
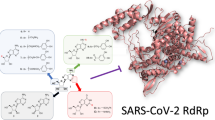
Abstract
RNA dependent RNA polymerase (RdRp), is an essential in the RNA replication within the life cycle of the severely acute respiratory coronavirus-2 (SARS-CoV-2), causing the deadly respiratory induced sickness COVID-19. Remdesivir is a prodrug that has seen some success in inhibiting this enzyme, however there is still the pressing need for effective alternatives. In this study, we present the discovery of four non-nucleoside small molecules that bind favorably to SARS-CoV-2 RdRp over the active form of the popular drug remdesivir (RTP) and adenosine triphosphate (ATP) by utilizing high-throughput virtual screening (HTVS) against the vast ZINC compound database coupled with extensive molecular dynamics (MD) simulations. After post-trajectory analysis, we found that the simulations of complexes containing both ATP and RTP remained stable for the duration of their trajectories. Additionally, it was revealed that the phosphate tail of RTP was stabilized by both the positive amino acid pocket and magnesium ions near the entry channel of RdRp which includes residues K551, R553, R555 and K621. It was also found that residues D623, D760, and N691 further stabilized the ribose portion of RTP with U10 on the template RNA strand forming hydrogen pairs with the adenosine motif. Using these models of RdRp, we employed them to screen the ZINC database of ~ 17 million molecules. Using docking and drug properties scoring, we narrowed down our selection to fourteen candidates. These were subjected to 200 ns simulations each underwent free energy calculations. We identified four hit compounds from the ZINC database that have similar binding poses to RTP while possessing lower overall binding free energies, with ZINC097971592 having a binding free energy two times lower than RTP.
Similar content being viewed by others

Introduction
The highly contagious coronavirus induced illness COVID-19 is now a global threat to human well-being, with the current case numbers rising near 220 million and over 4.5 million deaths as of September 2021. With the increase in rollouts of vaccinations around the world, there are now some signs of relief to even the most affected regions. However, there is still the potential for new vaccine resistant strains of the coronavirus are evolving often as seen in the recent deadly outbreak of the new B.1.617.2 “Delta” variant in Nepal1. Additionally, there is the possibility for some vaccinated individuals to not develop an adequate immune response to create neutralizing antibodies. Therefore, there is still an urgent need for effective drug therapy in order to slow down the progression of sickness in affected patients. The SARS-CoV-2 virus is a positive-strand RNA virus whose genomic structure expresses high similarity with previous severe acute respiratory syndrome coronavirus (SARS-CoV). Like other RNA viruses, the replication of SARS-CoV-2 requires RNA-dependent RNA polymerase (nsp 12), a key enzyme which regulates the viral genome replication and transcription within the viral life cycle. It is thus a key validated target for the development of COVID-19 disease small molecule therapeutics.
Currently, Gilead’s Remdesivir (RDV) otherwise known as VekluryⓇ has been approved by the FDA as an antiviral prodrug which targets the nucleotide addition process in RdRp. This prodrug is bio transformed in the body into the active-form remdesivir-triphosphate (RTP), a nucleoside mimic to adenosine triphosphate (ATP), which then undergoes nucleotide addition onto the growing RNA chain2. It is theorized that RDV inhibits RdRp by chain termination during the RNA translocation step3. Its action on treating SARS-CoV-2 infection has seen some success in reducing the time needed for recovery in very sick patients that require hospitalization4. However, this drug’s efficacy is also controversial, as its administration was recently halted due to the lack of significant improvement expected in COVID-19 infected patients. Therefore, because RDV is the only FDA approved drug for treating COVID-19 patients, there is interest in the discovery of alternative treatments. Additionally, there is interest in developing non-nucleoside mimetics with high RdRp affinity for this purpose, as these are thought to circumvent the polymerization reaction.
There is a remarkable effort underway from researchers around the globe to help shed more light on potential alternatives for the inhibition of RdRp. There have been several studies (Table 1)3,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42 that have reported the discovery of potential small molecule inhibitors of RdRp, mostly deriving from the in-silico approach coupled with virtual drug screening of numerous compound libraries. As of this study, few in-silico studies have considered the approach of using the massive ZINC library of approximately 17 million compounds using high-throughput virtual screening workflow (Fig. 1) paired with long, multi-system molecular dynamics (MD) simulations43. A previous study reported the discovery of two potential isoformic RdRp inhibitors: ZINC09128258 and ZINC09883305, however these were observed to have lower docked binding affinity than the compared standards21.

Virtual screening workflow designed to conduct this study.
In our present study, we have utilized long MD simulations (1.0 µs) each of SARS-CoV-2 RdRp in complex with free form active RTP and ATP and template-primer RNA starting from the recently solved crystal complex (PDBID: 7BV2)44. Using the most abundant conformations of RTP complex from 1.0 µs simulations, we performed pharmacophore-based screening of the ZINC 15 library against the binding site of RdRp, leading to fourteen compounds with top docking scores43. From these top fourteen compounds, we performed classical MD simulations (200 ns each) to observe the stability for each complex system. Lastly, we performed Molecular Mechanism-General Born Surface Area (MM-GBSA) energy predictions to observe their relative binding affinities to RdRp after simulation39,45,46,47. Four ZINC compounds showed stable binding to the RdRp-RNA complex. Additionally, these four systems showed improved MM-GBSA energies both the RTP and ATP complex systems as well as in reference to their initial docked complexes. This discovery helps introduce crucial knowledge of small molecule binding to RdRp in addition to the effort in discovering and development of anti-SARS-CoV-2 RdRp agents using in-silico methods.
Experimental section
Structure building and ligand preparation
The recent electron microscopy solved crystal structure of SARS-COV-2 nsp12-nsp7-nsp8 RdRp in complex with template-primer RNA and covalently bound RDV named PDB ID 7BV2 was retrieved from the RCSB protein database44,48. This structure was then prepared with the default charge state being optimized at pH 7 using Maestro’s built-in Protein Preparation Wizard49,50,51. The complex was then preprocessed, optimized, and minimized using the Protein Preparation Wizard within Maestro software49. A receptor docking grid was generated around the covalently bound RDV with a X: 7.07, Y: − 3.87 and Z: 1.27 spatial coordinates and a grid box size allowing for ligands with length ≤ 20 Å. Free form RTP was then generated by breaking phosphodiester bonds between U20 of primer strand and remdesivir. The phosphate tail was then added onto the 5′ carbon of remdesivir to render the triphosphate form bound within the i binding site. The ligand RTP was then redocked using the receptor grid file generated prior. The resulting docking grid was also applied for the docking of ATP towards RdRp. Before any docking procedures, the ligands were first prepared for docking by generating the ionization/tautomeric states at pH 7 ± 2 using Maestro’s Epik tool52,53. Each ligand was docked utilizing the Glide module within Maestro software and the GlideXP scoring function was employed50,51,54.
