
Abstract
Resistance to anthracnose caused by the fungal pathogen Colletotrichum lentis was explored through transcriptome sequencing over a period of 24 to 96 h post-inoculation (hpi) of the partially resistant recombinant inbred lines (RIL) LR-66-528 and susceptible LR-66-524 of the crop wild relative Lens ervoides population LR-66. The development of infection vesicles and primary hyphae by C. lentis were significantly higher on susceptible RIL LR-66-524 compared to partially resistant LR-66-528 at 24 and 48 hpi, but exponential trends in fungal growth were observed between 24 to 96 hpi in both RILs. Comparison of inoculated with mock-inoculated samples revealed 3091 disease responsive genes, among which 477 were differentially expressed between the two RILs. These were clustered into six expression clusters with genes that had either high or low expression in one of the RILs. Differentially expressed genes (DEGs) were functionally annotated and included genes coding LRR and NB-ARC domain disease resistance proteins, protein detoxification, LRR receptor-like kinase family proteins, and wall-associated Ser/Thr Kinases. DEGs were compared to genes in previously published anthracnose resistance QTLs mapped in LR-66 and revealed 22 DEGs located in 3 QTLs. Expression of 21 DEGs was validated using RT-qPCR confirming expression trends in RNA-seq.
Similar content being viewed by others


Introduction
Anthracnose caused by the hemibiotrophic fungal pathogen Colletotrichum lentis Damm is currently the most important foliar disease of cultivated lentil (Lens culinaris Medik.) in Canada, where it was first discovered in the province of Manitoba in 1987 before spreading into Saskatchewan in the 1990s1. It has also been reported from lentil crops in the USA, Bulgaria, Pakistan and New Zealand2. Yields losses of up to 100% have been encountered in worst case scenarios triggered by short crop rotations, frequent rainfall and high temperature3. Two pathogenic races of C. lentis infect the lentil crop in Canada4. Race 0 is dominating and more aggressive, and no high levels of resistance have been identified in cultivated lentil, whereas partial resistance to race 1 was identified and incorporated in lentil cultivars5. Wild relatives of crop species are repositories of allelic diversity for disease resistance, and accessions of Lens ervoides (Brign.) in the tertiary gene pool of Lens with superior resistance to anthracnose were discovered6. Using interspecific hybridization involving embryo rescue, transfer of resistance to C. lentis race 1 and race 0 from L. ervoides accession L01-827A to lentil cultivar Eston was achieved7,8. However, lack of understanding of the mechanisms and control of resistance has prevented the development of markers to trace the introgression of genes into breeding lines. The intraspecific recombinant inbred line (RIL) population LR-66 was developed from crossing L. ervoides accessions L01-827A and IG 72,8159 and allowed for the identification of QTLs conferring resistance to anthracnose through phenotyping and SNP-based genotyping10. In this study, the development of C. lentis on and in leaflets was investigated for two RILs of LR-66 with contrasting levels of resistance to identify specific stages of fungal growth for a transcriptome study. An RNA-seq time-series experiment was conducted on these two C. lentis-infected RILs to identify differentially expressed genes (DEGs) to gain an overview of host responses during infection.
Results
Quantification of fungal development
The development of C. lentis isolate CT-30 was studied in susceptible L. ervoides RIL LR-66-524 and partially resistant LR-66-528 through histopathological observations and an assessment of fungal biomass, estimated through quantification of genomic C. lentis ACTIN relative to the L. culinaris housekeeping gene Elongation factor 1-alpha (EF1-α) by qPCR. Appressoria formation was observed by 6 hpi in both genotypes but at variable numbers. Fungal biomass did not proliferated much from 6 to 72 hpi (Fig. 1a), most of which represented the biotrophic phase of C. lentis. Infection vesicle/primary hyphae were visible beneath some appressoria in susceptible LR-66-524 by 24 hpi, but not in partially resistant LR-66-528, where they had formed by 48 hpi (Fig. 1b). More infection vesicle/primary hyphae were observed in the susceptible than in the partially resistant RIL. A dramatic increase in fungal biomass was observed at 120 hpi in the susceptible RIL LR-66-524, whereas it peaked at a lower level at 96 hpi in the partially resistant RIL LR-66-528 (Fig. 1a). However, significant differences were only observed at 24 hpi. Secondary hyphae were not visible as they did not stain well during sample preparation. The percentage of dead tissue and the percentage of leaflet area covered by acervuli per leaflet due to infection increased with incubation time (Fig. 1c,d). There was significantly more dead leaf tissue in susceptible LR-66-524 than in partially resistant RIL LR-66-528 at 96, 120 and 144 hpi, and more leaflet area covered by acervuli at 120 and 144 hpi. Although fungal growth appeared to level off after 96 h in both RILs, there was a trend for higher biomass of C. lentis in susceptible RIL LR-66-524 as compared to partially resistant RIL LR-66-528 (Fig. 1a). Based on these observations, 24, 48, 72 and 96 hpi were selected for gene expression studies.

Quantitative parameters assessed for the infection process of Colletotrichum lentis race 0 isolate CT-30. Infection structures and symptoms were assessed in susceptible recombinant inbred lines LR-66-524 and partially resistant LR-66-528 of Lens ervoides population LR-66. (a) Relative fungal biomass of C. lentis expressed as the Log2 fold change of C. lentis ClACT (determined by qPCR) relative to mock (non- inoculated) samples. Error bars represent standard deviations of three biological replicates. (b) Proportion of infection vesicle (IV)/primary hyphae (PH) per 25 appressoria from 24 to 48 hpi. (c) Percentage of dead tissue per leaflet from 96 to 144 hpi. (d) Percentage of leaflet area covered with acervuli. (b–d) Error bars represent standard errors of the means of 3 biological replicates.
