
Abstract
Survival statistics, estimated using data from national cystic fibrosis (CF) registries, inform the CF community and monitor disease progression. This study aimed to estimate survival among people with CF in Australia and to identify factors associated with survival. This population-based cohort study used prospectively collected data from 23 Australian CF centres participating in the Australian CF Data Registry (ACFDR) from 2005–2020. Period survival analysis was used to calculate median age of survival estimates for each 5-year window from 2005–2009 until 2016–2020. The overall median survival was estimated using the Kaplan–Meier method. Between 2005–2020 the ACFDR followed 4,601 people with CF, noting 516 (11.2%) deaths including 195 following lung transplantation. Out of the total sample, more than half (52.5%) were male and 395 (8.6%) had undergone lung transplantation. Two thirds of people with CF (66.1%) were diagnosed before six weeks of age or by newborn/prenatal screening. The overall median age of survival was estimated as 54.0 years (95% CI: 51.0–57.04). Estimated median survival increased from 48.9 years (95% CI: 44.7–53.5) for people with CF born in 2005–2009, to 56.3 years (95% CI: 51.2–60.4) for those born in 2016–2020. Factors independently associated with reduced survival include receiving a lung transplant, having low FEV1pp and BMI. Median survival estimates are increasing in CF in Australia. This likely reflects multiple factors, including newborn screening, improvement in diagnosis, refinements in CF management and centre-based multidisciplinary care.
Similar content being viewed by others

Introduction
Cystic fibrosis (CF) is the most common autosomal recessive-inherited life-limiting condition, affecting approximately 90,000 individuals worldwide1. People with CF require support from healthcare services from diagnosis onwards; and respiratory failure is the commonest cause of premature death2. Although life expectancy for people with CF has increased substantially, the disease continues to result in reduced life expectancy, poorer quality of life, and a large burden of care for people with CF, their families and health care providers3. Prognosis continues to improve due to advancements in CF care4, such as the availability of new inhaled antibiotics, newborn screening (NBS), P. aeruginosa eradication therapy, mucolytic treatment, better growth and nutrition, lung transplantation and lifetime multidisciplinary care in specialised CF centres5.
Although CF survival estimates have greatly improved globally, survival continues to be influenced by various individual factors6. A recent study of UK registry data demonstrated that male sex was associated with better survival, as was later diagnosis in adulthood, but only in non-F508del homozygotes. Survival did not differ by genotype among individuals diagnosed in early infancy2. In the recent study of Durda-Masny et al7, the shortest life expectancy was observed in adult patients with a severe mutation on both alleles, Forced Expiratory Volume in one second percent predicted (FEV1pp) < 40%, patients infected with extensively drug-resistant P. aeruginosa, and body mass index (BMI) < 18.5 kg/m2. Most of the deaths in these people occurred between 30 and 40 years of age.
Providing up-to-date estimates of survival is helpful for counselling people with CF and their families on life expectancy, planning social and healthcare needs, optimising educational and occupational opportunities, guiding genetic counselling, the development of new therapies and evaluating the effectiveness of health interventions. Survival data is important in the development of evidence-based guidelines and standards of care for CF management and workforce planning, including screening, monitoring and management of age-related co-morbidities and complications. Further, comparisons of survival internationally promote equitable global health outcomes. Using a standardised approach to data processing and survival calculations will provide greater confidence in international comparisons and in the identification of factors that may contribute to the observed differences2,8.
The objectives of this study were to use a standardised approach for estimating survival among patients participating in the Australian CF registry. Specific aims of this study were to:1) estimate median survival for Australian people with CF, 2) identify factors associated with survival, and 3) estimate median age of survival in successive 5-year cohorts beginning with the period 2005–2009 up until 2016–2020.
Material and methods
Data source
This population-based cohort study used prospectively collected Australian Cystic Fibrosis Data Registry (ACFDR) data from 2005 to 2020 inclusive. The available data spanned 1998–2020, but analyses were limited to a cohort eligible from January 2005 through July 2021 to reflect completeness of dates of clinic visits used in the analysis.