MD system setup
The OPLS2005 force field was used to model the protein and the ligands55,56,57. When creating a MD simulation system for RDV-TP, ATP and the 14 ZINC compounds, its XP docked complex with the two crystal Mg2+ ions was placed into an orthorhombic water box with a buffer distance of 10 Å using the SPC water model58 and a salt concentration of 0.15 M NaCl was added to the system55,56.
Relaxation and production runs
The MD simulations for each system were performed using the Desmond simulation package59 and the default simulation protocols were followed just like our early work60,61,62. Here a brief description is given. Each system was relaxed using the default relaxation protocol for non-membrane proteins63. There are eight stages which constitute the protocol: (1) minimization of the system with a restraint of solute heavy atoms. (2) Minimization without any restraints. (3) Simulation with heating from 0 to 300 K, H2O barrier and gradual restraining. (4) Simulation under the NPT ensemble which is constant number of particles, pressure of 1 bar and constant temperature of 300 K with an H2O barrier and heavy atoms being restrained. (5) Simulation under the NPT ensemble with equilibration solvent. (6) Simulation under the NPT ensemble with protein heavy atoms annealing from 10.0 to 2.0 kcal/mol. (7) Simulation under the NPT ensemble with Cα atoms restrained at 2.0 kcal/mol; and (8) simulation of 1.5 ns under the NPT ensemble with no restraints. After this relaxation procedure, six 1000 ns production runs for RTP, ATP and four top zinc ligand systems, and ten 200 ns production runs were conducted under the NPT ensemble using the default protocol, leading to a total of 8000 ns. M-SHAKE64 was used to constrain bonds with hydrogen atoms, which allowed for 2.0 fs time steps during the simulation. Long-range electrostatic interactions were treated under periodic boundary conditions by the k-space Gaussian split Ewald method65. The charge grid spacing was ~ 1 Å and the direct sum tolerance was 10–9. Van der Waals interactions were based on a uniform density approximation. A distance of 9 Å was set as the cutoff for short range non-bonded interactions. Non-bonded forces were calculated by an r-REPA integrator66; the short range forces updated every 2 fs and the long range every 6 fs. The trajectories were saved every 50 ps for analysis. Pressure of 1 bar was controlled using the Martyna–Tobias–Klein chain coupling scheme67 (coupling constant = 2 ps), and temperature of 300 K by the Nosé–Hoover67 chain coupling scheme (coupling constant = 1 ps).
Conformational clustering of RdRp complexes
The Desmond trajectory clustering tool was used to group the complex structures from the last 100 ns of simulation for the ATP and RTP systems58. The merging distance cutoff was set to be 2.5 Å. The centroid representative (i.e. the structure having the largest number of neighbors in the structural family) was used to display the structural family. The most abundant conformations are identified as the cluster with the most occupancy. This was performed for the complexes containing ATP and RTP. Clustering was then performed on the top fourteen ZINC compounds selected from HTVS after 200 ns simulation.
Pharmacophore screening and hit prioritization
The pharmacophore screening was performed using the active Remdesivir against ZINC product molecules using LigandScout 4.368. In our model, we determined the seven pharmacophore features to include the two negative ionizable groups, one aromatic ring, and four hydrogen bonds acceptors with their corresponding positions. Before further experiments, we determined the ionizable/tautomeric states of each compound under pH 7 ± 2 using Maestro’s Epik tool52,53. The lowest ionization/tautomeric states were selected, and their bond geometries were minimized to the lowest energetic state. We then employed our prepared RdRp complex to generate a receptor grid file around the RTP binding site, or i site, and the ionization, tautomeric and bond states of ATP were generated at pH ± 2 using Maestro’s Epik tool52,53. This receptor grid file was additionally used for our docking of the ZINC database compounds using the Glide XP docking scoring function within Maestro software49,68. High-Throughput Virtual Screening (HTVS) was also performed using the Glide module on the most abundant cluster representative of the RTP system after simulation utilizing the ZINC15 compound database43,50,51. This process consists of screening at three levels with gradually increasing the computational cost. The first level is based on ligand ADME/Toxicity prediction using QikProp of Schrodinger and docking using Glide69. The second is performed using MD simulations of the docked complexes with explicit solvents (water)63. Lastly, the third is determined by the binding energy calculations. After HTVS, we narrowed down the library of compounds to the top fourteen based on their superior Glide scores and #star value50,51. We then subjected these fourteen to 200 ns simulations and performed MM-GBSA calculations to determine their relative binding affinities after the simulation39,45. From the fourteen compounds, four compounds were selected based on their favorable binding energies.
Confirmation of stability of hits and MM-GBSA calculation
The Desmond SID module was employed to analyze the interaction between proteins and ligands as well as to determine their stability in each MD simulation63. This includes the Root-Mean Square Deviation (RMSD) of the protein–ligand complexes and Root-Mean Square Deviation (RMSF) of both the protein and ligands. Additionally, secondary structure elements (SSE) were explored. Lastly, the protein–ligand contacts containing H-bond, ionic, hydrophobic interactions were explored.
In order to check the convergence of the simulations, we investigated the root mean square deviation (RMSD) values of protein Cɑ, RNA O5′ atoms, and ligand main atoms for each trajectory using the SID tool within Maestro software63. The root mean square fluctuation (RMSF) of each individual amino acid, RNA and ligand atom were also calculated to characterize the local movement of individual structural components of each complex.
The Molecular Mechanism-General Born Surface Area (MM-GBSA) binding energies were calculated on the frames for the whole duration of both systems as well as for each ZINC system45. The OPLS3 force field, VSGB 2.0 solvation model and the default Prime procedure was used for the MM-GBSA calculations46,47,55,56,57,70. The default procedure consists of three steps: Receptor alone (minimization), Ligand alone (minimization), Receptor-ligand complex (minimization). The total binding free energy equation is: ΔGBind = Ecomplex (minimized) − (Eligand (minimized) + Ereceptor(minimized)). To gain a more detailed understanding of binding nature, the original interaction terms (Coulombic + H-bond + GB solvation + van der Waals + π–π packing + self-contact + lipophilic) were merged into three components: Eelectrostatics, EvdW, and Elipophilic, where Eelectrostatics = (Hbond + Ecoulomb + EGBsolvation), EvdW = (EvdW + Eπ-π + Eself-contact) and Elipophilic.
Normal mode analysis by PCA
The trajectory of ATP, RTP and the four top zinc ligands was used in the Normal Mode Wizard in VMD71 to generate the top 5 normal modes by a principal component analysis (PCA).
