes were classified according to their functions, including photosynthesis-related genes, genetic systems, RNA genes, genes with other functions, and those with unknown functions (Table S2).

The organellar genome map of Sargassum fulvellum. The genes inside and outside of the circle are transcribed in clockwise and counterclockwise directions, respectively. The grey circle on the inside shows the GC content. The colored boxes represent gene functional groups. The thick lines in chloroplast genome map indicate the inverted repeats (IRa and IRb), which separate the genome into small (SSC) and large (LSC) single-copy regions. The chloroplast and mitochondrial genome maps of the other 5 species (S. confusum, S. horneri, S. macrocarpum, S. serratifolium, and S. siliquastrum) are shown in Fig. S2 and Fig. S3, respectively. Circular organellar genome maps were drawn using OGDRAW version 1.3.1 (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).

Comparison of the borders of large single copy (LSC), small single copy (SSC) and inverted repeat (IR) regions among the six Sargassum chloroplast genomes.
The lengths of the mitochondrial genomes ranged from 34,620 to 34,793 bp, with total GC contents ranging from 36.2% to 36.6% (Fig. 1b, Fig. S3, and Table 1). All mitochondrial genomes contained 37 unique PCGs and 3 rRNA genes. Twenty-five tRNA genes were encoded within all 6 Sargassum mitochondrial genomes, with 23 unique tRNA genes (except S. confusum with 24 unique tRNA genes) (Table 1). The mitochondrial genes involved in oxidative phosphorylation, genetic systems, RNA genes, genes with other functions, and genes whose functions are unknown are listed in Table S2.
Repeat and simple sequence repeat (SSR) analysis
In total, 39 (S. confusum), 49 (S. fulvellum), and 43 (S. horneri, S. macrocarpum, S. serratifolium, and S. siliquastrum) SSRs, respectively were detected in Sargassum chloroplast genomes. Among these, the most abundant SSRs were mononucleotide repeats (Table 2). In addition, A/T mono-nucleotide repeat units existed in all 6 Sargassum chloroplast genomes, but C/G repeats existed only in S. macrocarpum and S. serratifolium. The di-nucleotide repeat unit of AG/CT was only found in S. fulvellum, whereas the AT/AT repeat unit was found in more than 10 (10 to 14 repeats) repeats in all six species. The chloroplast genomes of the six species had under 10 repeats of tri-, tetra-, penta-, and hexa-nucleotide repeat units. Interestingly, the repeat units containing G or C occurred only once in all SSR types, with the exception of 2 repeats of AATG/ATTC tetra-nucleotide repeats in S. fulvellum. Dispersed long repeats of forward, reverse, complement, and palindrome were also detected in the chloroplast genomes (Fig. 3, the x-axis is the repeat length and the y-axis is the copy numbers). In Sargassum species, the most common repeat type was palindromic repeats, which accounted for 82% of the total repeats, followed by forward repeats (13%). Palindromic repeats with a range of 30–39 bp were the most abundant repeats. In particular, 12 copies of more than 100 bp palindromic repeat were abundant in S. fulvellum but only one to three copies were found in the other five species. Only six and three copies were found in the reverse and complement repeats, respectively over the six Sargassum chloroplast genomes.

The longer repeat analysis of chloroplast genomes of the six Sargassum species. Frequency of (a) forward, (b) reverse, (c) complete, (d) palindromic repeats. The x-axis: repeat length; the y-axis: copy number.
Mono-nucleotide SSRs with A/T repeats were found in Sargassum mitochondrial genomes (10–11 copies) and only one C/G repeat was found in S. siliquastrum (Table 2). There were no tri- and penta-nucleotide SSRs in any of the 6 Sargassum mitochondrial genomes. S. confusum and S. siliquastrum had one repeat of each unit of tetra- and hexa-nucleotide SSRs (AAAT/ATTT and AAAGGG/CCCTTT, respectively). S. fulvellum had one repeat of AC/GT, AAAC/GTTT, AAAT/ATTT, and AAAGGG/CCCTTT SSRs, and S. horneri had one repeat of tetra-nucleotide SSR (AAAT/ATTT). S. macrocarpum and S. serratifolium had one repeat of two SSR units of di-nucleotide (AC/GT and CG/CG), one tetra-nucleotide (AAAT/ATTT), and one hexa-nucleotide (AAAGGG/CCCTTT). In the mitochondrial genomes, REPuter detected only 1 forward repeat with a length of 34 bp in S. serratifolium (data not shown).
