
Abstract
Cardiovascular outcomes trials (CVOTs) with novel drugs to treat type 2 diabetes have uniformly chosen the composite “major adverse cardiovascular events (MACE)” as their primary endpoint, but they also report hazard ratios for individual cardiovascular outcomes (myocardial infarction, stroke, cardiovascular death, all-cause death, hospitalization for heart failure). We wanted to scrutinize the power to identify significant differences with respect to individual as compared to composite outcomes. We estimated post hoc the statistical power to detect significant differences of 10–25% for published studies, comparing the proportions of patients with an event (two-sided log-rank tests). For MACE, the power to detect a 15% difference ranged from 82.3 to 100.0% for larger trials, but was only 69.1 and 50.5 for smaller, preliminary trials (SUSTAIN-6 and PIONEER-6). For individual endpoints, the power, as a rule, was substantially lower. In conclusion, cardiovascular outcomes trials had appropriate power to detect significant reductions in hazard ratios with respect to the primary endpoint, but not for individual cardiovascular outcomes. This was particularly the case for small, preliminary studies. Our results call for caution when comparing results regarding individual endpoints between CVOTs, if the aim is to identify heterogeneity within or between medication classes.
Similar content being viewed by others


Introduction
Cardiovascular outcomes trials employing novel diabetes drugs (e.g., SGLT-2 inhibitors1,2,3,4, DPP-4 inhibitors5,6,7,8, and GLP-1 receptor agonists9,10,11,12,13,14,15,16) have been published between 2013 and today. These studies have established SGLT-2 inhibitors and GLP-1 receptor agonists as the two medication classes for the treatment of type 2 diabetes, which do not only provide glycaemic control and body weight reductions without provoking hypoglycaemic episodes, but also show a potential for reducing the risk for a composite endpoint composed of cardiovascular events like acute myocardial infarction, stroke, cardiovascular death (major adverse cardiovascular events or MACE) in the majority of studies1,2,3,10,11,13,14,16. In addition, SGLT-2 inhibitors (SGLT-2 Is) also reduce the vents of hospitalization for congestive heart failure1,2,3,4, and SGLT-2 Is, more than GLP-1 RAs, prevent renal endpoints2,3,4,11,16,17,18,19,20.
While meta-analyses have confirmed these general patterns of effects for SGLT-2 inhibitors 21,22,23 and GLP-1 RAs24, and, thus, have established “class effects”, on first sight, i.e., in the absence of head-to-head trials, some heterogeneity is obvious for results obtained with various SGLT-2 Is (effects on MACE reduction vs. placebo significant1,2,3 or not4), DPP-4 inhibitors (worsening of the risk for hospitalization because of congestive heart failure with saxagliptin5,25, and, as a trend, alogliptin6,26 but not with sitagliptin7 or linagliptin8) and, in particular, for GLP-1 RAs (not even a trend for improved MACE outcomes for lixisenatide9, highly significant reductions in MACE events with liraglutide10, semaglutide s.c.11, albiglutide13, dulaglutide14, and efpeglenatide16). The latter findings are well compatible with significant differences regarding pharmacokinetic behaviour and the selection of dosages for the obviously heterogeneous class of GLP-1 RAs27,28.
Current guidelines and treatment recommendations suggest to consider medications, which can provide evidence for cardiovascular benefits for those at risk for such events29,30,31. This concerns subjects with pre-existing atherosclerotic cardiovascular disease as well as patients with chronic kidney failure (typically associated with a high cardiovascular risk), and those with congestive heart failure29,30,31. Following this recommendation should prompt a comparison of reported cardiovascular outcomes between trials reporting beneficial effects in this respect, i.e., within the classes of SGLT-2 Is and GLP-1 RAs, based on the heterogeneity of results within one class of glucose-lowering medications (Fig. 1). A superficial inspection of such data may suggest that albiglutide is particularly suited to reduce acute myocardial infarction events13, while semaglutide (for once-weekly subcutaneous injection)11 or dulaglutide14,32 are particularly suited to prevent stroke events. Along the same lines, liraglutide10 and oral semaglutide15 may appear to be the drugs of choice to prevent cardiovascular or all-cause death. We have to be aware that such comparisons are indirect based on the general design of such trials as placebo-controlled trials on a background of the standard of care. One important prerequisite for meaningful comparisons is a sufficient power provided by reported trials to detect significant differences between active and placebo treatment. It is the purpose of the present analysis to provide post hoc estimates of the power to detect differences between 10 and 25°% (corresponding to the magnitude of relative changes observed in relevant cardiovascular outcomes trials1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16). Our estimates will provide important background information for the interpretation of such trials, in particular regarding the comparison between different classes (e.g., SGLT-2 Is and GLP-1 RAs) or within these classes, in order to identify medications that will best help individual patients to address their cardiovascular risk.