The ACFDR contains detailed demographic and clinical information about people with a confirmed diagnosis of CF, receiving clinical care at 23 CF centres in Australia9. It captures > 90% of all Australians with CF via enrolment in the registry, and at the end of 2020 there were 3,538 Australians diagnosed with CF whose records were registered with the ACFDR9.
Data including age, sex, age of diagnosis, symptoms at presentation, diagnosis by NBS/prenatal screening, date of transplantation, date and cause of death, as well as anthropometric measurements, lung function, microbiology, CF-related complications, and pancreatic status were collected. Data were censored at the time of transplantation.
A detailed description of the registry is provided elsewhere9,10,11.
Definitions of key variables
Diagnosis was categorized as: those detected with meconium ileus symptoms; diagnosed before 6 weeks of age or by NBS/prenatal screening; diagnosed 6 weeks– < 2 years of age; 2–17 years and ≥ 18 years of age. Genotype was classified as F508del homozygous or other. Lung function, specifically the forced expiratory volume in one second (FEV1), was recorded in litres and the percent predicted (FEV1pp) was calculated using Global Lung Function Initiative (GLI) reference equations12. BMI was calculated using weight/height2. Adult individuals (≥ 18 years) were classified into BMI categories based on World Health Organization guidelines as underweight (< 18.5 kg/m2), adequate weight status (18.5–24.9 kg/m2), or overweight (≥ 25.0 kg/m2)13. Children were classified into BMI percentile categories based on Australian New Zealand CF Nutrition guidelines as underweight (< 10th percentile), adequate weight (10–85th percentile), or overweight (> 85th percentile)14. Pancreatic insufficiency (PI) was determined by pancreatic enzyme use at the first data entry into the registry. Missing values were included as a separate category in the regression models.
Statistical analyses
Descriptive statistics were used to describe the study population. Demographic and clinical variables were summarised with categorical variables expressed as frequency and proportion and continuous variables summarised as mean, standard deviation (SD) and range.
Overall survival probability was estimated using Kaplan–Meier survival curve15. Period survival analysis was used to calculate median age of survival estimates over time16. Median age of survival was calculated for each 5-year window beginning with the period 2005–2009 and ending with 2016–2020. Univariate and multivariable Cox proportional hazard models with age as the underlying time were used to assess the associations between personal and clinical characteristics and mortality17.
Data were left truncated at the age on the 1 January of the year individuals were enrolled in the ACFDR and right censored at the date on which they were last seen. Death was defined as the event. Date of diagnosis was set at 30 days post date of birth for people with missing date of diagnosis. Any post-lung transplantation FEV1 and BMI measurements were removed from the analyses. The following personal and clinical characteristics were included in the model: sex (male vs female, time-independent variable), diagnosis category ((i) with meconium ileus symptoms, (ii) under < 6 weeks or by NBS/prenatal screening, (iii) 6 weeks– < 2 years, (iv) 2–17 years and (v) ≥ 18 years groups, time-independent variable), genotype (F508del homozygous or not, time-independent variable), pancreatic status (sufficient vs insufficient, due to uncertain quality of the earlier data, this variable was considered as time-independent), BMI (underweight, adequate weight status or overweight, categorical time-dependent variable), lung transplantation (binary, time-dependent variable), and FEV1pp (≥ 70, 40–69, or < 40), categorical time-dependent variable). FEV1 data were not recorded in those < 6 years of age.
All the analyses were performed in R version 4.1.1 and survival library 3.2 (https://www.R-project.org).
Ethics approval
The study had ethics approval from the Alfred Health Human Research Ethics Committees, Melbourne, Victoria, Australia (Project Number HREC/16/Alfred/187). Informed consent was obtained from individuals and parents/guardians of those < 18 years of age, and for sites where local ethics committee required this, and an opt-out model was applied to the other sites where patients had the opportunity to contact the registry and opt out from the registry. All methods were carried out in accordance with relevant guidelines and regulations.