Codon usage analysis
In the Sargassum chloroplast genomes, PCGs consisted of 31,778 (S. horneri) to 31,814 (S. confusum and S. siliquastrum) codons. The total GC content (GC, 0.31 to 0.312) with GC at the first (GC1, 0.421 to 0.423), second (GC2, 0.345 to 0.346), and third (GC3, 0.164 to 0.168) codon position, respectively, were calculated (Supplementary Table S3). All codons with RSCU > 1 end with A and U in the chloroplast genomes of the six species (Fig. 4a). The results for the six Sargassum species showed that the chloroplast genomes tend to have A or U bases at the third codon position. G or C was biased toward a lower nucleotide frequency than A or U at the third codon position, indicating that the mutation towards A + U is a strong driving force for the chloroplast genome.

RSCU of organellar genomes of six Sargassum species. (a) Chloroplasts, (b) mitochondrial genomes.
In the Sargassum mitochondrial genomes, the PCGs consisted of 8,983 (S. horneri) to 8,994 (S. confusum and S. fulvellum) codons in the mitochondria. The total GC content (GC, 0.353–0.359); GC content at the first (GC1, 0.414–0.421), second (GC2, 0.365–0.368), and third codon position (GC3, 0.279–0.289), respectively were calculated (Table S3). In the mitochondrial genomes, some codons with RSCU > 1 end with G (UUG in 6 Sargassum species) (Fig. 4b). However, all mitochondrial genomes tend to use A or U bases at the third codon position as chloroplast genomes.
Comparative analysis of the Sargassum organellar genomes
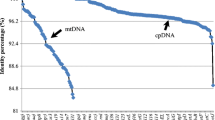
The similarity percentage between organellar genome sequences of six Sargassum species was calculated using MAFFT. The sequence similarities were ranged from 93.36% (between S. fulvellum and S. horneri) to 99.95% (between S. macrocarpum and S. serratifolium) in chloroplast genomes, and from 87.91% (between S. horneri and S. siliquastrum) to 99.81% (between S. macrocarpum and S. serratifolium) in mitochondrial genomes (Table 3).
We conducted a co-linear gene order analysis in the 44 Phaeophyceae reference chloroplast genomes deposited in the NCBI database (Fig. S4). The results showed that there were four gene order patterns in Phaeophyceae according to their taxonomic order, except Chorda asiatica with some inversion in the Laminariales gene order pattern. Furthermore, small variations in gene order were observed according to the taxonomic family. The analysis in mVISTA was conducted to identify the genome divergence of Sargassum species and six additional species from Phaeophyceae class, with S. horneri (NC_029856) as an alignment reference (Fig. 5). As illustrated in Fig. 5, the gene-coding regions (blue colored regions) and rRNAs (rns, rrn5, and rnl in skyblue colored regions) of all six Sargassum species had highly similar sequences, whereas tRNAs (the rest of skyblue colored regions) and spacer regions (red colored regions) showed large variations.

Alignment for sequence similarity among the 12 Phaeophyceae chloroplast genomes. Chloroplast genome of S. horneri (NC_02856) was used as the reference. Untranslated, conserved non-coding and coding regions were colored by skyblue, red, and blue, respectively.
The mitochondrial genomes of Phaeophyceae class species showed fewer or no gene rearrangements than the chloroplast genomes (data not shown). However, the mVISTA results showed that the sequences of the Phaeophyceae class, including Sargassum species, were more varied than the sequences of chloroplasts (Fig. S5).