Hazard ratios (± 95% confidence intervals) for major cardiovascular adverse events (MACE) as well as its components ([non-fatal] myocardial infarction and stroke, cardiovascular death) and all-cause death and hospitalization for congestive heart failure reported in cardiovascular outcomes studies comparing novel glucose-lowering medications with placebo treatment (both on a background of standard of care) in patients with type 2 diabetes. Asterisks indicate a significant difference to placebo treatment; †: p = 0.06; ‡: This nominally significant difference cannot claim significance due to hierarchical testing (difference in MACE not significant).
Results
Identification of suitable publications
Sixteen studies were identified as suitable for the present analysis1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16). Four studies each reported results from cardiovascular outcomes trials with SGLT-2 Is1,2,3,4 or DPP-4 Is5,6,7,8, respectively. Eight studies concerned treatment with various GLP-1 RAs9,10,11,12,13,14,15,16). Study and patient characteristics at baseline are compiled as Supplementary Tables 1 and 2, respectively. Table 1 compiles the most relevant information on study size (number of patients participating, study duration, and overall patient years of observation) and event numbers and the proportion of patients reporting a MACE event. It is obvious, that two studies (SUSTAIN-611 and PIONEER-615, examining semaglutide s.c. once weekly, and oral semaglutide administered daily, respectively) are much smaller in size (patient numbers, observation period, patient years of observation) as compared to all other studies (however, with considerable heterogeneity within the latter category). The reason is that the study objectives for SUSTAIN-6 and PIONEER-6 were different: Both were designed to provide preliminary evidence for safety pre-approval (with the necessity for additional post-approval studies in mind). Since the criteria for preliminary cardiovascular safety are more relaxed (hazard ratio for the MACE composite endpoint with upper 95% confidence interval < 1.80) than for the definite proof of cardiovascular safety (hazard ratio for the MACE composite endpoint with upper 95% confidence interval < 1.30) according to guidance by the food and drug administration of the United States (FDA) (https://www.fda.gov/media/71297/download), these criteria can be satisfied with fewer MACE events, allowing for fewer patients and a shorter observation period.
Power to detect differences in MACE between active drug and placebo treatment with the number of endpoints observed in individual studies
Significant differences in the number of MACE events between active drug and placebo treatment were detected in all but one studies with SGLT-2 Is (exception: VERTIS-CV trial using ertugliflozin), and in all but one publication on GLP-1 RAs (exception: ELIXA, examining lixisenatide effects), and in none of the DPP-4 inhibitor trials (Fig. 1). In those trials reporting significant reductions in the risk for MACE (Fig. 1), the power to detect this difference (ranging from 13 to 27% reduction in MACE) was 68.1 (DECLARE-TIMI 58; dapagliflozin) to 99.7 (AMPLITUDE-O; efpeglenatide; Fig. 2). 2 out of 4 SGLT-2 inhibitor trials, none of the DPP-4 I trials, and 6 out of 8 GLP-1 RA trials had an estimated power of > 80% to detect significant differences between active drug and placebo treatment as they were described in those studies (Fig. 2). Trials not describing a significant difference between active drug and placebo treatment typically showed small effect sizes (Fig. 1) associated with a low post hoc power estimate (Fig. 2).

Post hoc power estimation of for cardiovascular outcomes concerning trials comparing novel glucose-lowering medications with placebo treatment (both on a background of standard of care) in patients with type 2 diabetes for major adverse cardiovascular outcomes (MACE; upper row of panels) and non-fatal myocardial infarction (lower row of panels). Left hand panels display the power calculated regarding the hazard ratio as reported in each individual study. The second through fourth columns of panels illustrate the power to detect differences by 10, 15, 20, or 25% versus the proportion of patients with events reported with placebo treatment with SGLT-2 inhibitors (second column of panels), DPP-4 inhibitors (third column of panels) and GLP-1 receptor agonists (fourth column of panels). The power estimates for study results indicating a negligible difference to placebo treatment (≤ 10%) are highlighted with a dashed rectangle, because small differences are typically associated with low power.