Results
There were 4,601 people with CF observed over the period of 2005–2020 in the ACFDR. Table 1 shows basic demographic and clinical characteristics of the study sample.
There were more males (52.5%). Nearly half (45.6%) of all individuals with CF were F508del homozygous. Two thirds of people with CF (66.1%) were diagnosed < 6 weeks of age or by NBS/prenatal screening. Less than a third (23.5%) of the study population were pancreatic sufficient. Participants who survived were more likely to be diagnosed before 6 weeks of age, via NBS/prenatal screening (67.2% vs. 57.4%) and to have not received a transplant (95.1% vs. 62.2%).
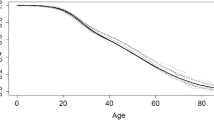
Mean BMI for adults and BMI percentile for children were similar in survivors compared to non-survivors. Overall median survival age of individuals with CF in Australia was estimated as 54.0 years (95% CI: 51.0–57.4) (Fig. 1A). Survival analysis using the Kaplan–Meier method shows the survival of people with CF is almost 100% up to the age of 12 and then starts to gradually decline. Of those people followed to the end of 2020, 648 (14.1%) were aged 40 years or more or had reached > 40 years at age of death. At the time of the data analysis, 69 (1.5%) of those followed were aged > 60 years or had reached > 60 years at death. Of those aged > 60 years, 38 (55%) were diagnosed when they were adults.

(A) Overall survival curve for people with cystic fibrosis, based on Kaplan–Meier estimates. (B) Probability functions depicting the age of people with cystic fibrosis by diagnosis category (with meconium ileus symptoms, under < 6 weeks or by NBS/prenatal screening, 6 weeks – < 2 years, 2–17 years and ≥ 18 years groups). (C) Probability functions depicting the age of people with CF by average lifetime lung function (FEV1 pp categories ≥ 70, 40–69, < 40). (D) Probability functions depicting the age of people with cystic fibrosis by BMI. Adult individuals (≥ 8 years) were classified into BMI categories based on World Health Organization guidelines as underweight (< 18.5 kg/m2), adequate weight (18.5–24.9 kg/m2), or overweight (≥ 25.0 kg/m2). Children were classified as underweight (< 10th percentile), adequate weight (10–85th percentile), or overweight (> 85th percentile).
The Kaplan–Meier curves varied significantly depending on individual characteristics such as age at diagnosis (p value of the log-rank score test = 0.001 (Fig. 1B), lung function (p value < 0.001) (Fig. 1C), and BMI (p value < 0.001) (Fig. 1D).
Table 2 displays the univariate and multivariable hazards ratios (HR) with 95% CIs, assessing the effects of the predictors of death.
In the multivariable model which was adjusted for individual characteristics, no difference in survival between males and females was observed (HR 1.00, 95% CI: 0.83–1.19). Compared to those who were diagnosed with meconium ileus symptoms, the risk of death was 37% higher in those who were diagnosed aged < 6 weeks or by NBS/prenatal screening (HR 1.37, 95% CI: 1.00–1.88). This effect was observed in the multivariable model only. The risk of death was highest in those who underwent lung transplant (HR 3.56, 95% CI: 2.84–4.47). In the model adjusted for both demographic characteristics and clinical factors, the risk of death remained significantly higher in those with lifetime average FEV1pp of 40–69% (HR 4.60 95% CI 3.33–6.36) and with lifetime average FEV1pp of < 40% (HR 9.77, 95% CI 6.84–13.96). In terms of BMI, those who were underweight (reference category) were at higher risk of death. In the multivariable model pancreatic status was not associated with death.
Table 3 (represented in Fig. 2), shows that the estimated median 5-year survival has increased over a 5-year period from 48.9 (95% CI: 44.7–53.5) years for people born in 2005–2009, to 56.3 (95% CI: 53.0–60.4) years for those born in 2016–2020.

Median survival of people with cystic fibrosis in Australia (5-year cohorts), 2005–2020. Black circles indicate median age data. Error bars represent 95% confidence intervals data. Summary statistics are provided in Table 3.