Phylogenetic analysis
For the phylogenic analysis, 50 Phaeophyceae organellar genome sequences, including six Sargassum species, were used with Dictyopteris divaricate as an outgroup. The phylogenetic tree was constructed based on concatenated nucleotide sequences of 132 chloroplast PCGs (orf501, petL, rbcR, rpl32, ycf34, ycf41, and ycf54 were excluded) and 33 mitochondrial PCGs (atp8, orf39, orf129, and rps11 were excluded), shared by 44 Phaeophyceae species and 6 Sargassum species sequenced in this study. According to the phylogenetic analysis, all the species were classified as taxonomic orders matching the Ectocarpales, Fucales, Laminariales, and Dictyotales for outgroups in both trees based on chloroplasts and mitochondria (Fig. 6). However, some species belonging to the Laminariaceae and Lessoniaceae families showed mixed branch clusters. 6 species belonging to the Laminariaceae family (A. bifidus, S. subsessilis, S. japonica, S. latissimi, M. integrifolia, and P. palmaeformis) were more closely clustered with 2 species belonging to the Lessoniaceae family (E. arborea and E. radicosa) than with the other 4 Laminaria species (L. ephemera, L. digitata, L. rodriguezii, and L. solidungula) of the Laminariaceae family. The 4 Laminaria species were more closely clustered with the 2 Lessonia species (L. spicata and L. flavicans) of the Lessoniaceae family. Mixed clustering was found in phylogenetic trees constructed from both chloroplast and mitochondrial PCGs.

Phylogenetic analysis of 50 Phaeophyceae species including the 44 species deposited in NCBI and the six newly sequenced Sargassum genomes using Maximum Likelihood (ML) methods based on the concatenated shared PCG sequences of (a) chloroplast and (b) mitochondria. Yellow colored species are newly sequenced species. The numbers at internal nodes indicated maximum likelihood (ML) bootstrap values.
Markers for identifying 6 Sargassum species
Although the SSRs and long repeats are dispersed in all organellar genomes of Sargassum species, the development of species-specific markers would be difficult because of their highly conserved border sequences and high AT content. Therefore, we focused on SNPs in PCG sequences for the development of species-specific markers. Based on the PCG sequence alignments, we developed 13 species-specific markers based on SNPs of chloroplast (five for S. horneri specific, five for S. confusum specific, and three for S. siliquastrum specific markers) and 14 markers based on SNPs of mitochondrial genomes (five for S. fulvellum and two each for S. confusum, S. horneri, S. serratifolium, and S. siliquastrum, and one for S. macrocarpum) (Fig. 7) with cut-off Ct ranging from 22 to 28 (Table S4).

Development of quantitative real-time PCR based species-specific markers using SNPs of chloroplast and mitochondrial genomes. Target species are (a) S. confusum, (b) S. fulvellum, (c) S. horneri, (d) S. macrocarpum, (e) S. serratifolium, and (f) S. siliquastrum. Lane 1: S. fulvellum; 2: S. horneri; 3: S. serratifolium; 4: S. confusum; 5: S. siliquastrum; 6: S. macrocarpum, M: DNA ladder. Full-length gels are presented in Supplementary Fig. S6.
Discussion
Comparative analysis of the complete chloroplast genomes of the 6 Sargassum species showed high conservation in their structure, genome size, GC content, gene composition, and gene order, which is consistent with previously published data34. The chloroplast genomes of the 6 sequenced species had 139 unique PCGs and 3 unique rRNA genes. The notable difference among the 6 Sargassum chloroplast genomes was the size of inverted repeats in S. fulvellum, which was 3,413 bp (the other 5 species had inverted repeat regions of size ranging from 5,373 bp to 5,446 bp). The mitochondrial genomes of the Sargassum species studied shared 37 unique PCGs and three unique rRNA genes, and had identical gene orders. The mean sequence similarities were 96.05% in chloroplast genomes and 92.25% in mitochondrial genomes. The conservation among Sargassum genus is supported by the phylogenic analysis which showed that all members of Sargassum were clustered into one clade (Fucales) according to their taxonomic order. The consistent clustering of the clade suggests that there has been no rapid evolution of the Sargassum genus with respect to organellar genomes. Especially, S. macrocarpum and S. serratifolium showed extremely high similarity in sequences and were sister-species phylogenetically in both chloroplast and mitochondrial genomes.
Repeat sequences, including SSRs and long repeats, have been widely used in taxonomic analysis and phylogenetic relationships; and are also used as valuable markers in comparative genomics35,36,37. In this study, we identified several types of repeat sequences in the chloroplast genomes. The most abundant repeats were mono-nucleotide SSRs of A or T, followed by di-nucleotide SSRs. Unlike chloroplast genomes, limited repeats in the mitochondrial genomes were found, even though a number of mono-nucleotide SSRs in mitochondrial genomes were found, and only a few long repeats were detected. In addition, most repeat sequences contained extremely high AT content in both the chloroplast and mitochondrial genomes.