Power to detect differences in individual cardiovascular endpoints between active drug and placebo treatment with the number of endpoints observed in individual studies
A sufficient power (> 80%) to detect significant differences, as they occurred in individual trials between active drug and placebo treatment, with respect to (non-fatal) myocardial infarction was estimated in 2 out of 4 SGLT-2 I studies, in none of the DPP-4 I trials, and in 4 out of 8 GLP-1 RA studies (Fig. 2). The respective numbers concerning (non-fatal) stroke were 2 out of 4 SGLT- I studies, none out of the 4 DPP-4 I trials, and 3 out of 8 GLP-1 RA trials (Fig. 3). For cardiovascular death, this criterion was fulfilled in 1 SGLT-2 I trial and 4 GLP-1 RA studies (Fig. 3). Regarding all-cause death, the figures were 2 (out of 4 SGLT-2 I trials), 1 (out of 4 DPP-4 I studies), and 4 (out of 8 GLP-1 RA studies; Supplementary Fig. 1). All SGLT-2 inhibitor studies were sufficiently powered to detect significant differences in hospitalization for congestive heart failure. This was the case in only 1 (out of 4) and 2 (out of 8) studies reporting results on DPP-4 Is or GLP-1 RAs, respectively (Supplementary Fig. 1). Thus, for the individual CV endpoints, sufficient power was estimated for a much smaller number of trials as compared to the composite MACE endpoint. Accordingly, fewer comparisons were significant for these individual endpoints (Fig. 1).

Post hoc power estimation of for cardiovascular outcomes concerning trials comparing novel glucose-lowering medications with placebo treatment (both on a background of standard of care) in patients with type 2 diabetes for non-fatal stroke (upper row of panels) and cardiovascular death (lower row of panels). Left hand panels display the power calculated regarding the hazard ratio as reported in each individual study. The second through fourth columns of panels illustrate the power to detect differences by 10, 15, 20, or 25% versus the proportion of patients with events reported with placebo treatment with SGLT-2 inhibitors (second column of panels), DPP-4 inhibitors (third column of panels) and GLP-1 receptor agonists (fourth column of panels). The power estimates for study results indicating a negligible difference to placebo treatment (≤ 10%) are highlighted with a dashed rectangle, because small differences are typically associated with low power.
Estimates of the power to detect significant differences of pre-defined magnitude (10–25%) between active drug and placebo treatment
Generally speaking, as expected, the estimated power to detect significant differences between active drug and placebo treatment rose considerably with the target difference increasing from 10 to 25% (Figs. 2 and 3, Supplementary Fig. 1). As displayed in Fig. 2, the power to detect differences by > 15% between active drug and placebo treatment in MACE endpoints was high (> 80%) for all SGLT-2 I and DPP-4 I trials, and for 5 out of 8 GLP-1 RA studies (Fig. 2). Notably, the AMPLITUDE-O trial (efpeglenatide) and SUSTAIN-6 (semaglutide s.c.) as well as PIONEER-6 (oral semaglutide, both designed to provide preliminary proof of safety), had a low power even for their primary endpoint MACE, unless the target difference was higher than 20% between placebo and active drug treatment (Fig. 2).
For (non-fatal) myocardial infarction, all SGLT-2 I and DPP-4 I studies provided an estimated power > 80% to detect significances differences between active drug and placebo treatment amounting to > 15% (Fig. 2). This was also the case in 5 out of 8 GLP-1 RA trials. A lower power was estimated for the AMPLITUDE-O, SUSTAIN-6, and PIONEER-6 trials, which only had sufficient power to detect differences ≥ 20 (AMPLITUDE-O) or ≥ 25% (SUSTAIN-6 and PIONEER-6 trials, Fig. 2).
For (non-fatal) stroke, 3 out of 4 SGLT-2 I and 2 out of 4 DPP-4 I studies provided an estimated power > 80% to detect significances differences between active drug and placebo treatment amounting to > 15% (Fig. 3). This was also the case in 2 out of 8 GLP-1 RA trials (Fig. 3). The majority of GLP-1 RA trials (7 out of 8) was, however, powered to detect differences approaching 25% (Fig. 3).