Results and discussion
The binding of remdesivir triphosphate and ATP to RdRp
Adenosine triphosphate was docked against the crystal structure of SARS-CoV-2 RdRp (PDBID: 7BV2) at the i-binding site which formed hydrogen pairing with the template strand of 14 nucleotide bases and the primer strand of 11 nucleotide bases44. The conformational pose of the docked RTP compared to the covalently bound RDV in the crystal structure was nearly identical (Fig. S2). We then generated a docking grid from the docking pose of RTP to dock ATP into the same catalytic site of RdRp. The conformational pose of ATP compared to the covalently bound crystal RDV was nearly identical. It was also seen that the triphosphate tail of RTP was positioned near one of the Mg+ ions. These conformations were then used to perform long (1.0 µs) simulations for each complex to further elucidate more about the relaxed conformations of each and to obtain a stable conformation for our high-throughput virtual screening portion of the study.
The trajectory convergence of the simulation for both systems was confirmed through analyzing the RMSD during the 1000 ns (1.0 µs) simulations shown in Fig. 2A,B. The trajectory RMSD is of the protein receptor backbone Cɑ atoms and the ligand main atoms for both the ATP and RTP complexes. It is clearly seen in Fig. 2A that the RTP system reached stability very early in the simulation (~ 50 ns) while the ATP system in Fig. 2B reached stability around 550 ns. Importantly, both protein complexes maintained average RMSD values of approximately 2.0 Å. This is indicative that the differences between ATP and RTP binding had no significant change in the overall RdRp conformation through the 1.0 µs simulation.

The RMSD values for the receptor Cα and ligand main atoms for the RTP, ATP and four ZINC systems. Protein Cα is shown in blue and ligand main atoms is shown in red. Figure (A) is the system containing RTP, (B) is the system containing ATP, (C) is the system containing ZINC097971592, (D) is the system containing ZINC002146610, (E) is the system containing ZINC069492350 and (F) is the system containing ZINC408592119.
The ligand RMSD average values for the ATP system are within 1 Å of the protein RMSD values and reached stability at approximately 550 ns. The ligand RMSD for the RTP system also matched the average distance as in the protein with a convergence very early in the simulation; approximately 50 ns. It is clearly seen that the complex containing ATP possessed higher RMSD values compared to the RTP complex indicating that the binding of RTP was more stable during the simulation.
We additionally wanted to confirm the stability of the template-primer RNA in the complex through simulation. The RMSD of the RNA main atoms is shown in Fig. S3, both ATP and RTP systems show clear stability and early convergence at around 50 ns with values both approximately 2.0 Å which is also consistent with the protein Cɑ RMSD. Likewise seen in the ligand main atoms RMSD plot, the complex containing RTP showed slightly lower average values compared to the ATP system indicating more stable binding occurred during the simulation. To observe the fluctuation of each individual amino acid in the protein structure, we recorded the RMSF of the RdRp complexes during the simulation.
Ligand main atoms RMSF (Fig. S5) was also recorded to observe the fluctuation of the individual atoms of each ligand in order to better understand the regions of stability of ATP and RTP. Clearly, RTP in complex saw less fluctuation per atom over the simulation as compared to ATP. Short peaks at atoms 1–4 indicate the slight instability seen in the gamma phosphate on the phosphate tail which is not abnormal. Interestingly, small peaks in atoms 16 and 30 indicate a slight increase in fluctuation seen at the 3′ and 4′ hydroxyl groups on the ribose ring of RTP. This specificity at only these two polar functional groups is also seen in atoms 29 and 30 of the ATP system and likely indicates positioning for nucleophilic attack during nucleotide addition.
Clustering analysis
We then performed cluster analysis of both MD systems after the simulation to generate the most abundant conformational pose during the 1000 ns duration. In the 2D residue interaction seen with the most abundant conformational poses of each simulation (Fig. 3A–F), it was observed that the adenine motif of both ATP and RTP maintained H-Bond pairing with U10 on the template strand and partial pi-pi stacking of A11 on the primer strand. The RTP system was more able to sustain salt bridge interactions with positive residues R555, R553, R624, K621. The RTP system also saw interaction with S759 on the nitrile group while Y619 was seen to form H-Bonds with negative oxygen on the gamma phosphate of ATP. In addition, the RTP system also saw stabilization of the 3′ hydroxyl group on the ribose ring by H-Bonds with residues N691 and T680.

Ligand–receptor interaction diagrams and representations of the most abundant conformational pose from MD simulation. (A,D) The 3D surface representation of SARS-CoV-2 RdRp colored in yellow. (B,E) Zoomed and rotated perspective of the template-primer RNA in complex with ATP and RTP, respectively. (C,F) The detailed receptor-RNA-ligand contacts of the most abundant conformational pose of ATP and RTP, respectively. Template and primer strands represented in blue and red licorice, respectively. Ligands are colored in green and magnesium ions are represented as pink balls.
During the 1.0 µs simulation of both the ATP and RTP systems, several amino acid contacts were observed to be maintained throughout with high occupancy (Table S7). For both the ATP and RTP systems, there was significant occupancy in the H-Bond interactions between the triphosphate tails and a positive region of amino acids such as K551, K621, K798, R553 and R555 of ≥ 30% of the simulation time. For the RTP complex, residues K621, R553 and R555 formed H-bonds with negatively charged oxygen on alpha, beta and gamma phosphate with all possessing an occupancy of above 90%, indicating that these contacts could be crucial for the stability and positioning of RTP in the catalytic site of RdRp. These interactions were seen in previous study where both ATP and RTP were docked to RdRp72. Both systems also maintained the maximum occupancy interaction with Mg+ ions with the triphosphate tails, suggesting that at least one ion is responsible for the stabilization and positioning of incoming nucleotides. There was modest occupancy of hydrogen bond formation with hydroxyl groups located on the ribose ring portions with residues D623, D760, and N691, indicating secondary stabilizing roles for these amino acids potentially related to the initiation of nucleotide addition. Negative residue D623 saw an average occupancy of 56% bonding with both hydroxyl groups of the ribose ring portion of RTP. In the ATP complex, D760 saw 34% occupancy with the 3′ hydroxyl group located on the ribose ring which is involved in nucleotide addition.
In Table S6 are the histogram of protein–ligand contacts occupancy during the simulations for ATP and RTP complexes. These show the smaller contacts seen with occurrence of ≤ 30% of the simulation as well as contacts that occurred at over 100% which indicates multiple ligand atoms contacting same amino acid. In the ATP complex, the ligand contacted several more protein residues with brief occupancy compared to the RTP complex which is likely due to lower relative ligand bound stability seen in the former. Also, more water bridge interactions occurred with the ATP complex indicating weaker overall affinity compared to the RTP complex. Furthermore, RTP saw higher occupancy of H-bonds compared to others confirming the relative favorability of bonding to RdRp.