Analyses of transcriptome variability between RILs
RNA sequencing from inoculated leaves of susceptible RIL LR-66-524 and partially resistant RIL LR-66-528 sampled 24, 48, 72 and 96 hpi generated approximately 10–14 million high-quality raw reads as paired-end sequences in a FASTQ format with a length of 125 bp per library. The STAR aligner output showed that approximately 80–85% of the filtered reads aligned to the annotated L. culinaris genome. Most of the remaining reads were assumed to be of fungal origin, and some were probably specific to L. ervoides. The read counts file generated during the mapping process had more than 30,000 genes. The gene read counts were used to analyze the transcriptome variability among the samples. Principal Component Analysis revealed increasing differences between the RILs as time after inoculation increased (Fig. 2). Principal Variance Component Analysis conducted on samples of the two RILs collected in replicates at 24, 48, 72 and 96 hpi indicated that they collectively accounted for 79.9% of the total variance. The largest variance proportion (69.8%) could be attributed to incubation time (hpi), followed by the hpi x RILs (1.8%) and block (0.8%), whereas RILs only contributed 0.4%.

Three-dimensional Principal Component Analysis plot representing the variability among sequencing samples. RNA-seq data were generated from partially resistant recombinant inbred lines RIL LR-66-528 and susceptible RIL LR-66-524 of Lens ervoides population LR-66 inoculated with race 0 isolate CT-30 of Colletotrichum lentis and incubated for 24, 48, 72 and 96 h. Data is represented in x-y-z coordinates with principal component axes (pca1, pca2 and pca3) representing genotypes, biological replicates and time-points.
Disease responsive DEGs between the two RILs were separated from other differentially expressed genes by comparing inoculated with mock-inoculated samples. As a result, 3091 genes were identified with a fold change larger than 2 (Padj < 0.05) compared to mock-inoculated samples (Supplementary Table S1). Interestingly, in the partially resistant RIL LR-66-528, 2511 disease-responsive genes were expressed each at 24 and 48 hpi compared to 352 and 561 in the susceptible LR-66-524, whereas that trend was reversed by 96 hpi when 2511 disease-response genes were expressed in LR-66-524 compared 1968 in LR-66-528. Among all of the 3091 disease responsive DEGs, 477 genes were differentially expressed between the two RILs (fold change > 2, Padj < 0.05, Supplementary Table S2) indicating that 85% of disease-responsive genes were commonly expressed by both RILs. Among the 477 genes, 80 genes were differentially expressed between the two RILs during the entire period lasting from 24 to 96 hpi (Fig. 3). Only two genes were differentially expressed between the two RILs exclusively at 24 hpi or at 96 hpi, whereas 108 were at 48 hpi and 38 genes at 72 hpi. Up to 185 genes were differentially expressed between the two RILs at more than one time point.

Venn diagram showing the number of genes differentially expressed between partially resistant recombinant inbred line LR-66-528 and susceptible RIL LR-66-524 of Lens ervoides population LR-66 in response to inoculation with race 0 isolate CT-30 of Colletotrichum lentis and incubated for 24, 48, 72 and 96 h. Violet color represents genes differentially expressed between LR-66-528 and RIL LR-66-524 exclusively at 24 hpi, yellow for genes at 48 hpi, green for genes at 72 hpi and pink for genes at 96 hpi. Genes in overlapping areas are differentially expressed between LR-66-528 and RIL LR-66-524 at more than one time-point.
The 477 DEGs clustered into six expression clusters based on K-mean clustering (Fig. 4). Cluster 1 contained 56 genes, 75% of which had high expression as indicated by z-scores higher than zero at 48, 72 and 96 hpi in susceptible RIL LR-66-524, compared to 21% in partially resistant LR-66-528. All of the 80 genes in Cluster 2 had z-scores higher than zero at 96 hpi in LR-66-524, whereas that ranged from 12 to 37 at the other time points for this genotype, and from six to 50 in LR-66-528. Among a total of 92 genes in Cluster 3, 86 and 87 had z-scores higher than zero in the partially resistant RIL LR-66-528 primarily at 72 and 96 hpi, respectively, compared to a high of 60 genes in LR-66-524 at 96 hpi. Cluster 4 contained 98 genes that were highly expressed in LR-66-524 primarily at 24 (90 genes with z-scores > 0) and 48 hpi (83 genes with z-scores > 0), compared to 34 genes with z-scores higher than zero in LR-66-528 at those time points. Cluster 5 was the smallest of all the clusters consisting of 52 genes, all of which had z-scores higher than zero in LR-66-528 at 24 hpi, and 45 genes at 48 hpi, compared to 17 and 2 genes for those two time points in LR-36 66-524. Cluster 6 included 96 genes highly expressed in the partially resistant RIL LR-66-528 with z scores higher than zero for all genes at 48 hpi and 64 genes at 72 hpi, whereas in LR-66-524 this was only the case for 43 at 48 hpi and 59 genes at 72 hpi.

Hierarchical cluster analysis of gene expression profiles of differentially expressed genes. Genes were differentially expressed between partially resistant recombinant inbred line LR-66-528 and susceptible RIL LR-66-524 of Lens ervoides population LR-66 in response to inoculation with race 0 isolate CT-30 of Colletotrichum lentis and incubated for 24, 48, 72 and 96 h. C1 to C6 represent different clusters based on the expression of genes. Heat map shows the normalized expression levels of transcripts represented by a color spectrum ranging from red (high expression levels) to blue (low expression levels). The dendrogram shows Pearson’s correlation with an average distance among clusters.