For cardiovascular death, all SGLT-2 I and 3 out of 4 DPP-4 I studies provided an estimated power > 80% to detect significances differences between active drug and placebo treatment amounting to > 15%. This was also the case in 4 out of 8 GLP-1 RA trials (Fig. 3). All but one study (PIONEER-6) had the estimated power to detect differences ≥ 25% (Fig. 3).
Regarding all-cause death, all SGLT-2 I and DPP-4 I studies provided an estimated power > 80% to detect significances differences between active drug and placebo treatment amounting to > 15% (Supplementary Fig. 1). This was also the case in 5 out of 8 GLP-1 RA trials. A lower power was estimated for the AMPLITUDE-O, SUSTAIN-6, and PIONEER-6 trials, which only had 80% power to detect differences ≥ 20 (AMPLITUDE-O) or ≥ 25% (Supplementary Fig. 1), respectively.
For hospitalization because of congestive heart failure, 2 out of 4 SGLT-2 I and 3 out of 4 DPP-4 I studies provided an estimated power > 80% to detect significances differences between active drug and placebo treatment amounting to > 15% (Supplementary Fig. 1). This was also the case in 4 out of 8 GLP-1 RA trials (Supplementary Fig. 1). 5 out of 8 trials had sufficient power to detect a difference of ≥ 20%, 7 out of 8 studies provided a power of > 80% to detect a ≥ 25% difference, but one study (PIONEER-6) was not even powered to detect a difference of 25% (Supplementary Fig. 1).
Relationship of study size with confidence intervals for hazard ratios comparing active drug vs. placebo treatment
Since confidence intervals spanning the line of unity are pivotal in determining the significance of differences between active drug and placebo treatment, we assessed the association of the width of the confidence intervals and parameters representing study size (either overall patient years of observation or numbers of events). Such an analysis is shown in Supplementary Fig. 2 for major adverse cardiovascular events (MACE). Regression analysis indicates a highly significant association (Supplementary Fig. 2), thus confirming an important role of study size (patient number) and duration, and, as a consequence, patient years of observations and (MACE) event rates, as determinants of statistical power, i.e. a pivotal prerequisite for detecting significant differences between active drug and placebo treatment.
Discussion
Our analysis provides evidence that the cardiovascular outcomes trials examining novel diabetes medications (SGLT-2 Is1,2,3,4, DPP-4 Is5,6,7,8, and GLP-1 RAs9,10,11,12,13,14,15,16) were well powered to detect significant differences with respect to the primary endpoint (major adverse cardiovascular events) in the range that really occurred as the result of such clinical studies (15–25%; Fig. 2). However, for individual endpoints, this was not uniformly the case. For acute myocardial infarction, stroke, cardiovascular death, all-cause death, and hospitalization for congestive heart failure, the estimated power was lower than for the main (composite) endpoint, explained by the lower numbers of events in these categories (Table 1, Supplementary Table 3).
To judge the consequences of the relatively lower power to detect individual cardiovascular endpoints, the true differences as found on the trials have to be taken into account: Regarding MACE, 4 out of 8 GLP-1 RA trials described a reduction by 21–27% (PIONEER-615, HARMONY Outcomes13, SUSTAIN-611, and AMPLITUDE-O16). Similar effect sizes were not observed with DPP-4 Is or SGLT-2 Is (Fig. 2). Regarding acute myocardial infarction, the difference amounted to 22–26% for three (AMPLITUDE-O16, HARMONY Outcomes13, SUSTAIN-611) out of eight studies9,10,11,12,13,14,15,16 . For stroke, the largest differences were 24–39% for three (REWIND14, PIONEER-615, SUSTAIN-611) out of eight studies9,10,11,12,13,14,15,16). Regarding cardiovascular death, the largest differences were 22–51% in GLP-1 RA studies (LEADER10, AMPLITUDE-O16, and PIONEER-615), and the SGLT-2 I trial EMPA-REG Outcomes1). All-cause death was reduced by up to 22–49% (GLP-1 RAs: AMPLITUDE-O16 and PIONEER-615; SGLT-2 I: EMPA-REG Outcomes1). For hospitalization because of congestive heart failure, a consistent 27–35% reduction was seen with all SGLT-2 Is1,2,3,4, but also with efpeglenatide (by 39%; AMPLITUDE-O16). Of note, not even all these large differences were significant (due to small numbers of events and sample sizes; Table 1 and Supplementary Table 3). Thus, for the majority of study results concerning the individual endpoints of interest, the power was not sufficient to detect the magnitude of differences as the have been described with respect to acute myocardial infarction, stroke, cardiovascular death, all-cause death, and hospitalization for congestive heart failure in the majority of studies, even for medications belonging to classes which in principle have proven the potential to reduce cardiovascular endpoints selected for the present analysis. The latter can be concluded from meta-analyses of cardiovascular outcomes trials examining SGLT-2 Is and GLP-1 RAs, which found significant effects on all endpoints, composite or individual, which have been explored in the present analysis, with the exception of stroke in trials employing SGLT-2 inhibitors21,22,23,24.