Previous studies have also reported similar findings on the critical amino acids involved in RDV binding. Koulgi et al. described an ensemble approach of the free form binding of prodrug RDV in complex with SARS-CoV-2 RdRp contacted amino acids Y451, T540, M542, R548, K551, R553, R555, A558, D618, S674, D761 and E81173. Amino acid R555 was observed to form H-bonds with the alpha phosphate group in RDV in all five ensemble representatives73. Other studies have aimed to observe the active triphosphate form interaction upon RdRp binding. Zhang et al. found that the nitrile group was positioned in a pocket including K545, Y546 and A547 with the phosphate tail interacting with K551, R553 and R555. Additionally, positive amino acids near the palm subdomain were found to be crucial for RTP activity74.
Normal mode analysis
To probe the dynamics of the two systems (ATP vs RTP), the trajectory was analyzed by a PCA analysis to generate top five low frequency normal modes of ATP (Fig. S6a) and RTP (Fig. S6b). By visual inspection, although the two systems undergoes a similar opening and closing motion in their normal modes, subtle differences in magnitude between the two systems was also identified in some modes. For example, the APT system appears to have larger inter-domain movement than the RTP system among the three key RNA binding domains: Thumb domain, Palm domain and Finger domain. To show this, the mode 5 of the ATP system and the mode 1 of the RTP are shown in Fig. 4 and the movie files are provided in the supporting info. Because the ATP and RTP in these complex have already in a closed active site conformation, the translocation is the next critical step for growing the primer strand after incorporating AMP/RMP into the primer stand, this larger opening-closing mode of the ATP system than the RTP might forecast some critical difference in the translation step between the two systems. Interestingly, RTP has been found to halt RNA extension at the i + 3 site after RMP is incorporated into the RNA primer strand, through stalling the translocation of the primer strand75. In contrast, ATP does not have this translocation problem. In Fig. S6a, it can be observed that each mode undergoes a different opening and closing motion.

The top view (top) and the side view (bottom) of a selected normal mode from the ATP (model 5) and RTP system (model 1). RNA (yellow) and Thumb domain (Red), Palm domain (Green), and Finger domain (Blue) of RdRp is shown ribbon.
High-throughput virtual screening finds 14 compounds
Once we validated that our simulations with ATP and RTP bound to RdRp met convergence during the simulation, we then used these complexes to perform our initial screening. Firstly, pharmacophoric screening of the ZINC database was performed by selecting seven pharmacophoric features, i.e. two negative ionizable, one aromatic ring, and four H-bond acceptors of RTP. Shown in Table 2 is the results from HTVS docking against RdRp to retrieve the top fourteen hits with the highest GlideXP scores and lowest #stars as the determining factors. The GlideXP scores measured in kcal/mol are tabulated based on decreasing favorability. A more negative GlideXP score typically indicates a good initial binding affinity to RdRp. The initial conformational pose of each ligand in complex with RdRp and RNA as well as their detailed 2D ligand contacts is shown in Table S2. These fourteen compounds were then subjected to classical 200 ns MD simulations and further MM-GBSA energy calculations for each.
The MM-GBSA energies and average protein–ligand RMSD of all fourteen MD simulations are summarized in Table S3. Within Fig. 5A,B show the ligand RMSD and RMSF values of all fourteen ZINC candidates with ATP and RTP systems calculated over the last 100 ns of simulation. The free energy of binding gives a more accurate insight into both the simulation efficacy and flexible binding affinity of ligands to their target as opposed to rigid ligand docking. The summary of the 200 ns simulation runs and MM-GBSA energy calculation in Table S3 shows that several compounds possessed more favorable free energy of binding compared to the ATP and RTP systems. From this subset of ligands, we selected the top four with the most favorable MM-GBSA calculated energies for further convergence analysis to ensure that the conformational stability was reached and maintained. The molecular structures of the four candidates are shown in Fig. 6. Furthermore, in Fig. 2C–F shows the RMSD of protein Cα atoms and ligand main atoms projected over the duration for these selected compounds. The protein portions reached convergence early in the simulation for each system containing ZINC097971592, ZINC002146610, ZINC069492350 and ZINC408592119. It was observed that the RMSD values of the protein Cα of the four ZINC systems were lower than that of the RTP system. Also, the ligand RMSD values for ZINC097971592 and ZINC408592119 were higher than that of the RTP complex.

Average ligand RMSD values of all tested systems over the last 100 ns of simulation time (A). Average ligand RMSF of SARS-CoV-2 RdRp for all tested systems (B).

The 2D structures of RTP, ATP and the top four selected ZINC compounds.
We additionally wanted to observe the RMSF of the RNA main atoms during simulation to gain some more insight into the individual fluctuation of each nucleotide in the RNA sequence. In Fig. 7A, this displays the template-primer RNA RMSF obtained from the O5′ atoms on the RNA backbone for ATP and RTP systems and the four ZINC systems with Fig. 7B indicating the nucleotide sequence indexing used during the recording. The RMSF values were high at nucleotides G1-U3 on the 5′ end of the primer strand as well as high peaks around A22–C25 towards the 3′ end of the template strand for all 6 systems. This indicates high fluctuation induced from the exposure to solvent. In the middle of the plot is a slight peak around U12 on the template strand indicating that this portion did not interact with either ligand. Nucleotides U13-A15 on the template strand show a sharp decrease in fluctuation which is most likely due to the binding and hydrogen pairing of both ATP and RTP.

RMSF values of the template-primer RNA O5′ atoms of the ATP, RTP and four ZINC compound systems (A) and a guide displaying nucleotide indexing values respective to the nucleotide positions on the RNA chain (B).