The results of gene ontology showed that a total of 370 out of 477 DEGs had homology with the M. truncatula genome (Supplementary Table S2), among which 159 could be assigned to GO terms in different hierarchical clusters and were involved in different biological processes (Fig. 4, Supplementary Table S3). The GO terms for genes in Cluster 1 showed that these were enriched in cell death related processes such as “programmed cell death”, “regulation of cell death”, and in cell wall organization or biogenesis such as “cell wall macromolecule metabolic process”. Enriched GO terms for Cluster 2 were involved in responses to stress such as “response to water”, “response to water deprivation”, and cell morphogenesis involved in differentiation such as “pollen tube growth” and “cell tip growth”. GO terms for genes in Cluster 3 were enriched in protein phosphorylation processes such as “peptidyl-tyrosine dephosphorylation” and RNA processing such as “mRNA splicing, via spliceosome”, RNA splicing, via transesterification reactions” and “RNA splicing, via transesterification reactions with bulged adenosine as nucleophile”. GO mapping of genes in Cluster 4 showed that these were enriched for genes with function in cell growth and differentiation processes such as “cellular developmental process”, cellular biogenesis such as “plant-type secondary cell wall biogenesis”, “regulation of cellular component biogenesis”, and metabolic processes such as “hormone metabolic process” and “regulation of hormone levels”. GO terms for genes in Cluster 5 were enriched in carbohydrate and amino acid metabolic processes such as “oligosaccharide biosynthetic process” and “aspartate family amino acid metabolic process”. Go terms for genes in Cluster 6 were enriched with stress-related process such as “response to water”, DNA metabolic process such as “DNA recombination”, regulation of hydrolase activity such as “regulation of GTPase activity”, DNA packaging processes such as “chromatin assembly” and “nucleosome organization”.
Comparison of differentially expressed genes with QTLs
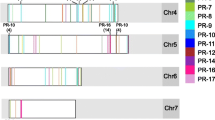
The projection of anthracnose resistance QTLs qANTH0-2, qANTH1-2.1 and qANTH1-2.2 present on linkage group 210 of the L. culinaris linkage map onto the physical map led to the identification of 3078 genes. The QTL intervals qANTH0-3, qANTH1-3.1 and qANTH1-3.2 on linkage group 3 contained 605 genes, and those of qANTH0-5.1, qANTH0-5.2, and qANTH1-5.2 present on linkage group 5 had 1437 genes. The projection of qANTH0-7 localized on linkage group 7 had a total of 713 genes. Comparison of these genes with DEGs identified by RNA-seq led to the identification of 22 DEGs, of which 9 DEGs were found on chromosome 2, 10 DEGs on chromosome 5 and 3 DEGs on chromosome 7 (Table 1). Two of these genes, Lc23518 and Lc09295, were included in the gene expression validation through RT-qPCR. Based on RNAseq analysis, Lc23518 had low expression at 24 hpi and was highly expressed at 96 hpi in the partially resistant RIL and was found in QTL interval qANTH0-5.1 localized on linkage group 5. Lc09295 was highly expressed at 96 hpi in the partially resistant RIL and was located in the QTL interval qANTH0-2 on linkage group 2.
Validation of gene expression using RT-qPCR
Based on functional annotations indicating a putative role in disease responses, 21 DEGs were selected for RT-qPCR at one, two or three time points, for a total of 28 RT-qPCR reactions. Expression trends estimated through RT-qPCR were the same as those estimated through RNA-seq; however, for nine out of the 28 RT-qPCR tests, or 32%, differences in gene expression were not significant (Supplementary Table S4). Fold changes for the expression of eight DEGs, assessed through RT-qPCR, in the partially resistant LR-66-528 and the susceptible LR-66-524 compared to the mock-inoculated controls of both RILs are presented in Fig. 5.

Fold changes in gene expression of selected genes and time points (hpi) after inoculation with Colletotrichum lentis in partially resistant Lens ervoides LR-66-528 and susceptible RIL LR-66-524 compared to mock-inoculated controls based on RT-qPCR. Fold changes (2-ΔΔCT) were calculated as described by Schmittgen and Livak40, using EF1-α for normalization. Bars indicate standard deviations. Lc20454: LRR receptor-like kinase, LC34856: F-box/LRR protein, Lc34550: LRR and NB-ARC domain disease resistance protein, Lc13986: Adenosylhomocysteinase (AdoHcyase) (3.3.1.1) (S-adenosyl-L-homocysteine hydrolase), Lc34767: TMV resistance protein N, Lc14149: LRR and ubiquitin-like domain plant-like protein, Lc33978: LRR receptor-like kinase.
Discussion
The hemibiotroph C. lentis, causal agent of anthracnose, is the most important foliar pathogen of lentil in Canada due to limited resistance in the cultivated species. Resistance to the dominating and aggressive race 0 of C. lentis has been identified in L. ervoides6 and interest in introgressing useful genes from L. ervoides into elite cultivars has increased facilitated by embryo rescue techniques that overcome interspecific reproductive barriers between L. ervoides and L. culinaris7. Nevertheless, progress in developing partially resistant varieties has been slow, to a large part because an understanding of resistance mechanisms and their underlying genes are lacking. Previously, five QTLs were associated with anthracnose resistance on a SNP-based linkage map of the F9 recombinant L. ervoides inbred line population LR-6610. However, the direct use of these QTLs for marker-assisted selection has been limited and identification of candidate genes associated with resistance has been difficult because of the relatively large size of QTL intervals and linkage drag in interspecific progeny.
