
Abstract
Type 1 diabetes (T1D) incidence is increased after COVID-19 infection in children under 18 years of age. Interferon-α-activated oligoadenylate synthetase and downstream RNAseL activation degrade pathogen RNA, but can also damage host RNA when RNAseL activity is poorly regulated. One such regulator is PDE12 which degrades 2′-5′ oligoadenylate units, thereby decreasing RNAseL activity. We analyzed PDE12 expression in islets from non-diabetic donors, individuals with newly (median disease duration 35 days) and recently (5 years) diagnosed T1D, and individuals with type 2 diabetes (T2D). We also analyzed PDE12 single-nucleotide polymorphisms (SNPs) relative to T1D incidence. PDE12 expression was decreased in individuals with recently diagnosed T1D, in three of five individuals with newly diagnosed T1D, but not in individuals with T2D. Two rare PDE12 SNPs were found to have odds ratios of 1.80 and 1.74 for T1D development. We discuss whether decreased PDE12 expression after COVID-19 infection might be part of the up to 2.5-fold increase in T1D incidence.
Similar content being viewed by others


Introduction
Recent research has shown that the incidence of type 1 diabetes (T1D) is increased up to 2.5-fold after coronavirus disease 2019 (COVID-19) infection in children under 18 years of age1,2. Similar increases in new-onset T1D have also been reported in adults3. One theory that explains how viral infections may lead to T1D involves the interferon (IFN)-α-activated latent ribonuclease (RNAseL) signaling pathway4. When IFN-α mediated cell stimulation induces downstream activation of 2′-5′ oligoadenylate synthetases (OASs), the high levels of 2′-5′ oligoadenylate (2-5A) produced bind to and activate RNaseL. Excessive RNaseL activity may lead to the degradation of both pathogen and host RNA, thereby causing cellular damage5,6. This activity is regulated by phosphodiesterases such as PDE12, which degrade 2-5A molecules, suppressing RNaseL activation. In fact, a direct link between PDE12 and OAS has been described in a PDE12-null HeLa cell line7. PDE12-null cells were also resistant to infection with encephalomyocarditis virus, human rhinovirus and respiratory syncytial virus, highlighting a protective effect that is associated with decreased PDE12 activity and thereby increased RNaseL activity. In addition, a separate study on inflammatory pathways in patients with T1D found that PDE12 levels are decreased in the peripheral blood of individuals with new-onset T1D (i.e., mean diabetes duration of 0.22 years)8.
Results
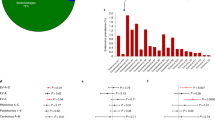
From the Affymetrix analysis (Fig. 1), we observed significant decreases in PDE12 expression for the islets of individuals with recently diagnosed T1D (median disease duration, 5.0 years) and for islets from biopsies originating from donors with recurrent T1D after pancreas transplantation. PDE12 expression was also decreased in autoantibody-positive individuals, but not significantly. Furthermore, three of the five individuals with newly diagnosed T1D (median disease duration, 35 days) exhibited low levels of PDE12 expression. However, PDE12 expression was not altered in individuals with type 2 diabetes (median disease duration, 2.0 years) (Table 1).

Phosphodiesterase 12 (PDE12) gene expression. CTR: non-diabetic controls (n = 18); AB + : non-diabetic autoimmune antibody-positive donors (n = 12); T1D (median disease duration, 35 days): donors with newly diagnosed type 1 diabetes (n = 5); T1D (median 5 years): donors with recently diagnosed type 1 diabetes (n = 20); T2D (median 2 years): donors with type 2 diabetes (n = 8); T1D Tx: biopsies from donors with recurrent T1D (n = 4). Boxes indicate 25% and 75% quartiles, whiskers 1.5 × interquartile ranges, and squares mark outliers. The p-values shown were calculated using unpaired two-sided t-tests relative to CTR. Test statistics for CTR vs T1D (5 years): t-statistic 6.054, 95%CI 7.74;15.59, degrees of freedom 31.997, mean of CTR 35.95, mean of T1D (5 years) 24.29. Test statistics for CTR vs T1D Tx: t-statistic 3.43, degrees of freedom 5.87, 95%CI 2.78;16.81, mean of T1D Tx 26.16.
The single-nucleotide polymorphism (SNP) analysis revealed that individuals with the two rare PDE12 SNP variants shown in Table 2 had an odds ratio of 1.80 and 1.74 for developing T1D.