The average RMSD values over the last 50 ns of simulation for receptor and ligand is also summarized in Table 3. The four ZINC systems maintained similar average receptor RMSD values were approximately 3.9 Å which is near the value of the ATP system of 2.4 Å. This result suggests the RdRp complex system remained in similar conformational state regardless of their structure dissimilarity against ATP. Table 3 is also a breakdown of the individual MM-GBSA terms such as the Van der Waals (ΔGVDW), electrophilic (ΔGELE) and lipophilic (ΔGHYD) energies of the top four selected ZINC compounds as well as ATP and RTP for comparison. The summation of these term equates to the relative free energy of binding (ΔGTotal) measured in kcal/mol. Clearly, the four selected compounds from the ZINC database resulted in lower overall MM-GBSA values over the ATP and RTP systems of − 35.9 ± 3.1 kcal/mol and − 21.3 ± 5.9 kcal/mol, respectively. The RdRp complex containing ZINC069492350 had the most negative MM-GBSA value of − 43.8 ± 4.1 kcal/mol, with ZINC002146610, ZINC097971592 and ZINC408592119 having values of − 32.4 ± 5.1, − 37.4 ± 8.1 and − 31.4 ± 4.9 kcal/mol, respectively. A conserved trend was seen in the decomposition of each contributing energy term with the Van der Waals ΔGVDW contributing the most towards the overall ΔGTotal especially for the case of the ZINC compounds. This is likely attributed simply to the high contract between nucleotides and the ZINC compounds in molecular mass, atom count and therefore a larger electron cloud to induce fluctuation to adjacent residues in the enzyme pocket. The average RMSD was also provided to confirm the stability off each system compared to the standard. The four ZINC systems maintained a similar average receptor RMSD value of approximately 3.3 Å compared to 2.4 Å of the ATP system. This result suggests the RdRp complex system of each ZINC system remained in a slightly more unstable conformational state against the ATP system.
The detailed 2D receptor-RNA-ligand contacts diagrams of the most abundant conformational pose for each of the top four ZINC candidates are shown in Fig. 8A–L. The 2d ligand–protein interactions diagram for the most abundant conformational pose of ZINC097971592, or estriol 3 glucuronide, in complex with RdRp is shown in Fig. 8C. There were several residue contacts conserved from the ATP and RTP pose. Positive residues R624, R555, and R553 formed H-bonds with mainly the polar negative carboxylate portion of the molecule, indicating that this compound is situated with the polar end in the positive catalytic region of RdRp. Additionally, ZINC097971592 also came into H-Bond contact with S682 on the hydroxyl groups on the aromatic portion. The ligand–protein contacts summary diagrams for these four ZINC candidates are displayed in Table S7 along with the total simulation protein–ligand contact heatmap to supplement in Table S11. In this, several of the residues seen in contact with the most abundant pose were also maintained with large occupancy through the simulation. Residues R555 and R624 established interactions with ZINC097971592 with 89% and 74% occupancy, respectively. Interestingly, K798 formed a salt-bridge with the negative oxygen with a relatively high occupancy value of 85%. Contacts with lesser occupancy were S682 with a 44% occupancy and R551, R553 of 50% or less. This compound’s enantiomers were identified in a previous VirtualFlow in that there was activity towards Papain-like protease (plpro), nucleoprotein, nsp10, nsp14 and orf7a in addition to nsp1276.

Ligand–receptor interaction diagrams and representations of the most abundant conformational pose from MD simulation. (A,D,G,J) The 3D cartoon representation of SARS-CoV-2 RdRp colored in gray. (B,E,H,K) Zoomed and rotated perspective of the template-primer RNA in complex with four ZINC candidates. (C,F,I,L) Detailed receptor-RNA-ligand contacts of the most abundant conformational pose of four ZINC candidates. Template and primer strands represented in blue and red licorice, respectively. Ligands are colored in green and magnesium ions are represented as pink balls.
In Fig. 8F, the psoralen derivative ZINC002146610 also shows some similarity with the RTP pose and with the other ZINC hits in that the polar carboxylate group is facing inwards towards the positive amino acid region where triphosphate tail usually lays. Also, this compound formed pi–pi stacking interactions on the psoralen ring with nucleotide U20 on the primer strand and A11 of the template strand of RNA. Additionally, the polar end formed salt bridge and H-Bond interactions with R555 and the nearby Mg+ ion. The ligand–protein contacts summary for ZINC002146610 shows similar contacts seen in the most abundant pose. Positive amino acid R555 maintained an occupancy of 113% with the polar region of the molecule, meaning there were additional contacts seen with residue in addition to the full simulation time. Like the most abundant pose, the magnesium ion maintained full contact with the polar end via salt-bridge interactions which was seen to be further stabilized by D618. Additional stabilizing water-bridges were established with occupancies of 47% and 48% with D623 and R555, respectively. Interestingly, this compound as well as its derivatives have seen previous attention in being low toxicity potential selective and reversable inhibitors of (B5i) or the chymotrypsin-like subunit of human immunoproteasome which is associated with the treatment of autoimmune diseases and various types of cancer77.
In Fig. 8I, conserved residue R555 is seen in contact with the polar end of ZINC69492350 in similar fashion to the previous ZINC compounds in addition to the RTP system. Amino acid R555 has been seen to be crucial in the activity of small molecule binding to RdRp in this study as well as others7,8,9,10,11,31,39,40,41,73,78,79. In this binding pose of ZINC69492350, R555 forms a salt bridge to the negatively charged oxygen on the carboxyl group, with an additional hydrogen bond contact with the partially negative doubly bonded oxygen on the carboxyl end. Likewise, the aromatic portion of the molecule faces in direction with the hydrogen pairing nucleotide U10 on the template strand RNA. Surrounding amino acids are also conserved which includes D760, D623, S682 and K545. Lastly, the magnesium ions are seen to stabilize ZINC69492350 with pi-cation interactions with the thiophene portion. In the ligand–protein contacts summary diagram in the supporting information, R555 maintained full occupancy with the addition of other interactions less seen with a value of 150% with the polar carboxyl end of the molecule. Like the other selected ZINC compounds, D623 formed stabilizing water-bridge interactions with the same polar end with a value of 33%. Polar residue T687 contacted ZINC69492350 55% of the simulation, unlike the other compounds where it served as a spectator.
In Fig. 8L, ZINC408592119 lies in a similar binding pose to ATP and RTP. According to the amino acid contacts seen in the most abundant conformational pose of the simulation. Positive residues K551, K545, K621 and R555 were all seen to form hydrogen bonds and pi-cation interactions with partially positive groups on the polar ends of the molecule. Negative residues such as D618 and D760 are seen to form hydrogen bonding with partially positive portions such as on the imidizolidine group. The significance of D618 is consistent with previous studies39,40,41,73,78,79. In the ligand–protein contacts summary diagram in the supporting information, positive residue K551 maintained an occupancy of 34% in the same fashion as the most abundant pose. Magnesium also contributed full occupancy with the polar carboxyl end of the molecule consistent in Table S7 and other ZINC candidates. Lastly, in addition to forming hydrogen bonds with the imidazolidine portion, also formed a water-bridge 76% of the simulation. Table 4 is the receptor-ligand contacts with an occurrence of greater than 30% tabulated for the ATP, RTP and four ZINC hit complexes. Clearly, positive residues K621, R555, R553 and K551 all induced contact with more than two-thirds of the ligands selected, with R555 meeting all 5 ligands. Likewise, negative residues D623, D798 and D760 saw interaction with at least half of the ligands. Lastly, hydrophobic residue N691 interacted with all the selected ligands apart from ZINC408592119.