Codon usage is known to play an important role in gene expression levels and translation38,39. It provides valuable means for evolution by selection and mutation at the molecular level40. In the organellar genomes of the 6 Sargassum species, codon usage bias and RSCU based on the PCG sequences were calculated. In chloroplast genomes, AAA is the most common synonymous codon across species. Furthermore, UUA, GUU, and AGA in leucine, valine, and arginine, respectively, showed higher RSCU values (> 2.0), indicating that these synonymous codons were used more frequently than expected. The patterns of synonymous codon usage were more stable in Sargassum chloroplast genome, rather than in the mitochondrial genomes. Compared to other Phaeophyceae species, variations in synonymous usage patterns could be detected in both chloroplast and mitochondrial genomes. These specific patterns can be used to investigate the evolution of the brown algal family in future studies.
In this study, to further understand the Sargassum specific chloroplast genome characteristics, the complete chloroplast genomes of 44 Phaeophyceae species were aligned using the progressiveMauve program. The alignments revealed five patterns of gene rearrangements in the chloroplast genomes of Phaeophyceae species, according to taxonomic orders. Overall, comparative genomic analysis revealed that the Sargassum chloroplast genomes were relatively more conserved than those of the other species. In particular, the gene coding regions were highly conserved within the Sargassum species. The mitochondrial genomes of Phaeophyceae species used in the comparative study showed less variation in gene rearrangement than chloroplast genomes, whereas the sequence divergence in coding regions was relatively higher than that in the chloroplast genomes. Phylogenetic analysis also showed results similar to those of the comparative analysis, especially for gene rearrangements, except for Chorda asiatica, wherein the gene order patterns were clustered according to the taxonomic orders and even some families. However, our results showed that some species in the Laminariaceae and Lessoniaceae families are mixed in phylogenetic clustering. Previous reports have classified the species based on their morphological characteristics and phylogenetic analysis with a few marker genes41. In this study, we used 165 PCGs (132 chloroplast PCGs and 32 mitochondrial PCGs) shared in 50 species to reconstruct the phylogenetic tree, but we did not consider their morphological characteristics or other genetic factors. To verify their classifications, further studies should be conducted on the order Laminariales.
The results of the genome structures, codon usage analysis and comparative studies indicated that Sargassum species had highly conserved organelle genome sequences. Although, the repeat sequences showed some variation within the species, they showed high AT content, and these results were problematic for the development of species-specific molecular markers. Consequently, we focused on the SNPs in PCG sequences within the 6 Sargassum species, and developed 13 chloroplast and 14 mitochondrial genome based species-specific markers. As the genome sequence divergence results by mVISTA showed that mitochondrial genomes had more variations in their genome sequences, the development of mitochondrial markers was easier and more specific to the target species than the development of chloroplast markers. As the seaweed market is growing owing to its potential value, identifying precise target species has become a very important and sensitive issue, especially in Asia. The developed PCR markers are fast, easy, and specific for detecting target species. The Sargassum species are difficult to distinguish because of the similarities in both genetic and morphological characteristics. Additionally, many Sargassum species are the invasive species, and it makes the identification of accurate species more difficult. The species-specific patterns of genomic characteristics and molecular markers developed in this study can be used as valuable methods to define a target Sargassum species, and thus protecting consumers’ health and rights.
Data availability
The complete organelle sequences generated and analyzed during the current study are uploaded in NCBI GenBank (ON660588 and ON675439–ON675449).
References
Godoy, E. A. S. & Coutinho, R. Can artificial beds of plastic mimics compensate for seasonal absence of natural beds of Sargassum furcatum?. ICES J. Mar. Sci. 59, S111–S115 (2002).
Wang, S. et al. First draft genome assembly of the seaweed Sargassum fusiforme. Front. Genet. 11, 590065 (2020).
Cui, Y., Liu, T., Wang, X., Qu, J. & Jia, X. The complete chloroplast genome of Sargassum horneri and its phylogenetic analysis. Mitochond. DNA B Resour. 4, 3312–3313 (2019).
Yende, S. R., Harle, U. N. & Chaugule, B. B. Therapeutic potential and health benefits of Sargassum species. Phcog. Rev. 8, 1–7 (2014).