Discussion
The observed decrease in PDE12 expression seems to have a protective effect against viral infections because it upregulates RNaseL activity in beta cells and other cells7; however, it may have the unfortunate side effect of triggering beta-cell damage and subsequent diabetes pathogenesis. Vaccines against COVID-19 should not activate the RNaseL cascade and therefore should not increase the incidence of T1D. Prolonged RNaseL activity may damage and kill cells9. Therefore, RNaseL activity must be carefully regulated to protect against viruses without compromising cellular function. Consequently, any treatments that inhibit PDE12 activity and thereby stimulate antiviral defenses should only be given for short durations, to prevent damage to cells. In fact, we found that PDE12 expression levels are decreased in individuals with recently diagnosed T1D (median disease duration, 5.0 years). During viral infection, which may initiate T1D development, individuals have high levels of PDE12 activity which makes combating the virus difficult. Then, in the post-virus phase there is a decrease in PDE12 expression which leads to beta-cell damage. Here, stimulating PDE12 expression might have inhibited T1D development.
The link between COVID-19 and T1D supports the theory that viruses can act as pathogenic triggers for T1D1,3. Recent research has shown that severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) decreases insulin expression and induces transdifferentiation of beta cells from COVID-19-infected and deceased donors10,11. Furthermore, beta cells readily express the angiotensin converting enzyme 2 (ACE2) receptor12 used by SARS-CoV-2 for host entry, and βTC3 cells and isolated rat beta cells show substantially higher 2-5A activity upon IFN-α stimulation when compared to αTC3 cells or rat alpha cells13. These observations may explain why beta cells are at increased risk of RNaseL-mediated cellular damage upon viral challenge, even though the virus itself is not toxic. Together, these data might support the increased incidence of T1D after COVID-19 infection and provide valuable insight into the pathogenesis of T1D. However, several other mechanisms for the comorbidity has been suggested including the ACE2-receptor and pro-inflammatory cytokine changes14. Since our study is fairly small, it is not possible at this point to have a firm conclusion of the relationship between COVID-19 and T1D. However, the PDE12 hypothesis seems not to be in conflict with the other mechanisms just mentioned.
Methods
Human tissue
Pancreatic tissue from donors was collected in the Diabetes Virus Detection (DiViD)15 and Network for Pancreatic Organ Donors with Diabetes (nPOD)16 studies, with informed consent obtained from all participants. Briefly, DiViD donors with diabetes had a surgical resection of the pancreatic tail, between three and nine weeks after their type 1 diabetes diagnosis, while nPOD material originates from cadaveric organ donors (see Table 1). The procedures were approved by The Norwegian Government’s Regional Ethics Committee (reference 2009/1907); nPOD donors with approval by the University of Tennessee Health Science Center (UTHSC) local Institutional Review Board (reference 10–00848-XM). All experiments were performed in accordance with relevant guidelines and regulations.
Microdissection of pancreatic islets
Acquired pancreatic samples were laser microdissected as described previously17. Briefly, frozen tissue sections from nPOD and DiViD was microdissected with the Arcturus Pixcell II laser capture microdissection system (Arcturus Bioscience, Mountain View, CA, USA). Islets from 2 to 5 sections per donor were detected by autofluorescence and pooled together, and afterwards subjected to RNA extraction with the Arcturus PicoPure RNA Isolation Kit (Applied Biosystems, Grand Island, NY, USA). RNA quality and quantity was validated with the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA), and samples underwent gene expression analysis with the Affymetrix expression arrays (Thermo Fisher, Santa Clara, CA, USA) as described previously18.
SNP analysis
Genotyping data were retrieved from the UCSD T1D GWAS meta-analysis19 which includes samples from 501,638 control individuals and 18,942 patients with T1D. Similarly, the T2D multi-ethnic meta-analysis20 includes samples from nearly 1.2 million control subjects and 228,499 T2D cases.
Statistics
PDE12 expression statistics were calculated using Welch’s t-test and visualized with R software (ver. 4.1.2; R Development Core Team, 2021) using the tidyverse (ver. 1.3.1), ggplot2 (ver. 3.3.5), and ggpubr (ver. 0.4.0) packages.
Ethical approval
DiViD and nPOD studies were approved by The Norwegian government’s regional ethics committee (reference 2009/1907) and by the University of Tennessee Health Science Center’s local institutional review board (reference 10-00848-XM).