Based on the findings of our present analysis, we do not believe that it is justified to compare the effectiveness in reducing individual cardiovascular endpoints for the treatment with various compounds belonging to one class of glucose-lowering medications (e.g., within GLP-1 RAs), or even between different classes (e.g., comparing SGLT-2 Is and GLP-1 RAs). The main reason is that the power has not been found sufficient to detect typical event reductions observed in such trials, which mainly range from 10 to 15% (Fig. 1).
This is not to say that the studies which we have analysed are ill-powered for the purpose that they were designed for, e.g. the provision of evidence sufficient to support the claim of non-inferiority (safety) and with a chance to detect superiority, if the difference was, e.g., ≥ 15%, depending on the hypothesis guiding sample size calculation. Differences are obvious between studies aiming at the final proof of safety according to FDA criteria, and trials designed to satisfy criteria suggesting preliminary safety for getting a novel diabetes medication approved, however, with the commitment to perform a definitive post-marketing cardiovascular trial. The latter applies mainly to the two semaglutide trials, SUSTAIN-611 (semaglutide s.c. once weekly) and PIONEER-615 (oral semaglutide for once-daily administration). As mentioned in the results section, these trials in many respects lack a power that is sufficient to conclude superiority regarding several endpoints. As a result, non-significant differences sometimes had to be concluded even in the presence of impressive effect sizes (e.g., MACE in PIONEER-615). Needless to say, a final determination of robustly estimated effect sizes is difficult under these circumstances. Similar considerations apply to AMPLITUDE-O (efpeglenatide) because of its relatively short duration16.
The results of cardiovascular outcomes trials show widely heterogeneous results based on the medication class employed, but also comparing results with glucose-lowering medications belonging to the same class (Figs. 1). This heterogeneity, most likely, is the result of differences which can be ascribed to the drugs themselves, but also to features related to the design, in- and exclusion criteria, patient characteristics (Supplementary Tables 1 and 2) and sample sizes (Table 1) of the clinical trials assessing clinical effects.
As can be derived from the effect sizes for the influence on various cardiovascular outcomes, there appears to be little heterogeneity with respect to SGLT- Is, in particular with respect to their effect on hospitalizations because of heart failure (Fig. 1)1,2,3,4. Likewise, there seems to be little heterogeneity with respect to differences between various DPP-4 inhibitors (with the potential exception of their influence on heart failure-related hospitalizations, Fig. 1). However, as has been previously noted, the class of GLP-1 RAs seems to be highly heterogeneous. In particular, lixisenatide has been almost without any hint for cardiovascular benefits (Fig. 1)9, which has been explained by the limited exposure to effective drug levels for a minor proportion of a 24 h period27,28,33. The development of other GLP-1 RAs also markedly differed with respect to pharmacokinetic properties and the scrutiny of defining effective, but tolerable doses27,28. These differences have proven relevant for the resulting effect sizes regarding glycaemic control and body weight reduction, which seem to be related to those reported for cardiovascular effects27,28, perhaps because both effects are related to optimized target engagement.
It is obvious from the present data, that smaller studies more likely yield erratic results (larger effect sizes compared to larger studies, which, nevertheless, often are not significant, or unexpectedly small effect sizes; Fig. 1). Therefore, when attempting to characterize cardiovascular properties of glucose-lowering medications, the emphasis should be on larger studies accruing sufficient numbers of events of interest. Along the same lines, larger, definite trial are underway to characterize cardiovascular effects of oral semaglutide in type 2 diabetes (SOUL trial, ClinicalTrials.gov: NCT03914326) and in populations characterized by obesity and related cardiovascular complications (SELECT trial, ClinicalTrials.gov: NCT03574597).