In the current study, anthracnose development and gene expression in responses to infection were further explored on the most resistant and most susceptible RILs of LR-66. The differentiation of appressoria, infection vesicles and primary hyphae are the first crucial steps in the invasion of the host plant by C. lentis. Histopathological examination revealed that infection vesicles and primary hyphae were still absent in the partially resistant LR-66-528 at 24 hpi, but had already differentiated in the susceptible LR-66-524, indicating that penetration of C. lentis isolate CT-30 may have been delayed or the success rate may have been much lower in the partially resistant LR-66-528 compared to the susceptible RIL. A similar observation was made in the Arabidopsis thaliana—Colletotrichum higginsianum host-pathogen system where more than 50% of appressoria initiated successful penetrations through the leaf surface to form primary hyphae in susceptible accession Ler-0, compared to 10% of appressoria on partially resistant accessions Ws-0, Gifu-2, Can-0 and Kondara11. As a hemibiotroph, this initial biotrophic phase is essential for pathogenesis, after which C. lentis switches to necrotrophic growth, visually evident here in the development of cell death starting at 96 hpi. Compared to the partially resistant RIL LR-66-528, cell death was significantly more pronounced in the susceptible LR-66-524 and more leaflet area was covered with acervuli, the fruiting structures of C. lentis, signaling that C. lentis was more successful at completing its life cycle on LR-66-524 than on LR-66-528. Differences in cell death, lesion number, lesion size and colonization efficiency of moderately virulent C. lentis isolate JPPTNL 882 were previously observed on susceptible L. culinaris cv. Eston compared to partially resistant lentil line PI 320,937 at 72 to 144 hpi, whereas there were no differences for highly virulent isolate 95S29, which was probably a race 0 isolate similar to CT-30 used in the current study12. Adding to histopathological observations, determination of fungal biomass indicated a dramatic increase after 48 hpi in both RILs, with a trend in the partially resistant LR-66-528 to accommodate less C. lentis than the susceptible RIL LR-66-524. It was previously observed that C. lentis switched from the biotrophic to the necrotrophic phase after 48 hpi12,13, so exponential growth of C. lentis based on qPCR would correlate with that switch from biotrophy to necrotrophy.
A total of 477 genes among 3091 disease-responsive genes (15%) were found to be differentially expressed upon pathogen infection between partially resistant LR-66-528 and susceptible LR-66-524. The large number of common genes in both RILs responding to infection by C. lentis are probably involved in basal defense reactions, whereas the genes differentially expressed between the two lines are associated with the different levels of resistance in the RILs, which was much higher in LR-66-528 (5% disease severity) compared to LR-66-524 (72%)10. Separation of sequencing samples in the PCA plot were evident at 48 and 96 hpi and indicated that transcriptome responses of LR-66-528 and LR-66-524 to C. lentis infection diverged after 24 hpi. Samples of both genotypes from 24 hpi were positioned far from those collected at 48, 72 and 96 hpi and correlated with the progression from biotrophy to necrotrophy. PVCA showed that the highest proportion of the variance (69.8%) could be attributed to incubation time. The percentage of the variance attributed to genotypes was the lowest (0.4%) among all principal components, indicating that difference in the host responses were subtle. A much higher proportion of the variance (15%) could be attributed to two other RILs of LR-66 responding to the necrotrophic pathogen S. bortryosum14.
Two DEGs, Lc33118 and Lc27093 were expressed exclusively at 24 hpi and were highly expressed in the partially resistant RIL LR-66-528. Functional annotations of these genes determined that Lc33118 encodes a cytokinin receptor histidine kinase. Cytokinin and their receptors, histidine kinases, are involved in diverse developmental functions in plants, but also play a role in responses to pathogens where they were shown to positively and negatively regulate the development of biotrophs15. Treatment of Arabidopsis plants with exogenous cytokinin benzyl adenine (BA) 48 h prior to inoculation with the biotrophic pathogen Hyaloperonospora arabidopsidis at low concentrations increased susceptibility of the plants whereas high levels of BA increased resistance16. Cytokinins can also be secreted by certain pathogens to manipulate the host cell environment to its advantage thereby functioning as a virulence factor17. This was shown for C. graminicola and was associated with typical symptoms for high cytokinin referred to as green islands on senescing maize leaves17,18. A search of the C. lentis genome revealed that the enzyme isopentenyltransferases, which catalyses the first step in cytolkinin synthesis, is also present in this pathogen; however, high expression of the putative cytokinin receptor Lc33118 at 24 hpi in the partially resistant LR-66-528 here indicates that an increase in endogenous rather than pathogen-derived cytokinin may contribute to resistance in this RIL. This was further supported by the simultaneous high expression levels of Lc37093 in this RIL, which showed features of a CAP/cysteine rich secretory/antigen 5 protein. These proteins belong to the CAP protein superfamily (Cysteine-rich secretory proteins (CRISPs), Antigen 5 (Ag5), and Pathogenesis-related 1 (PR-1) proteins), proteins of which have been identified in animals, plants and fungi19. The most studied CAP proteins in plant defense are pathogenesis-related 1 (PR1) proteins20, and indeed, Lc37093 had the highest similarity with MtSTS14 (a PR-1 protein) in Medicago. In Arabidopsis, it was demonstrated that cytokinin and SA together strongly activate PR-1 expression through the transcription factor TGA321.
The highest number of DEGs (108) exclusively expressed at any one time point were identified at 48 hpi, which coincides with the biotrophy-necrotrophy switch. Many of these genes were highly expressed in the partially resistant RIL LR-66-528 and were associated with stress-related processes, DNA metabolic processes, the regulation of hydrolase activity and DNA packaging processes. Among these DEGs were also genes that regulated or coordinated the SA and ET/JA pathways, both of which are typically engaged in response to hemibiotrophs. Among the 38 DEGs exclusively expressed at 72 hpi, Lc29695, a disease resistance protein (TIR-NBS-LRR), and Lc33733, a Pentatricopeptide-repeat proteins (PPRs) containing plant protein, were highly expressed in the partially resistant RIL LR-66-528. PPRs were previously shown to be upregulated in rice in response to inoculation with Magnaporthe oryzae22. At 96 hpi, among numerous disease-responsive LRR receptor-like kinase family proteins, one (Lc34740) was expressed at high level at this time point in LR-66-528. Among many other roles, some LRR receptor-like kinase family proteins function as pattern recognition receptors (PRRs) that recognize pathogen associated molecular patterns (PAMPs) thereby regulating plant immune responses against invasive microorganisms23.