Data availability
Data have been deposited with datadryad.org https://doi.org/10.5061/dryad.d7wm37q4b. The protocols used can be obtained upon request to the corresponding author. Researchers interested in acquiring biological sample from the donors can apply through the DiViD and nPOD programs.
Code availability
The code used to produce visuals and statistics for Fig. 1 can be obtained upon request from the corresponding author.
References
Barrett, C. E. et al. Risk for newly diagnosed diabetes >30 days after SARS-CoV-2 infection among persons aged <18 years - United States, March 1, 2020-June 28, 2021. MMWR Morb. Mortal. Wkly. Rep. 71, 59–65 (2022).
Unsworth, R. et al. New-onset type 1 diabetes in children during COVID-19: Multicenter regional findings in the U.K.. Diabetes Care 43, e170–e171 (2020).
Ebekozien, O. A., Noor, N., Gallagher, M. P. & Alonso, G. T. Type 1 diabetes and COVID-19: Preliminary findings from a multicenter surveillance Study in the US. Diabetes Care 43, 83–85 (2020).
Pedersen, K. et al. Genetic predisposition in the 2′-5′A pathway in the development of type 1 diabetes: Potential contribution to dysregulation of innate antiviral immunity. Diabetologia 64, 1805–1815 (2021).
Poulsen, J. B. et al. Characterization of human phosphodiesterase 12 and identification of a novel 2′-5′ oligoadenylate nuclease – The ectonucleotide pyrophosphatase/phosphodiesterase 1. Biochimie 94, 1098–1107 (2012).
Banerjee, S. et al. OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc. Natl. Acad. Sci. 116, 5071–5076 (2019).
Wood, E. R. et al. The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 290, 19681–19696 (2015).
Santos, A. S. et al. Prevalence of inflammatory pathways over immuno-tolerance in peripheral blood mononuclear cells of recent-onset type 1 diabetes. Front. Immunol. 12, 765264 (2021).
Ezelle, H. J. & Hassel, B. A. Pathologic effects of RNase-L dysregulation in immunity and proliferative control. Front. Biosci. Sch. Ed. 4, 767–786 (2012).
Tang, X. et al. SARS-CoV-2 infection induces beta cell transdifferentiation. Cell Metab. 33, 1577-1591.e7 (2021).
Wu, C.-T. et al. SARS-CoV-2 infects human pancreatic β cells and elicits β cell impairment. Cell Metab. 33, 1565-1576.e5 (2021).
Tikellis, C. et al. Improved islet morphology after blockade of the Renin-angiotensin system in the ZDF rat. Diabetes 53, 989–997 (2004).
Bonnevie-Nielsen, V., Buschard, K. & Dyrberg, T. Differential responsiveness to interferon-alpha in beta-cells and non-beta cells. Diabetes 45, 818–821 (1996).
Montefusco, L., Bolla, A. M. & Fiorina, P. Should we expect a wave of type 1 diabetes following SARS-CoV-2 pandemic?. Diabetes Metab. Res. Rev. 38, e3529 (2022).
Krogvold, L. et al. Pancreatic biopsy by minimal tail resection in live adult patients at the onset of type 1 diabetes: Experiences from the DiViD study. Diabetologia 57, 841–843 (2014).
Campbell-Thompson, M. et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): Developing a tissue biobank for type 1 diabetes. Diabetes Metab. Res. Rev. 28, 608–617 (2012).
Richardson, S. J. et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 59, 2448–2458 (2016).
Wu, J. et al. Molecular phenotyping of immune cells from young NOD mice reveals abnormal metabolic pathways in the early induction phase of autoimmune diabetes. PLoS ONE 7, e46941 (2012).
Chiou, J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021).
Vujkovic, M. et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 52, 680–691 (2020).
Funding
The study is funded by the Axius and Bagger Sørensen foundations. The funding sources had no role in conceiving this study or preparing the manuscript.
Author information
Authors and Affiliations
Contributions
K.B. conceptualized the project and together with H.T. and K.J. wrote the original manuscript draft. L.K., K.D.J., and I.G. provided the analyzed material and performed the RNA expression analysis. F.P. performed the SNP analysis. All authors edited, reviewed, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tekin, H., Josefsen, K., Krogvold, L. et al. PDE12 in type 1 diabetes. Sci Rep 12, 18149 (2022). https://doi.org/10.1038/s41598-022-22890-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-22890-x
- Springer Nature Limited