Our analysis has shown that cardiovascular outcomes studies performed as part of the development of novel glucose-lowering drugs are typically adequately powered for the main (primary) endpoint MACE, but that they have variable power, depending on sample size and the number of cardiovascular events accrued, to provide significance for the effect size of differences that are typically observed with SGLT-2 Is and GLP-1 RAs. As a result, indirect comparisons between placebo-controlled cardiovascular outcomes trials with different glucose-lowering drugs do not appear uniformly suitable to detect differences in their ability to elicit a unique pattern of effects on various cardiovascular endpoints.
Materials and methods
Study design
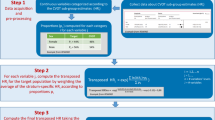
The present study is a systematic analysis of published cardiovascular outcomes trials examining glucose-lowering medications, belonging to the medication classes SGLT-2 Is1,2,3,4, DPP-4 Is5,6,7,8, and GLP-1 RAs9,10,11,12,13,14,15,16, in patients with type 2 diabetes and a high risk to develop cardiovascular complications, which used major adverse cardiovascular outcomes (“MACE”) as their primary endpoint, but also reported individual cardiovascular outcomes including MACE components (non-fatal acute myocardial infarction or stroke, cardiovascular death), all-cause death, and hospitalization for congestive heart failure. Studies reporting other populations (e.g., at high risk for renal endpoints (e.g., the CREDENCE trial20) or heart failure (DAPA HF trial34, EMPEROR reduced trail35), were not used for the present analysis. After a systematic literature search identifying suitable publications, data regarding the sample size (number of participants) and on cardiovascular outcomes (number of patients experiencing primary and secondary endpoints with active or placebo treatment (on a background of “standard of care”) were extracted. The power to detect 10, 15, 20 or 25% differences between active and placebo treatment was estimated. In addition, the power to detect a difference equivalent to that observed between active and placebo treatment in any particular study was also estimated.
Literature search
The PubMed database was searched using Endnote 7.0. Search terms were lixisenatide, liraglutide, semaglutide, exenatide, albiglutide, dulaglutide, and efpeglenatide (GLP-1 RAs), saxagliptin, alogliptin, sitagliptin and linagliptin (DPP-4 Is), and empagliflozin, canagliflozin, dapagliflozin, and ertugliflozin (SGLT-2 Is) in the title and “major adverse cardiovascular outcomes” or MACE, myocardial infarction, stroke, cardiovascular death, all-cause death or total mortality, and hospitalization for (congestive) heart failure in the abstract. Retrieved literature was checked regarding suitability by analysing the abstracts, and—if found potentially suitable—the full manuscript. Reference lists of all retrieved publications were search for additional publications that could potentially be useful.
Data extraction
Data from individual publications (including their online supplementary files) were transferred to a paper form systematically listing items of interest for the present analysis, independently by SB and MAN. Discrepancies were resolved by discussion with JJM. The main focus was on parameters characterizing the dimensions of the study with respect to participant numbers, and the number of primary and secondary outcomes and the resulting hazard ratios (with 95% confidence intervals) for MACE, acute myocardial infarction, stroke, cardiovascular death, all-cause death, hospitalization for heart failure reported with placebo and active treatment with SGLT-2 Is, DPP-4 Is, and GLP-1 RAs. In addition, patient characteristics at baseline were extracted in order to provide information on the selection of patient populations. These data were transferred to Excel spreadsheets for further use (e.g., for the generation of Tables and Figures).
Study quality was assessed applying the Jadad score36 and the Risk of Bias tool (https://www.riskofbias.info/)37. All publications turned out to be suitable for our analysis.
Power calculation
Statistica 13.3 (TIBCO Software Inc. (2017). Statistica (software system for data analysis), version 13. http://statistica.io) was used to estimate the power of individual trials to detect differences by 10, 15, 20, or 25% vs. event rates published for placebo-treated patients. Input variables were the proportions of patients developing the endpoint of interest, and the total number of patients in this particular trial (sum of numbers treated with active drug and placebo). The output variable was the power estimated based on a log-rank test to compare survival curves between patients treated with active drug and placebo. A power of > 80% was tentatively defined as a meaningful prerequisite for drawing conclusions from significant differences.