Other genes primarily highly expressed in the partially resistant RIL LR-66-528 were associated with RNA splicing (Cluster 3), the primary metabolic and transportation processes (Cluster 5) and processes such as “regulation of GTPase activity” (Cluster 6). While the up-regulation of the primary metabolism to modulate signal transduction cascades and enhances plant defense responses has been well studied24, the role of RNA splicing, in particular of long non-coding RNAs, in plant defense has only emerged more recently25. In a recent differential transcriptome study of walnut infected with the hemibiotroph C. gloeosporioides, approximately 5% of transcripts were non-coding and almost 24% were long non-coding RNAs, many of which were differentially expressed between a partially resistant and susceptible genotype at various time points26. Much better understood is the role of GTPase in plant defense. GTPase functions as molecular switch downstream of immune receptors, triggering immune responses and therefore leads to enhanced disease resistance27. Based on expressed sequence tags, 13 discrete unigenes encoding G-proteins including small GTPases were previously detected in C. truncatum (reclassified to C. lentis Damm, sp. nov.)—infected L. culinaris leaves13.
Highly expressed genes were also identified in the susceptible RIL LR-66-524 and clustered primarily in Clusters 1 (associated with cell-death functions), 2 (enriched for genes involved in water deprivation) and 4 (many genes related to cell growth and development processes). Cell death in the form of a hypersensitive response (HR) or programmed cell death (PCD) is a highly effective defense strategy of plants against biotrophic pathogens as it deprives the pathogen of essential viable host plant cells28. However, thriving in the presence of cell death, this mechanism can also be manipulated by necrotrophic pathogens to effectively increase susceptibility29. In hemibiotrophs, HR could be fatal during the biotrophic phase, but may be promoted by the pathogen during the transition to the necrotrophic phase. This was shown in C. lentis (previously C. trunatum) where constitutive overexpression of the effector CtNUDIX in isolates arrested their development in the biotrophic phase, during which they induced an HR, whereas in wild-type isolates CtNUDIX was only expressed towards the end of the biotrophic phase30.
Comparison of the 477 DEGs identified with the five QTL intervals significantly associated with resistance to C. lentis race 0 previously identified in a SNP-based linkage map of the L. ervoides RIL population LR-6610 revealed 22 genes that had high or low expression in the partially resistant RIL LR-66-528 at 24, 48, 72 and 96 hpi. Among them, two genes with high expression during the biotrophic as well as the necrotrophic phases had the most obvious involvement in host resistance. Lc23518 was identified as an LRR receptor-like kinase, genes of which form one of the largest family of plant receptors, and its up-regulation at the necrotophic phase (96 hpi) in the partially resistant RIL was also validated by qPCR (Supplementary Table S4). The gene Lc05911 contained a motif that encodes for a DEAD-box ATP-dependent RNA helicase. Overexpression of the DEAD-box ATP-dependent RNA helicase OsBIRH1 in transgenic Arabidopsis showed enhanced disease resistance against Alternaria brassicola (necrotroph) and Pseudomonas syringae (biotroph)31. Overall, low numbers of overlapping genes between these two approaches may be a reflection of the different incubation times associated with QTL mapping (168 hpi) and the transcriptome study (24 to 96 hpi), but may also have been affected by using the L. culinaris genome for alignment.
Whether and to what degree these genes represent targets for resistance breeding remains to be determined. The ideal candidate would be a host receptor that interacts with a pathogen molecule during the biotrophic phase and arrests further development of C. lentis at this stage, or reduces the number of infections that successfully transition into the destructive necrotrophic phase. A candidate of interest for further explorations would be Lc33118, the cytokinin receptor histidine kinase exclusively expressed at 24 hpi in the partially resistant RIL LR-66-528, and its potential interaction with PR-1 gene Lc37093. Other targets would be the numerous LRR receptor-like kinase, in particular those detected early during the infection process. Further exploration of the filtered RNA-seq reads that did not align with the L. culinaris or will not align with the C. lentis genome, but may align to the L. ervoides genome could also reveal unique resistance gene candidates not present in the L. culinaris genome, considering that the latter appears to lack effective resistance against race 0 of C. lentis.
Materials and methods
Plant materials and inoculations
Lens ervoides RILs LR-66-524 with an average anthracnose severity of 72% and LR-66-528 with a severity of 5% were selected based on their reactions to inoculation with C. lentis isolate CT-30 in a previous study10. Four seedlings were grown in each 10 cm pot filled with a 3:1 mix of Sunshine #4 (Sun Gro® Horticulture, Alberta, Canada) and perlite (Special Vermiculite Canada, Winnipeg, Canada) in a growth chamber (Conviron GR48, Controlled Environments Limited, Winnipeg, Manitoba, Canada) at 22 °C (day)/16 °C (night) with a 16 h photoperiod. Plants were fertilized with a complete fertilizer solution (PlantProd® 20-20-20 plus micronutrients, Premier Tech Home &Garden Inc., Brantford, Ontario) 11 days after planting at a rate of 3 g L−1 H2O. The experiment was conducted in a randomized complete block design with three biological replications each represented by one pot with four seedlings. These were spray-inoculated with C. lentis isolate CT-30 (race 0) at a concentration of 5 × 104 conidia mL−1. Due to space and sampling time constraints, replicates were blocked over time. Sampling was conducted on mock (water)-inoculated plants and on inoculated plants 6, 12, 24, 48, 72, 96, 120 and 144 h post-inoculation (hpi) and leaflets were fixed and stored in ethanol (histopathology) or stored in − 80 °C (RNA-seq).
Histopathological study
Sampled leaves were fixed and stored in CMAA fixative (30% chloroform, 60% methanol, 10% acetic acid) at room temperature. Inoculated leaflets were treated with CMAA fixative twice or thrice until the leaflets were cleared before storage in 95% ethanol at room temperature. Six arbitrary leaflets rehydrated through decreasing ethanol concentrations of 70% (1 h), 50% (1.5 h) and 30% (1.5 h), stained with 0.05% Trypan blue and stored in 50% glycerol. Leaflets were mounted and were examined visually under a Zeiss Axioskop 40 microscope (Zeiss, Oberkochen, Germany) at 400X magnification. Pictures were taken using a Pixelink A686C camera (Pixelink A Navitor Company, Ottawa, ON, Canada) and Zeiss Axiovision software (Version 4.8.2 plus measurement module). The number of infection vesicle (IV)/primary hyphae (PH) formed per 25 appressoria, the percentage of leaflet area covered by acervuli per leaflet and the percentage area of dead tissue per leaflet were visually estimated.