The top 5 normal modes were obtained for the four top zinc compound systems (ZINC000002146610/Fig. S6c, ZINC000069492350/Fig. S6d, ZINC000097971592/Fig. S6e, and ZINC000408592119/Fig. S6f). The visual inspection suggest that although these modes of the four compounds also undergo an opening to closing motion, the magnitude is small which is more similar to RTP than ATP. Figure 9 show the mode 1 from each system. Figure S7 show the mode that is most similar to the mode 5 of the ATP system. It can be noted that the ZINC000408592119 undergoes similar opening to closing motions in comparison to the ATP system. It was also observed that ZINC000002146610, ZINC000069492350, and ZINC000097971592 have similar opening and closing motions in comparison to the RTP system.

The top view (left) and the side view (right) of a selected normal mode from the top 4 ZINC systems that is most similar to the mode 1 of RTP system. RNA (yellow) and, Thumb domain (Red), Palm domain (Green), Finger domain (Blue) of RdRp is shown ribbon.
The absorption, distribution, metabolism and excretion (ADME) properties were also explored for these four compounds in addition to ATP and prodrug RTP using the SwissADME webserver75 (Table 5). Within the four non-nucleoside analogs, three of them were reported to have high predicted GI absorption and cytochrome P450 inhibition for CYP1A2, CYP2C19, CYP2C9 and CYP3A4. With these compounds being very early drug hits selected using in silico methods, obviously there is the need for experimental evaluation needed to evaluate their biological attributes. Additionally, it is still unclear how these compounds would behave and be metabolized when introduced to a biological system. Based on the predicted bioavailability, three of the four compounds possess high (56%) availability in rats.
Conclusion
In this study, we aimed to determine the binding of the active-form of remdesivir to SARS-CoV-2 RdRp in complex with template-primer RNA and magnesium ions. Using the model constructed from PDB 7BV244, we then constructed a complex with ATP for further validation. We confirmed that these models were stable during the 1.0 µs MD simulation by observing the receptor/ligand RMSD and RMSF values. We then performed HTVS of 1179 compounds from the ZINC database against the most abundant conformation obtained from the RTP system. Based on their best docking score and low #stars, we selected the best 14 compounds form this list. We then subjected these 14 to an additional 200 ns relaxation MD simulation as well as MM-GBSA binding energy calculations to further elucidate more about their binding behavior and comparability to the classical nucleotides ATP and RTP. From these results, we ultimately selected the four ZINC candidates: ZINC097971592, ZINC002146610, ZINC69492350 and ZINC408592119 based on their lower MM-GBSA binding energy values than that of ATP and RTP. We also observed the detailed ligand-receptor contacts for each ZINC compounds pre and post simulation to compare their similarity in conformational pose to ATP and RTP. It was observed that R551, R553 and R555 have significant importance relating to negative motif hydrogen bond stabilization of both nucleotide phosphate tails and the ZINC polar ends. Additionally, negative amino acids D618 and D760 were seen to establish metal coordination bridges with Mg+ ions to further stabilization, as well as other water bridges and hydrogen bonding interactions with nearly full occupancies. These discoveries suggest that the four selected ligands from HTVS of the ZINC database could display similar or better binding affinity and inhibition activities of towards RdRp in-vitro compared to traditional nucleoside analogues.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information document.
References
Lopez Bernal, J. et al. Effectiveness of covid-19 vaccines against the B.1.617.2 (Delta) variant. N. Engl. J. Med. 385, 585–594. https://doi.org/10.1056/NEJMoa2108891 (2021).
Kokic, G. et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 12, 279. https://doi.org/10.1038/s41467-020-20542-0 (2021).
Jockusch, S. et al. Sofosbuvir terminated RNA is more resistant to SARS-CoV-2 proofreader than RNA terminated by remdesivir. Sci. Rep. https://doi.org/10.1038/s41598-020-73641-9 (2020).
Beigel, J. H. et al. Remdesivir for the treatment of covid-19—Final report. N. Engl. J. Med. 383, 1813–1826. https://doi.org/10.1056/NEJMoa2007764 (2020).
Chien, M. C. et al. Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res. 19, 4690–4697. https://doi.org/10.1021/acs.jproteome.0c00392 (2020).
Ju, J. Y. et al. Nucleotide analogues as inhibitors of SARS-CoV polymerase. Pharmacol. Res. Perspect. 8, 674. https://doi.org/10.1002/prp2.674 (2020).
Aftab, S. O. et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. https://doi.org/10.1186/s12967-020-02439-0 (2020).
Ahmad, J., Ikram, S., Ahmad, F., Rehman, I. U. & Mushtaq, M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)—A drug repurposing study. Heliyon 6, e04502. https://doi.org/10.1016/j.heliyon.2020.e04502 (2020).
Aktas, A., Tuzun, B., Aslan, R., Sayin, K. & Ataseven, H. New anti-viral drugs for the treatment of COVID-19 instead of favipiravir. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1806112 (2020).
Alexpandi, R., De Mesquita, J. F., Pandian, S. K. & Ravi, A. V. Quinolines-based SARS-CoV-2 3CLpro and RdRp inhibitors and spike-RBD-ACE2 inhibitor for drug-repurposing against COVID-19: An in silico analysis. Front. Microbiol. 11, 1796. https://doi.org/10.3389/fmicb.2020.01796 (2020).
Al-Masoudi, N. A., Elias, R. S. & Saeed, B. Molecular docking studies of some antiviral and antimalarial drugs via bindings to 3CL-protease and polymerase enzymes of the novel coronavirus (SARS-CoV-2). Biointerface Res. Appl. Chem. 10, 6444–6459. https://doi.org/10.33263/briac105.64446459 (2020).
Ao, S. et al. Identification of potential key agents for targeting RNA-dependent RNA polymerase of SARS-CoV-2 by integrated analysis and virtual drug screening. Front. Genet. 11, 581668. https://doi.org/10.3389/fgene.2020.581668 (2020).
Calligari, P., Bobone, S., Ricci, G. & Bocedi, A. Molecular investigation of SARS-CoV-2 proteins and their interactions with antiviral drugs. Viruses-Basel 12, 40445. https://doi.org/10.3390/v12040445 (2020).
da Silva, F. M. A. et al. Flavonoid glycosides and their putative human metabolites as potential inhibitors of the SARS-CoV-2 main protease (Mpro) and RNA-dependent RNA polymerase (RdRp). Mem. Inst. Oswaldo Cruz. https://doi.org/10.1590/0074-02760200207 (2020).
da Silva, J. K. R., Figueiredo, P. L. B., Byler, K. G. & Setzer, W. N. Essential oils as antiviral agents, potential of essential oils to treat SARS-CoV-2 infection: An in-silico investigation. Int. J. Mol. Sci. 21, 3426. https://doi.org/10.3390/ijms21103426 (2020).