Statistics
Descriptive statistics use means ± standard deviations for continuous variables and numbers fulfilling certain criteria and the proportions (percentages) of the total population for categorical variables.
Ethical approval and consent to participate
Not applicable, since the present study analyses published data on cardiovascular outcomes trials, not individual patients’ data. All the procedures were performed in accordance with the relevant guidelines and regulations.
Data availability
All data analysed and presented in the present manuscript have been taken from published manuscripts. No original data have been used.
References
Zinman, B. et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 373, 2117–2128. https://doi.org/10.1056/NEJMoa1504720 (2015).
Neal, B. et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 377, 644–657. https://doi.org/10.1056/NEJMoa1611925 (2017).
Wiviott, S. D. et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 380, 347–357. https://doi.org/10.1056/NEJMoa1812389 (2019).
Cannon, C. P. et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N. Engl. J. Med. 383, 1425–1435. https://doi.org/10.1056/NEJMoa2004967 (2020).
Scirica, B. M. et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 369, 1317–1326. https://doi.org/10.1056/NEJMoa1307684 (2013).
White, W. B. et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 369, 1327–1335. https://doi.org/10.1056/NEJMoa1305889 (2013).
Green, J. B. et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 373, 232–242. https://doi.org/10.1056/NEJMoa1501352 (2015).
Rosenstock, J. et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: The CARMELINA randomized clinical trial. J. Am. Med. Ass. 321, 69–79. https://doi.org/10.1001/jama.2018.18269 (2019).
Pfeffer, M. A. et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N. Engl. J. Med. 373, 2247–2257. https://doi.org/10.1056/NEJMoa1509225 (2015).
Marso, S. P. et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 375, 311–322. https://doi.org/10.1056/NEJMoa1603827 (2016).
Marso, S. P. et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 375, 1834–1844. https://doi.org/10.1056/NEJMoa1607141 (2016).
Holman, R. R. et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 377, 1228–1239. https://doi.org/10.1056/NEJMoa1612917 (2017).
Hernandez, A. F. et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 392, 1519–1529. https://doi.org/10.1016/S0140-6736(18)32261-X (2018).
Gerstein, H. C. et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 394, 121–130. https://doi.org/10.1016/S0140-6736(19)31149-3 (2019).
Husain, M. et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 381, 841–851. https://doi.org/10.1056/NEJMoa1901118 (2019).
Gerstein, H. C. et al. Cardiovascular and renal outcomes with efpeglenatide in type 2 diabetes. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2108269 (2021).
Mann, J. F. E. et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med 377, 839–848. https://doi.org/10.1056/NEJMoa1616011 (2017).
Gerstein, H. C. et al. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomised, placebo-controlled trial. Lancet 394, 131–138. https://doi.org/10.1016/S0140-6736(19)31150-X (2019).
Wanner, C. et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 375, 323–334. https://doi.org/10.1056/NEJMoa1515920 (2016).
Perkovic, V. et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 380, 2295–2306. https://doi.org/10.1056/NEJMoa1811744 (2019).
Salah, H. M. et al. Effects of sodium-glucose cotransporter 1 and 2 inhibitors on cardiovascular and kidney outcomes in type 2 diabetes: A meta-analysis update. Am. Heart J. 233, 86–91. https://doi.org/10.1016/j.ahj.2020.12.007 (2021).
Salah, H. M. et al. Effect of sodium-glucose cotransporter 2 inhibitors on cardiovascular and kidney outcomes-Systematic review and meta-analysis of randomized placebo-controlled trials. Am. Heart J. 232, 10–22. https://doi.org/10.1016/j.ahj.2020.10.064 (2021).
Zelniker, T. A. et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 393, 31–39. https://doi.org/10.1016/S0140-6736(18)32590-X (2019).
Kristensen, S. L. et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 7, 776–785. https://doi.org/10.1016/S2213-8587(19)30249-9 (2019).
Scirica, B. M. et al. Heart failure, saxagliptin, and diabetes mellitus: Observations from the SAVOR-TIMI 53 randomized trial. Circulation 130, 1579–1588. https://doi.org/10.1161/CIRCULATIONAHA.114.010389 (2014).