Data were analyzed using SAS statistical package (Version 9.4, SAS Institute, Cary, NC, USA). Homogeneity of variances were assessed with the Levene’s test and normaility of error distribution with the Shapiro test. Data was analyzed with a repeated measures model where RILs and time points were considered fixed effects, time points were identified as repeated measure, and replications were considered random effects. At different time points, RILs were compared by multiple comparisons of means using Fisher’s least significant difference.
Assessment of fungal biomass
For in planta fungal biomass determination, genomic DNA was extracted from 100 mg finely ground samples of inoculated and non-inoculated plants at 6, 12, 24, 48, 72, 96, 120 and 144 hpi using Qiagen DNeasy Plant Mini Kit®. The purity and quantity of DNA was determined using a NanoDrop ND8000 (Thermo Fisher Scientific, Wilmington, USA) (A260/280 < 2.0). For qPCR, previously designed primers were used to amplify the housekeeping gene Elongation factor 1-alpha of L. culinaris (LcEF1-α: forward TGTCGACTCTGGGAAGTCAA, reverse CTCTTTCCCTTTCAGCCTTG)13 and ACTIN of C. lentis (ClACT: forward CACGCTCTACTACGACGGAC, reverse GAAGACGAAGTTGTCGGGA)32. Primers were validated for their specificity using genomic DNA of IG 72,815, L01-827A, LR-66-528 and LR-66-524 in a C1000TM Thermal Cycler (Bio-rad Laboratories, Inc., Hercules, CA, USA) and by testing amplification efficiency using qPCR in a QuantStudio 3 System (Applied Biosystems Inc., Foster City, CA, USA) for a total of 5 dilutions for three biological replicates of mock samples of both RILs. Genomic DNA was adjusted to 25 ng µL−1. Each qPCR reaction contained 2 µL DNA template, 5 µL SYBR® Green (Catalog no. 4309155, Thermo Scientific), 0.2 µL of each 10 µM forward and reverse primers, and 2.6 µL sterilized water. The qPCR amplifications were performed in a QuantStudio 3 System using a fast-run program with default settings. The qPCR data of ClActin were normalized using LcEF1-α as a reference gene and log2 fold change (2−ΔCT) were used for statistical analyses following the method of Livak & Schmittgen33. The Student’s t test was used to determine the differences among the genotypes at different time-points (p-value < 0.05).
Identification of differentially expressed genes
Total RNA was extracted and purified from three biological replicates per time-point (24, 48, 72 and 96 hpi) of CT-30-inoculated plants of LR-66-528 and LR-66-524, and from three biological replicates of non-inoculated plants using the Qiagen RNeasy Plant Mini Kit (with on-column DNAse treatment). The RNA integrity was determined by denaturing agarose gel electrophoresis and its quality and quantity were determined using NanoDrop ND8000 and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Strand-specific RNA libraries (adapted from the NEB Ultra-Directional RNA Library Prep protocol) were generated and sequenced using the Illumina PE125 HiSeq 2500 (v4 chemistry) at the Michael Smith Genome Sciences Center (BCGSC), BC, Canada. Raw fastq reads were filtered in Trimmomatic (version 0.36)34 to remove Illumina adaptors (TruSeq3-PE-2.fa:2:30:10) and leading and trailing low quality or N bases (below quality 3) (leading:3 and trailing:3), to cut when the average quality per base dropped below 15 after scanning of read with a 4-base wide sliding window (slidingwindow:4:15) and to drop reads of less than 36 bases (minlen:36). Removal of low-quality reads and adaptors was confirmed using FASTQC. Cleaned reads were submitted to Spliced Transcripts Alignment to a Reference (STAR version 2.6.1a; default settings)35 for mapping against the pre-release version of the L. culinaris genome version 1.236. The read counts per gene were determined using STAR during the mapping process.
Variability of gene expression among the sequencing samples was estimated through Principal Component Analysis (PCA) by feeding gene read counts as the input file into JMP genomics 8.0. The proportion of variability accounted by RILs, incubation time, their interaction and replication was estimated through Principal Component Variance Analysis (PVCA) in the same program.
Comparisons were then made between non-inoculated mock and inoculated sample data from 24, 48, 72 and 96 hpi for each RIL to identify genes that responded to CT-30 infection using the R package DESeq2 at Padj < 0.05 and gene expression fold change > 2 with the raw counts file for all genes obtained using the STAR aligner as an input file. Pair-wise comparisons between LR-66-528 and LR-66-524 at 24, 48, 72 and 96 hpi were conducted on disease-responsive genes to identify DEGs between the two RILs for each time-point, using a threshold Padj < 0.05 and gene expression fold change > 2, and were visualized in a Venn diagram generated with jvenn37.
Comparison of transcriptome and QTL mapping data
Previously identified QTLs (2-LOD) intervals associated with anthracnose resistance10 were projected onto the physical map of L. culinaris36 using the software Strudel (v 1.15.08.25)38. The main region on the physical map with high density projections was focused, whereas minor regions projecting to other chromosome were not taken into consideration in order to restrict the distance. Using the GTF file of the L. culinaris genome, the markers spanning the highly dense region were used to identify the genes present in those intervals. These were compared with DEGs identified by RNA-seq.