Liu, L., Heinrich, M., Myers, S. & Dworjanyn, S. A. Towards a better understanding of medicinal uses of the brown seaweed Sargassum in Traditional Chinese Medicine: A phytochemical and pharmacological review. J. Ethnopharmacol. 142, 591–619 (2012).
Lim, S. et al. Evaluation of antioxidant activities of various solvent extract from Sargassum serratifolium and its major antioxidant components. Food. Chem. 278, 178–184 (2019).
Mattio, L. & Payri, C. E. 190 Years of Sargassum taxonomy, facing the advent of DNA phylogenies. Bot. Rev. 77, 31–70 (2011).
Huang, C. et al. Molecular analysis of Sargassum from the Northern China seas. Phytotaxa 319, 71–83 (2017).
Bruno de Sousa, C. et al. Improved phylogeny of brown algae Cystoseira (Fucales) from the Atlantic-Mediterranean region based on mitochondrial sequences. PLoS ONE 14, e0210143 (2019).
Bringloe, T. T. et al. Phylogeny and evolution of the brown algae. CRC. Crit. Rev. Plant Sci. 39, 281–321 (2020).
Cheang, C. C., Chu, K. H. & Ang, Put O. Morphological and genetic variation in the populations of Sargassum hemiphylum (Phaeophyceae) in the Northwestern pacific(1). J. Phycol. 44, 855–865 (2008).
Smith, D. Updating our view of organelle genome nucleotide landscape. Front. Genet. 3, 00175 (2012).
Cai, C. et al. Screening and verification of extranuclear genetic markers in green tide algae from the Yellow Sea. PLoS ONE 16, e0250968 (2021).
Liu, F., Pan, J., Zhang, Z. & Moejes, F. W. Organelle genomes of Sargassum confusum (Fucales, Phaeophyceae): mtDNA vs cpDNA. J. Appl. Phycol. 30, 2715–2722 (2018).
Hwang, E. K. & Park, C. S. Seaweed cultivation and utilization of Korea. Algae 35, 107–121 (2020).
Ok, J. H. & Lee, I. K. Taxonomy of the genus Sargassum (Fucales, Phaeophyceae) from Korea II. subgenus Bactrophycus section Halochloa and Repentia. Algae 21, 393–4056 (2006).
Liu, J. et al. Therapeutic and nutraceutical potentials of a brown seaweed Sargassum fusiforme. Food Sci. Nutr. 8, 5195–5205 (2020).
Sanjeewa, K. K. A. et al. Bioactive potentials of sulfated polysaccharides isolated from brown seaweed Sargassum spp in related to human health applications: A review. Food Hydrocoll. 81, 200–208 (2018).
Jin, J. J. et al. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Tillich, M. et al. GeSeq: Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11 (2017).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14 (2019).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64 (2019).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147 (2010).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Zhang, D. et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Li, R. et al. The complete plastid genomes of seven Sargassaceae species and their phylogenetic analysis. Front. Plant Sci. 12, 747036 (2021).
Provan, J. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 9, 2183–2185 (2000).
Flannery, M. L. et al. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 113, 1221–1231 (2006).
Park, I., Yang, S., Choi, G., Kim, W. J. & Moon, B. C. The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum Subgenus Lycoctonum. Molecules 22, 22112012 (2017).
Zhipeng, Z. et al. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. U.S.A. 113, E6117–E6125 (2016).
Sharp, P. M. & Matassi, G. Codon usage and genome evolution. Curr. Opin. Genet. Dev. 4, 851–860 (1994).
Plotkin, J. B. & Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 (2011).
Silberfeld, T., Rousseau, F. & de Reviers, B. An updated classification of brown algae (Ochrophyta, Phaeophyceae). Cryptogam. Algol. 35, 117–156 (2014).
Acknowledgements
This research was supported by Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries, Korea (20210656).
Author information
Authors and Affiliations
Contributions
Y.J.L. analyzed the data; Y.J.L. and C.S.J. wrote the manuscript; Y.D.K., Y.R.U., and Y.M.K. designed and performed experiments; T.-H. S. and S.C. collected samples. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, Y.J., Kim, Y.D., Uh, Y.R. et al. Complete organellar genomes of six Sargassum species and development of species-specific markers. Sci Rep 12, 20981 (2022). https://doi.org/10.1038/s41598-022-25443-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25443-4
- Springer Nature Limited