Elfiky, A. A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 248, 117477. https://doi.org/10.1016/j.lfs.2020.117477 (2020).
Elfiky, A. A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1761882 (2020).
Indu, P. et al. Raltegravir, Indinavir, Tipranavir, Dolutegravir, and Etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: A molecular docking and drug repurposing approach. J. Infect. Public Health 13, 1856–1861. https://doi.org/10.1016/j.jiph.2020.10.015 (2020).
Kandeel, M., Kitade, Y. & Almubarak, A. Repurposing FDA-approved phytomedicines, natural products, antivirals and cell protectives against SARS-Cov-2 (COVID-19) RNA-dependent RNA polymerase. Peerj 8, e10480. https://doi.org/10.7717/peerj.10480 (2020).
Mohamed, T. A. et al. Carotane sesquiterpenes from Ferula vesceritensis: In silico analysis as SARS-CoV-2 binding inhibitors. RSC Adv. 10, 34541–34548. https://doi.org/10.1039/d0ra06901a (2020).
Parvez, M. S. A. et al. Prediction of potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2 using comprehensive drug repurposing and molecular docking approach. Int. J. Biol. Macromol. 163, 1787–1797. https://doi.org/10.1016/j.ijbiomac.2020.09.098 (2020).
Pokhrel, R., Chapagain, P. & Siltberg-Liberles, J. Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. J. Med. Microbiol. 69, 864–873. https://doi.org/10.1099/jmm.0.001203 (2020).
Ruan, Z. J. et al. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 93, 389–400. https://doi.org/10.1002/jmv.26222 (2021).
Tchesnokov, E. P. et al. Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J. Biol. Chem. 295, 16156–16165. https://doi.org/10.1074/jbc.AC120.015720 (2020).
Wu, C. R. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 10, 766–788. https://doi.org/10.1016/j.apsb.2020.02.008 (2020).
Zhao, Z. & Bourne, P. E. Structural insights into the binding modes of viral RNA-dependent RNA polymerases using a function-site interaction fingerprint method for RNA virus drug discovery. J. Proteome Res. 19, 4698–4705. https://doi.org/10.1021/acs.jproteome.0c00623 (2020).
Ahmed, S. et al. Investigating the binding affinity, interaction, and structure-activity-relationship of 76 prescription antiviral drugs targeting RdRp and Mpro of SARS-CoV-2. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1796804 (2020).
Barage, S. et al. Identification and characterization of novel RdRp and Nsp15 inhibitors for SARS-COV2 using computational approach. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1841026 (2020).
Borquaye, L. S. et al. Alkaloids from Cryptolepis sanguinolenta as potential inhibitors of SARS-CoV-2 viral proteins: An in silico study. Biomed Res. Int. 2020, 1–14. https://doi.org/10.1155/2020/5324560 (2020).
Dwarka, D., Agoni, C., Mellem, J. J., Soliman, M. E. & Baijnath, H. Identification of potential SARS-CoV-2 inhibitors from South African medicinal plant extracts using molecular modelling approaches. S. Afr. J. Bot. 133, 273–284. https://doi.org/10.1016/j.sajb.2020.07.035 (2020).
El Hassab, M. A., Shoun, A. A., Al-Rashood, S. T., Al-Warhi, T. & Eldehna, W. M. Identification of a new potential SARS-COV-2 RNA-dependent RNA polymerase inhibitor via combining fragment-based drug design, docking, molecular dynamics, and MM-PBSA calculations. Front. Chem. 8, 584894. https://doi.org/10.3389/fchem.2020.584894 (2020).
Gul, S. et al. In silico identification of widely used and well-tolerated drugs as potential SARS-CoV-2 3C-like protease and viral RNA-dependent RNA polymerase inhibitors for direct use in clinical trials. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1802346 (2020).
Gutierrez-Villagomez, J. M., Campos-Garcia, T., Molina-Torres, J., Lopez, M. G. & Vazquez-Martinez, J. Alkamides and piperamides as potential antivirals against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). J. Phys. Chem. Lett. 11, 8008–8016. https://doi.org/10.1021/acs.jpclett.0c01685 (2020).
Kar, P., Sharma, N. R., Singh, B., Sen, A. & Roy, A. Natural compounds from Clerodendrum spp. as possible therapeutic candidates against SARS-CoV-2: An in silico investigation. J. Biomol. Struct. Dyn. 39, 4774. https://doi.org/10.1080/07391102.2020.1780947 (2020).
Khan, A. et al. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARs-CoV-2 with natural products. Interdiscip. Sci.-Comput. Life Sci. 12, 335–348. https://doi.org/10.1007/s12539-020-00381-9 (2020).
Mutlu, O. et al. Targeting SARS-CoV-2 Nsp12/Nsp8 interaction interface with approved and investigational drugs: An in silico structure-based approach. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1819882 (2020).
Narayanan, N. & Nair, D. T. Vitamin B12 may inhibit RNA-dependent-RNA polymerase activity of nsp12 from theSARS-CoV-2 virus. IUBMB Life 72, 2112–2120. https://doi.org/10.1002/iub.2359 (2020).
Ribaudo, G. et al. A computational approach to drug repurposing against SARS-CoV-2 RNA dependent RNA polymerase (RdRp). J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1822209 (2020).
Sharma, A. et al. Identification of natural inhibitors against prime targets of SARS-CoV-2 using molecular docking, molecular dynamics simulation and MM-PBSA approaches. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1846624 (2020).
Singh, J., Malik, D. & Raina, A. Computational investigation for identification of potential phytochemicals and antiviral drugs as potential inhibitors for RNA-dependent RNA polymerase of COVID-19. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1847688 (2020).
Singh, S., Sk, M. F., Sonawane, A., Kar, P. & Sadhukhan, S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2viaRNA-dependent RNA polymerase (RdRp) inhibition: An in-silico analysis. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1796810 (2020).
Elkarhat, Z., Charoute, H., Elkhattabi, L., Barakat, A. & Rouba, H. Potential inhibitors of SARS-cov-2 RNA dependent RNA polymerase protein: Molecular docking, molecular dynamics simulations and MM-PBSA analyses. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1813628 (2020).
Sterling, T. & Irwin, J. J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model 55, 2324–2337. https://doi.org/10.1021/acs.jcim.5b00559 (2015).
Yin, W. C. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368, 1499. https://doi.org/10.1126/science.abc1560 (2020).
Kollman, P. A. et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. https://doi.org/10.1021/ar000033j (2000).
Jacobson, M. P., Friesner, R. A., Xiang, Z. & Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608. https://doi.org/10.1016/S0022-2836(02)00470-9 (2002).