Gene expression profile analysis
Read counts per gene data procured using the STAR aligner were normalized by transforming to Reads Per Kilobase of exon per Million reads (RPKM). The genes with read counts less than 10 were removed before normalization. The resulting file was submitted to JMP genomics 8.0 for K-mean clustering analysis and GO enrichment analysis of genes in each of the resulting clusters. The L. culinaris gene IDs were transformed to the M. truncatula gene IDs because the L. culinaris was not completely annotated. The Entrez IDs of the corresponding genes were used as the input file for the gene enrichment analysis specifying Medicago truncatula as the input species (http://plantregmap.cbi.pku.edu.cn/go.php). The biological process aspect was selected for analysis at threshold False Discovery Rate (FDR) value (< 0.001) calculated with the method developed by Benjamini and Hochberg39.
Validation of gene expression using quantitative PCR
Reproducibility of results was assessed by validating the expression of 21 selected DEGs (based on their potential role in disease resistance) at 24, 48, 72 and/or 96 hpi (depending on significant fold changes in the RNA-seq analysis, Supplementary Table S4) through RT-qPCR. Samples of LR-66-528 and LR-66-524 from an additional, independent experiment conducted as described above were generated, and total RNA was extracted from samples collected at 24, 48, 72 and 96 hpi, and non-inoculated RILs as described earlier. RNA of all the samples were adjusted to 50 ng L−1 to synthesize cDNA using the High-Capacity cDNA Reverse Transcription kit (Thermo Fischer Scientific, Wilmington, USA). Elongation factor 1-alpha was used as the reference gene. Primers were designed with Primer3 (web version 4.1.0) using default settings from the exonic regions of the selected genes based on the lentil genome. Amplification efficiencies were tested as described above using cDNA (50 ng/μl) serially diluted 1:2 (V/V) 4 times in nuclease-free ultra pure water (Invitrogen Life Technologies, Carlsbad, CA, USA) and were calculated based on the slope of the standard curve with equation E = − 1 + 10 (− 1/slope). Each RT-qPCR reaction consisted of 2 μL cDNA template, 5 μL SYBR green, 0.8 μL of each forward and reverse primers and 1.4 μL of nuclease-free water. RT-qPCR was performed in QuantStudio 3 System using a fast-run program with default settings. Fold changes in gene expression in LR-66-528 and LR-66-524 compared to that in mock-inoculated samples were calculated as described by Schmittgen and Livak using EF1-α as the reference gene40. Student t tests were used to determine significant differences at P < 0.05 in gene expression (2−ΔCT) between partially resistant and susceptible RILs after confirmation of significant differences with respective mock-inoculated samples.
Guideline statement
All experimental research complies with relevant institutional, national, and international guidelines and legislation.
Data availability
RNA-seq data of this study can be found in the NCBI BioProject Repository accession id: PRJNA784980.
References
Morrall, R. A. A. Evolution of lentil diseases over 25 years in western Canada. Can. J. Plant Pathol. 19, 197–207 (1997).
Kaiser, W. J., Ramsey, M. D. & Makkouk, K. M. Foliar diseases of cold season food legumes and their control. In Linking Research and Marketing Opportunities for Pulses in the 21st Century: Proceedings of the Third International Food Legume Research Conference (ed. Knight, R.) 437–456 (Kluwer Academic Publishers, 2000).
Morrall, R. A. A. & Pedersen, E. A. Discovery of anthracnose in Saskatchewan in 1990. Can. Plant Dis. Surv. 71, 105–106 (1991).
Buchwaldt, L., Anderson, K. L., Morrall, R. A. A., Gossen, B. D. & Bernier, C. C. Identification of lentil germplasm resistant to Colletotrichum truncatum and characterization of two pathogen races. Phytopathology 94, 236–243 (2004).
Vandenberg, A. et al. ‘CDC Robin’ lentil. Can. J. Plant Sci. 82, 111–112 (2002).
Tullu, A., Banniza, S., Tar’an, B., Warkentin, T. & Vandenberg, A. Sources of resistance to ascochyta blight in wild species of lentil (Lens culinaris Medik.). Genet. Resour. Crop Evol. 57, 1053–1063 (2010).
Fiala, J. V., Tullu, A., Banniza, S., Séguin-Swartz, G. & Vandenberg, A. Interspecies transfer of resistance to anthracnose in lentil (Lens culinaris Medik.). Crop Sci. 49, 825–830 (2009).
Vail, S. L., Tullu, A. & Vandenberg, A. Field evaluation of resistance to Colletotrichum truncatum in Lens culinaris, Lens ervoides, and Lens ervoides x Lens culinaris derivatives. Field Crops Res. 126, 145–151 (2011).
Gujaria-Verma, N. et al. Genetic mapping of legume orthologs reveals high conservation of synteny between lentil species and the sequenced genomes of Medicago and chickpea. Front. Plant Sci. 5, 676 (2014).
Bhadauria, V. et al. QTL mapping reveals genetic determinants of fungal disease resistance in the wild lentil species Lens ervoides. Sci. Rep. 7, 3231 (2017).
Birker, D. et al. A locus conferring resistance to Colletotrichum higginsianum is shared by four geographically distinct Arabidopsis accessions. Plant J. 60, 602–613 (2009).
Chongo, G., Gossen, B. D. & Bernier, C. C. Infection by Colletotrichum truncatum in resistant and susceptible lentil genotypes. Can. J. Plant Pathol. 24, 81–85 (2002).
Bhadauria, V. et al. Identification of Lens culinaris defense genes responsive to the anthracnose pathogen Colletotrichum truncatum. BMC Genet. 14, 31–40 (2013).
Cao, Z. et al. Using a transcriptome sequencing approach to explore candidate resistance genes against stemphylium blight in the wild lentil species Lens ervoides. BMC Plant Biol. 19, 399 (2019).
Shigenaga, A. M. & Argueso, C. T. No hormone to rule them all: Interactions of plant hormones during the responses of plants to pathogens. Semin. Cell Dev. Biol. 56, 174–189 (2016).
Argueso, C. T. et al. Two-component elements mediate interactions between cytokinin and salicylic acid in plant immunity. LoS. Genet. 8, e1002448 (2012).