Figure 2 shows median survival of people with CF in Australia. Each dot and line represent the estimated median survival age and 95% CI, respectively, shown from 2005 to 2020.
Discussion
This study described estimated survival of people with CF in Australia with survival in those born in 2016–2020 emerging as 56.3 years (95% CI: 51.2–60.4) utilising the data from 2005–2020. Our data demonstrates the increase in median estimated survival among Australian people with CF over the past 15 years, with survival estimates that are comparable with international estimates. According to our study findings, the greatest risk factors for worse survival were low lung function and poorer nutritional status, emphasising the need for ongoing targeted interventions that slow the progression of lung disease and improve nutrition.
National CF registries are valuable tools for performing quality survival analyses and have been instrumental in demonstrating improved survival6. Many CF registries show the median age at death, supplemented by a graph representing the distribution of ages at death or the time trends in this median age at death. The Canadian, Irish, UK and US registries determine the estimated median age of survival based on the period approach2,8,16,18.
Our results present overall and period survival of people with CF in Australia and the factors associated with survival. In 2012, the median age of survival varied between countries: 47.0 years in Australia (95% CI: 43.8–51.5), 49.7 years in Canada (95% CI: 46.1–52.2)5, 43.5 years in the UK (95% CI: 37.6–49.9)19 and 41.1 years in the US (95% CI: 37.4–43.1)20. In recent years the estimated median age of survival in Australia was not dissimilar to that reported by other registries. For example, in 2019 the median age of survival was estimated to be 54.3 years of age in Canada21 and the Cystic Fibrosis Foundation Registry Report from the US calculated the predicted median survival age of a child born that year with CF to be 53 years22.
Factors associated with worse survival in the Australian CF population include receiving a lung transplant, lower lung function, and low BMI. In contrast to previous studies, an unexpected association between survival and earlier age of diagnosis was observed in our study. This effect could be explained by a potentially higher proportion of those with severe genotypes identified via NBS or presenting very early, the poorer quality of the diagnosis data captured in earlier years of the data registry, or a chance finding.
Sex and pancreatic status were not found to be independently associated with the survival probability. This finding is different from the previously reported registry survival studies2. The study by MacKenzie et al23 reported that sex, F508del status and increasing age at diagnosis were independently associated with survival in the US, and female sex has long been associated with worse survival in CF compared to males2,24. It is possible that the gender gap in Australia is narrowing through improvements in treatment, diagnosis and lung function trajectory in females25,26, although further studies are required to confirm this.
Published data suggest that receiving a lung transplant is associated with earlier death27. This finding was also observed in our study. Similarly, poorer lung function and lower BMI were also found to be associated with increased odds of mortality6,7,8,27,28,29. The results of our study and previous studies6,27,30 confirm the impact of lung function and nutritional status on the survival of people with CF, showing that severe and moderate pulmonary impairment and undernutrition have a major impact on survival. These findings justify the emphasis on interventions aimed at optimising pulmonary function and nutritional status in CF guidelines and standards of care31.
Improving median survival age in CF can be attributed to many different factors including NBS, nutritional interventions, proactive disease surveillance at both individual and population levels (including via the ACFDR), management of respiratory infections, access to novel therapies and improved standards of care16,32,33. The impact of NBS on survival estimates would not be evident for several decades until babies screened at birth reached an age where they would be at risk for death. NBS programmes for CF were first implemented in the early 1970s in the Royal Gwent Hospital in UK34. In Australia, NBS started in 1981and was progressively implemented across different states/jurisdictions becoming universal in 200135, potentially explaining increased age of survival in recent years. Those identified by NBS have improved nutritional status and growth compared to people who are diagnosed by symptoms later in life36, and some studies indicate improved pulmonary function37. People identified at an early age benefit from early interventions to optimize nutrition, prevent and treat lung disease, and early monitoring for liver disease and other complications38.