Jacobson, M. P. et al. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct. Funct. Bioinform. 55, 351–367. https://doi.org/10.1002/prot.10613 (2004).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242. https://doi.org/10.1093/nar/28.1.235 (2000).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 27, 221–234. https://doi.org/10.1007/s10822-013-9644-8 (2013).
Friesner, R. A. et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. https://doi.org/10.1021/jm0306430 (2004).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem. 49, 6177–6196. https://doi.org/10.1021/jm051256o (2006).
Greenwood, J. R., Calkins, D., Sullivan, A. P. & Shelley, J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24, 591–604. https://doi.org/10.1007/s10822-010-9349-1 (2010).
Shelley, J. C. et al. Epik: A software program for pKaprediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691. https://doi.org/10.1007/s10822-007-9133-z (2007).
Halgren, T. A. et al. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759. https://doi.org/10.1021/jm030644s (2004).
Harder, E. et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296. https://doi.org/10.1021/acs.jctc.5b00864 (2016).
Shivakumar, D., Harder, E., Damm, W., Friesner, R. A. & Sherman, W. Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J. Chem. Theory Comput. 8, 2553–2558. https://doi.org/10.1021/ct300203w (2012).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236. https://doi.org/10.1021/ja9621760 (1996).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105, 9954–9960. https://doi.org/10.1021/jp003020w (2001).
Jorgensen, W. L., Maxwell, D. S. & TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236. https://doi.org/10.1021/ja9621760 (1996).
Montgomery, D., Campbell, A., Sullivan, H.-J. & Wu, C. Molecular dynamics simulation of biased agonists at the dopamine D2 receptor suggests the mechanism of receptor functional selectivity. J. Biomol. Struct. Dyn. 37, 3206–3225. https://doi.org/10.1080/07391102.2018.1513378 (2019).
Sader, S., Cai, J., Muller, A. C. G. & Wu, C. Can human allergy drug fexofenadine, an antagonist of histamine (H1) receptor, be used to treat dog and cat? Homology modeling, docking and molecular dynamic simulation of three H1 receptors in complex with fexofenadine. J. Mol. Graph. Model. 75, 106–116. https://doi.org/10.1016/j.jmgm.2017.05.010 (2017).
Sader, S. & Wu, C. Computational analysis of Amsacrine resistance in human topoisomerase II alpha mutants (R487K and E571K) using homology modeling, docking and all-atom molecular dynamics simulation in explicit solvent. J. Mol. Graph. Model. 72, 209–219. https://doi.org/10.1016/j.jmgm.2016.11.019 (2017).
Bowers, K. J. et al. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (Association for Computing Machinery, 2006).
Bailey, A. G. & Lowe, C. P. MILCH SHAKE: An efficient method for constraint dynamics applied to alkanes. J. Comput. Chem. 30, 2485–2493. https://doi.org/10.1002/jcc.21237 (2009).
Shan, Y. B., Klepeis, J. L., Eastwood, M. P., Dror, R. O. & Shaw, D. E. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys. 122, 054101doi:https://doi.org/10.1063/1.1839571 (2005).
Stuart, S. J., Zhou, R. H. & Berne, B. J. Molecular dynamics with multiple time scales: The selection of efficient reference system propagators. J. Chem. Phys. 105, 1426–1436. https://doi.org/10.1063/1.472005 (1996).
Ikeguchi, M. Partial rigid-body dynamics in NPT, NPAT and NP gamma T ensembles for proteins and membranes. J. Comput. Chem. 25, 529–541. https://doi.org/10.1002/jcc.10402 (2004).
Wolber, G. & Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45, 160–169. https://doi.org/10.1021/ci049885e (2005).
QikProp v. Schrödinger Release 2021-4 (2021).
Li, J. et al. The VSGB 20 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 79, 2794. https://doi.org/10.1002/prot.23106 (2011).
Bakan, A., Meireles, L. M. & Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577. https://doi.org/10.1093/bioinformatics/btr168 (2011).
Wu, Y. et al. In silico identification of drug candidates against COVID-19. Inform. Med. Unlocked 21, 100461. https://doi.org/10.1016/j.imu.2020.100461 (2020).
Koulgi, S., Jani, V., Uppuladinne, M. V. N., Sonavane, U. & Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 10, 26792–26803. https://doi.org/10.1039/d0ra04743k (2020).
Zhang, L. & Zhou, R. Structural basis of the potential binding mechanism of remdesivir to SARS-CoV-2 RNA-dependent RNA polymerase. J. Phys. Chem. B 124, 6955–6962. https://doi.org/10.1021/acs.jpcb.0c04198 (2020).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. https://doi.org/10.1038/srep42717 (2017).
Gorgulla, C. et al. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. iScience 24, 102021–102021. https://doi.org/10.1016/j.isci.2020.102021 (2021).
Sosič, I. et al. Nonpeptidic selective inhibitors of the chymotrypsin-like (β5 i) subunit of the immunoproteasome. Angew. Chem. Int. Ed. 55, 5745–5748. https://doi.org/10.1002/anie.201600190 (2016).
Arba, M. et al. Virtual screening of the Indonesian medicinal plant and zinc databases for potential inhibitors of the RNA-dependent RNA polymerase (RdRp) of 2019 novel coronavirus. Indones. J. Chem. 20, 1430. https://doi.org/10.22146/ijc.56120 (2020).
Arba, M., Wahyudi, S. T., Brunt, D. J., Paradis, N. & Wu, C. Mechanistic insight on the remdesivir binding to RNA-dependent RNA polymerase (RdRp) of SARS-cov-2. Comput. Biol. Med. 129, 104156. https://doi.org/10.1016/j.compbiomed.2020.104156 (2021).
Acknowledgements
C.W acknowledges the support by the New Jersey Health Foundation (PC 76-21) and the National Science Foundation under Grants NSF ACI-1429467/RUI-1904797, and XSEDE MCB 170088. The Anton2 machine at the Pittsburgh Supercomputing Center (MCB170090P) was generously made available by D. E. Shaw Research.
Author information
Authors and Affiliations
Contributions
C.W. designed and ran the experiments. D.B. and P.L. analyzed the data. D.B. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brunt, D., Lakernick, P.M. & Wu, C. Discovering new potential inhibitors to SARS-CoV-2 RNA dependent RNA polymerase (RdRp) using high throughput virtual screening and molecular dynamics simulations. Sci Rep 12, 19986 (2022). https://doi.org/10.1038/s41598-022-24695-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24695-4
- Springer Nature Limited
This article is cited by
-
Structure-Based Drug Design of RdRp Inhibitors against SARS-CoV-2
Topics in Current Chemistry (2023)