Spallek, T., Gan, P., Kadota, Y. & Shirasu, K. Same tune, different song—cytokinins as virulence factors in plant–pathogen interactions?. Curr. Opin. Plant Biol. 44, 82–87 (2018).
Eisermann, I. et al. CgIPT1 is required for synthesis of cis-zeatin cytokinins and contributes to stress tolerance and virulence in Colletotrichum graminicola. Fungal Genet. Biol. 143, 103436 (2020).
Tadokoro, T., Modahl, C. M., Maenaka, K. & Aoki-Shioi, N. Cysteine-rich secretory proteins (CRISPs) from venomous snakes: An overview of the functional diversity in a large and underappreciated superfamily. Toxins 12, 175. https://doi.org/10.3390/toxins12030175 (2020).
Pečenková, T. et al. Immunity functions of Arabidopsis pathogenesis-related 1 are coupled but not confined to its C-terminus processing and trafficking. Mol. Plant Pathol. 23, 664–678 (2022).
Choi, J. et al. The cytokinin-activated transcription factor ARR2 promotes plant immunity via TGA3/NPR1-dependent salicylic acid signaling in Arabidopsis. Dev. Cell 19, 284–295 (2010).
Chen, G. et al. Genome-wide analysis of the rice PPR gene family and their expression profiles under different stress treatments. BMC Genom. 19, 720 (2018).
Tang, D., Wang, G. & Zhou, J. M. Receptor kinases in plant-pathogen interactions: More than pattern recognition. Plant Cell 29, 618–637 (2017).
Rojas, C., Kumar, M. S., Tzin, V. & Mysore, K. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 5, 17–29 (2014).
Zaynab, M. et al. Long non-coding RNAs as molecular players in plant defense against pathogens. Microb. Pathog. 121, 277–282. https://doi.org/10.1016/j.micpath.2018.05.050 (2018).
Feng, S. et al. Genome-wide identification and characterization of long non-coding RNAs conferring resistance to Colletotrichum gloeosporioides in walnut (Juglans regia). BMC Genom. 22, 15 (2021).
Kawano, Y., Takako, K.-K. & Ko, S. Rho family GTPase-dependent immunity in plants and animals. Front. Plant Sci. 5, 522 (2014).
Balint-Kurti, P. The plant hypersensitive response: Concepts, control and consequences. Mol. Plant Pathol. 20, 1163–1178 (2019).
Leisen, T. et al. Multiple knockout mutants reveal a high redundancy of phytotoxic compounds contributing to necrotrophic pathogenesis of Botrytis cinerea. PLoS Pathog. 18, e1010367 (2022).
Bhadauria, V., Banniza, S., Vandenberg, A., Selvaraj, G. & Wei, Y. Overexpression of a novel biotrophy-specific Colletotrichum truncatum effector, CtNUDIX, in hemibiotrophic fungal phytopathogens causes incompatibility with their host plants. Eukaryot. Cell 12, 2–11 (2013).
Li, D., Liu, H., Zhang, H., Wang, X. & Song, F. OsBIRH1, a DEAD-box RNA helicase with functions in modulating defence responses against pathogen infection and oxidative stress. J. Exp. Bot. 59, 2133–2146 (2008).
Bhadauria, V., McLachlan, R., Pozniak, C. & Banniza, S. Candidate effectors contribute to race differentiation and virulence of the lentil anthracnose pathogen Colletotrichum lentis. BMC Genom. 16, 628–649 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 25, 402–408 (2001).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. & Gingeras, T. R. Optimizing RNA-Seq mapping with STAR. Methods Mol. Biol. 1415, 245–262 (2016).
Bett, K.E. & Cook, D.R. KnowPulse. The Lentil Genome Sequencing (LenGen) Project. https://knowpulse.usask.ca/study/2653517. Accessed 22 April 2019 (2013).
Bardou, P., Mariette, J., Escridie, F., Djemiel, C. & Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinf. 15, 293–297 (2014).
Bayer, M. et al. Comparative visualization of genetic and physical maps with strudel. Bioinformatics 27, 1307–1308 (2011).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B 57, 289–300 (1995).
Schmittgen, T. & Livak, K. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 (2008).
Acknowledgements
We respectfully acknowledge that this research was conducted on Treaty 4 and Treaty 6 territories. Research presented here is part of the academic theses of the University of Saskatchewan of Preeni Bawa on “Identification of candidate genes associated with resistance against race 0 of Colletotrichum lentis in Lens ervoides”, and Karan Kapoor on “Screening and quantification of disease responses in Lens ervoides against multiple fungal pathogens”. Technical support was provided by members of the Pulse Crop Pathology research group. We would like to thank Dr. Zhe Cao for critically reviewing the manuscript and assistance in the PCA analysis. This project was funded by the National Science and Engineering Research Council—Collaborative Research and Development (NSERC-CRD) grant to Sabine Banniza with Industry contributions by the Saskatchewan Pulse Growers and the Western Grains Research Foundation, and additional funds from the Saskatchewan Agriculture Development Fund.
Author information
Authors and Affiliations
Contributions
S.B. conceived the project and was P.I., supervised J.H., P.B. and K.K., contributed to and revised the manuscript. K.K. conducted pathogenicity tests, histopathological studies and contributed those sections to the manuscript. P.B. conducted fungal biomass, RNA-seq and RT-qPCR experiments and together with J.H. analyzed the data, and contributed those sections to the manuscript. J.H. provided technical supervision, contributed to the discussion and critically reviewed the manuscript. All authors have read and approved the manuscript for final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bawa, P.K., Halliday, J., Kapoor, K. et al. Identification of candidate genes associated with resistance against race 0 of Colletotrichum lentis in Lens ervoides. Sci Rep 12, 18447 (2022). https://doi.org/10.1038/s41598-022-23175-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23175-z
- Springer Nature Limited