Zannad, F. et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: A multicentre, randomised, double-blind trial. Lancet 385, 2067–2076. https://doi.org/10.1016/S0140-6736(14)62225-X (2015).
Nauck, M. A. & Meier, J. J. Management of endocrine disease: Are all GLP-1 agonists equal in the treatment of type 2 diabetes?. Eur. J. Endocrinol. 181, R211–R234. https://doi.org/10.1530/EJE-19-0566 (2019).
Nauck, M. A., Quast, D. R., Wefers, J. & Meier, J. J. GLP-1 receptor agonists in the treatment of type 2 diabetes - state-of-the-art. Mol. Metab. https://doi.org/10.1016/j.molmet.2020.101102 (2020).
Davies, M. J. et al. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 61, 2461–2498. https://doi.org/10.1007/s00125-018-4729-5 (2018).
Buse, J. B. et al. 2019 Update to: Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD. Diabetes Care 43, 487–493. https://doi.org/10.2337/dci19-0066 (2020).
Cosentino, F. et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 41, 255–323. https://doi.org/10.1093/eurheartj/ehz486 (2020).
Gerstein, H. C. et al. The effect of dulaglutide on stroke: An exploratory analysis of the REWIND trial. Lancet Diabetes Endocrinol. 8, 106–114. https://doi.org/10.1016/S2213-8587(19)30423-1 (2020).
Becker, R. H., Stechl, J., Msihid, J. & Kapitza, C. Lixisenatide resensitizes the insulin-secretory response to intravenous glucose challenge in people with type 2 diabetes-a study in both people with type 2 diabetes and healthy subjects. Diabetes Obes. Metab. 16, 793–800. https://doi.org/10.1111/dom.12278 (2014).
McMurray, J. J. V. et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 381, 1995–2008. https://doi.org/10.1056/NEJMoa1911303 (2019).
Packer, M. et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 383, 1413–1424. https://doi.org/10.1056/NEJMoa2022190 (2020).
Jadad, A. R. et al. Assessing the quality of reports of randomized clinical trials: Is blinding necessary?. Control. Clin. Trials 17, 1–12 (1996).
Higgins, J. P. T. et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 343, 5928. https://doi.org/10.1136/bmj.d5928 (2011).
Acknowledgements
We thank Ms. Sonja Bartholomé, Diabetes Division, St. Josef Hospital, Ruhr-University Bochum, for help retrieving literature.
Funding
Open Access funding enabled and organized by Projekt DEAL. We acknowledge support by the DFG Open Access Publication Funds of the Ruhr-Universität Bochum. There has not been any specific funding for the present study.
Author information
Authors and Affiliations
Contributions
S.B., J.J.M. and M.A.N. designed the study, S.B. and M.A.N. searched literature, S.B. and M.A.N. analyzed the data, performed the statistical analysis, and wrote the draft manuscript. All authors have seen an approved the final draft of this manuscript and have decided to submit it for publication. MAN is the guarantor who takes full responsibility for the work as a whole, including study design, access to data, and the decision to submit and publish the manuscript. All authors have seen the final version of the manuscript and have consented to the submission for publication in its present form.
Corresponding author
Ethics declarations
Competing interests
MAN has been member on advisory boards or has consulted with Boehringer Ingelheim, Eli Lilly & Co., Menarini/Berlin Chemie, Merck, Sharp & Dohme, and NovoNordisk. He has received grant support from AstraZeneca, Eli Lilly & Co., Menarini/Berlin-Chemie, Merck, Sharp & Dohme, and NovoNordisk, outside the submitted work. He has also served on the speakers’ bureau of AstraZeneca, Boehringer Ingelheim, Eli Lilly & Co., Menarini/Berlin Chemie, Merck, Sharp & Dohme, and NovoNordisk. JJM reports personal fees from Astra Zeneca, Eli Lilly, and Servier; grants and personal fees from Boehringer Ingelheim, MSD, Novo Nordisk, and Sanofi, outside the submitted work. SB has nothing to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Birker, S., Meier, J.J. & Nauck, M.A. Statistical power for MACE and individual secondary endpoints in cardiovascular outcomes trials for type 2 diabetes: a systematic review. Sci Rep 12, 21069 (2022). https://doi.org/10.1038/s41598-022-25296-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25296-x
- Springer Nature Limited