CFTR modulator therapies have the potential to reduce symptoms and increase survival for an increasing number of people with CF6. In Australia, the first CFTR modulator (ivacaftor) was approved for use from December 2014, with lumacaftor/ivacaftor (OrkambiR) tezacaftor/ivacaftor (SymdekoR) and elexacaftor/tezacaftor/ivacaftor (TrikaftaR) available for patients from October 2018, December 2019 and April 2022 respectively9. It is too soon to see the impact of these novel therapies on survival although the estimated median age of survival of people with CF in Australia is expected to continue to improve. The Australian CF registry data is an essential tool to evaluate the impact of CFTR modulator therapies on future clinical outcomes including survival long-term6.
Strengths and limitations
The strengths of our study include the large sample size, the longitudinal data within the ACFDR, the consistency of our results across multiple sub-groups and the unified approach to the analysis. There is a very high participation rate at the centre level as well as in the registry as since mid-2019 participating centres receive payment for data submission which results in a comprehensive national picture of the CF population in Australia11. In addition, this study is of the longest running cohort of patients diagnosed via NBS35.
As the ACFDR does not capture identifiable data, some death and transplant data could not be verified through national linkages. In addition, data pertaining to CF co-morbidities, such as CF-related diabetes and chronic infections with P. aeruginosa were not analysed separately, and could influence survival outcomes. Certain data elements were incompletely recorded for the earlier years for this analysis (i.e. microbiology, CF-related diabetes and socioeconomic status) thus we were unable to account for these factors in our analyses. The differentiation of NBS diagnosis and clinical diagnosis based on meconium ileus /failure to thrive in first few weeks is not absolutely clear and could affect the quality of the diagnosis data. A database redesign conducted in 2018, and a new format of the registry will enable better quality and completeness of the registry records and greater accuracy of future analyses arising from the registry data9,10.
Conclusions
We have demonstrated successive improvements in survival among people with CF in Australia over the last 15 years and identified factors influencing survival. Further research is needed to understand the complex interactions between biological and epidemiological factors not examined in this current study on the severity of disease and survival.
The increase in survival and longevity requires an evolution in models of CF care towards prevention and management of age-related comorbidities such as diabetes, metabolic and cardiovascular disease, malignancies and osteoporosis alongside nutritional and respiratory care39. Increasing longevity must be coupled with increasing quality of life and the ability for people with CF to actively participate in a broad range of personal, community and work-based activities. Accordingly, these survival data presented highlight the need for future care models to incorporate a greater focus on the psychological, social, educational and occupational potential of people with CF as they live into old age.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on request and pending approval to the ACFDR Data Access and Research Publishing Committee.
References
Schmidt, B. Z., Haaf, J. B., Leal, T. & Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: current perspectives. Clin. Pharmacol. 8, 127–140. https://doi.org/10.2147/cpaa.S100759 (2016).
Keogh, R. H., Szczesniak, R., Taylor-Robinson, D. & Bilton, D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 17, 218–227. https://doi.org/10.1016/j.jcf.2017.11.019 (2018).
Bell, S. C. et al. The future of cystic fibrosis care: A global perspective. Lancet. Respir. Med. 8, 65–124. https://doi.org/10.1016/s2213-2600(19)30337-6 (2020).
Corriveau, S., Sykes, J. & Stephenson, A. L. Cystic fibrosis survival: the changing epidemiology. Curr. Opin. Pulm. Med. 24, 574–578. https://doi.org/10.1097/mcp.0000000000000520 (2018).
Stephenson, A. L. et al. A contemporary survival analysis of individuals with cystic fibrosis: A cohort study. Eur. Respir. J. 45, 670–679. https://doi.org/10.1183/09031936.00119714 (2015).
Scotet, V., L’Hostis, C. & Férec, C. The changing epidemiology of cystic fibrosis: incidence, survival and impact of the CFTR gene discovery. Genes Basel 11, 3390. https://doi.org/10.3390/genes11060589 (2020).
Durda-Masny, M. et al. The determinants of survival among adults with cystic fibrosis-a cohort study. J. Physiol. Anthropol. 40, 19. https://doi.org/10.1186/s40101-021-00269-7 (2021).
Stephenson, A. L. et al. Survival comparison of patients with cystic fibrosis in Canada and the United States: A population-based cohort study. Ann. Intern. Med. 166, 537–546. https://doi.org/10.7326/m16-0858 (2017).
Ahern, S. et al. The ACFDR registry annual report, 2020. Monash university, Department of epidemiology and preventive medicine, July 2021, Report No 22. (2021).
Ahern, S. et al. Redesign of the Australian cystic fibrosis data registry: A multidisciplinary collaboration. Paediatr. Respir. Rev. 37, 37–43. https://doi.org/10.1016/j.prrv.2020.03.001 (2020).
Ahern S. et al. on behalf of the ACFDR. The australian cystic fibrosis data registry, annual report, 2019. Monash university, department of epidemiology and preventive medicine, January 2021, Report No 21. (2021).
Quanjer, P. H. et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur. Respir. J. 40, 1324–1343. https://doi.org/10.1183/09031936.00080312 (2012).
World Health Organization. BMI Classification. Geneva, Switzerland: 2013. p. 2012 Ref Type: Online Source.
van der Haak, N. et al. Highlights from the nutrition guidelines for cystic fibrosis in Australia and New Zealand. J. Cyst. Fibros. Official J Eur. Cyst. Fibros. Soc. 19, 16–25. https://doi.org/10.1016/j.jcf.2019.05.007 (2020).
Goel, M. K., Khanna, P. & Kishore, J. Understanding survival analysis: Kaplan-Meier estimate. Int. J Ayurveda Res. 1, 274–278. https://doi.org/10.4103/0974-7788.76794 (2010).
Keogh, R. H. & Stanojevic, S. A guide to interpreting estimated median age of survival in cystic fibrosis patient registry reports. J. Eur. Cyst. Fibros. Soc. 17, 213–217. https://doi.org/10.1016/j.jcf.2017.11.014 (2018).
Muggeo, V. M. Estimating regression models with unknown break-points. Stat. Med. 22, 3055–3071. https://doi.org/10.1002/sim.1545 (2003).
Ramos, K. J. et al. Survival and lung transplant outcomes for individuals with advanced cystic fibrosis lung disease in the United States and Canada: An analysis of national registries. Chest https://doi.org/10.1016/j.chest.2021.04.010 (2021).
Cystic Fibrosis: our focus. Cystic Fibrosis Trust; Kent: 2013. UK Cystic Fibrosis Registry Annual data report 2012.
Cystic Fibrosis Foundation Patient Registry. 2012 Annual Data Report to the Center Directors (Cystic Fibrosis Foundation; Bethesda, Maryland, 2013).
Canadian Cystic Fibrosis Registry. Annual data report 2019. Available online: https://www.cysticfibrosis.ca/uploads/RegistryReport2019/2019RegistryAnnualDataReport.pdf.
Cystic Fibrosis Foundation Patient Registry. 2021 Cystic fibrosis foundation patient registry highlights cystic fibrosis foundation. https://www.cff.org/medical-professionals/patient-registry.
MacKenzie, T. et al. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: Survival analysis of the Cystic Fibrosis Foundation patient registry. Ann. Intern. Med. 161, 233–241. https://doi.org/10.7326/m13-0636 (2014).
Calella, P., Valerio, G., Brodlie, M., Donini, L. M. & Siervo, M. Cystic fibrosis, body composition, and health outcomes: A systematic review. Nutr. Burbank Los Angel. Ctry. Calif. 55–56, 131–139. https://doi.org/10.1016/j.nut.2018.03.052 (2018).
Lai, H. C., Kosorok, M. R., Laxova, A., Makholm, L. M. & Farrell, P. M. Delayed diagnosis of US females with cystic fibrosis. Am. J. Epidemiol. 156, 165–173. https://doi.org/10.1093/aje/kwf014 (2002).
Viviani, L., Bossi, A. & Assael, B. M. Absence of a gender gap in survival. An analysis of the Italian registry for cystic fibrosis in the paediatric age. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 10, 313–317. https://doi.org/10.1016/j.jcf.2011.03.007 (2011).
Stephenson, A. L. et al. Clinical and demographic factors associated with post-lung transplantation survival in individuals with cystic fibrosis. J. Heart Lung Transplant. 34, 1139–1145. https://doi.org/10.1016/j.healun.2015.05.003 (2015).
Keogh, R. H., Seaman, S. R., Barrett, J. K., Taylor-Robinson, D. & Szczesniak, R. Dynamic prediction of survival in cystic fibrosis: A landmarking analysis using UK patient registry data. Epidemiology 30, 29–37. https://doi.org/10.1097/ede.0000000000000920 (2019).
McKone, E. F. et al. Survival estimates in European cystic fibrosis patients and the impact of socioeconomic factors: A retrospective registry Cohort study. Eur. Respir. J. https://doi.org/10.1183/13993003.02288-2020,10.1183/13993003.02288-2020 (2021).
Szwed, A. et al. Survival of patients with cystic fibrosis depending on mutation type and nutritional status. Adv. Exp. Med. Biol. 1023, 65–72. https://doi.org/10.1007/5584_2017_66 (2018).
Cystic Fibrosis Australia. Cystic Fibrosis Standards of Care, Australia (Cystic Fibrosis Australia, 2008).
Goss, C. H. et al. Comparison of nutrition and lung function outcomes in patients with cystic fibrosis living in Canada and the United States. Am. J. Respir. Crit. Care Med. 197, 768–775. https://doi.org/10.1164/rccm.201707-1541OC (2017).
Zampoli, M. et al. Trends in cystic fibrosis survival over 40 years in South Africa: An observational cohort study. Pediatr. Pulmonol. https://doi.org/10.1002/ppul.25810 (2021).
Prosser, R. et al. Screening for cystic fibrosis by examination of meconium. Arch. Dis. Child. 49, 597–601. https://doi.org/10.1136/adc.49.8.597 (1974).
Waters, D. L. et al. Clinical outcomes of newborn screening for cystic fibrosis. Arch. Dis. Child Fetal. Neonatal. Ed. 80, F1-7. https://doi.org/10.1136/fn.80.1.f1 (1999).
Farrell, P. M. et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth wisconsin cystic fibrosis neonatal screening study group. Pediatrics 107, 1–13. https://doi.org/10.1542/peds.107.1.1 (2001).
Grosse, S. D. et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm. Rep. 53, 1–36 (2004).
McBennett, K. A., Davis, P. B. & Konstan, M. W. Increasing life expectancy in cystic fibrosis: Advances and challenges. Pediatr. Pulmonol. 57(Suppl 1), S5-s12. https://doi.org/10.1002/ppul.25733 (2022).
McDonald, C. M. et al. Academy of nutrition and dietetics: 2020 cystic fibrosis evidence analysis center evidence-based nutrition practice guideline. J. Acad. Nutr. Diet. 121, 1591-1636.e1593. https://doi.org/10.1016/j.jand.2020.03.015 (2021).
Acknowledgements
The authors thank the participating CF centres, data entry personnel, survey participants, patients and their families.
Funding
This study was supported by Cystic Fibrosis Australia.
Author information
Authors and Affiliations
Contributions
R.R., S.A., A.E. and S.B. conceived the study. R.R., A.E. and F.S. performed data processing and statistical analysis. R.R., F.S. and S.A. were responsible for the creation of the original draft of the manuscript that was revised and approved by all authors for important intellectual content. T.D., K.F., L.K., S.K., T.K., P.G.M., S.M., S.M., A.S., C.W., N.W. and P.W. contributed to the development of the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruseckaite, R., Salimi, F., Earnest, A. et al. Survival of people with cystic fibrosis in Australia. Sci Rep 12, 19748 (2022). https://doi.org/10.1038/s41598-022-24374-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24374-4
- Springer Nature Limited
This article is cited by
-
Transition Care in Cystic Fibrosis
Indian Journal of Pediatrics (2023)