
Abstract
Although substantial progress has been made in managing COVID-19, it is still difficult to predict a patient’s prognosis. We explored the epigenetic signatures of COVID-19 in peripheral blood using data from an ongoing prospective observational study of COVID-19 called the Norwegian Corona Cohort Study. A series of EWASs were performed to compare the DNA methylation profiles between COVID-19 cases and controls three months post-infection. We also investigated differences associated with severity and long-COVID. Three CpGs—cg22399236, cg03607951, and cg09829636—were significantly hypomethylated (FDR < 0.05) in COVID-19 positive individuals. cg03607951 is located in IFI44L which is involved in innate response to viral infection and several systemic autoimmune diseases. cg09829636 is located in ANKRD9, a gene implicated in a wide variety of cellular processes, including the degradation of IMPDH2. The link between ANKRD9 and IMPDH2 is striking given that IMPDHs are considered therapeutic targets for COVID-19. Furthermore, gene ontology analyses revealed pathways involved in response to viruses. The lack of significant differences associated with severity and long-COVID may be real or reflect limitations in sample size. Our findings support the involvement of interferon responsive genes in the pathophysiology of COVID-19 and indicate a possible link to systemic autoimmune diseases.
Similar content being viewed by others


Introduction
Coronavirus disease 2019 (COVID-19) is caused by the highly contagious severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and is marked by a wide spectrum of symptoms. Although most patients with COVID-19 experience a mild disease course, up to 35% report long-term sequelae, including memory problems, depression, dyspnea, anosmia, ageusia, and fatigue lasting months after the initial infection1,2. These are among the symptoms that characterize a syndrome referred to as Post-Acute Sequelae of SARS-CoV-2 infection (PASC) or “long-COVID”. The causes and persistence of long-COVID are not well understood, although several hypotheses have been proposed, including damage to the autonomic nerve system caused by inflammation3, the formation of autoantibodies in susceptible individuals4, and viral persistence. The lack of detectable antibodies in half of the patients presenting symptoms attributed to SARS-CoV-2 in another study5 points to a possible confounding etiology such as pandemic stress.
Evidence points to subtle differences in the immunological profiles of individuals with long-COVID compared to those who recover quickly. The syndrome seems to occur regardless of the development of long-term immunity, as antibody levels have been found to be similar four months after COVID-19 symptom onset to that of convalescents6. However, participants with long-COVID appear to have a rapidly waning number of interferon gamma (IFN-γ) producing CD8+ cells four months after COVID-19 symptom onset6. This is interesting, as cytotoxic lymphocytes, particularly CD8+ lymphocytes and Natural Killer (NK) cells, affect viral clearance directly or through the release of IFN-γ7. An improved understanding of the role of host factors in COVID-19 is necessary to improve the diagnosis of long-COVID and develop efficient treatments.
DNA methylation (DNAm) is a stable epigenetic mark that regulates gene expression without altering the DNA sequence itself. It plays a pivotal role in normal biological processes as well as in the pathogenesis of diseases8,9,10. An epigenome-wide association study (EWAS) entails studying DNAm differences associated with a phenotype at hundreds of thousands of cytosine-phosphate-guanine (CpG) sites across the epigenome. The human epigenome harbors an estimated 28 million CpG sites11, of which 60–70% are located near transcription start sites12. Differentially methylated regions (DMRs) represent genomic regions with different DNAm levels between a case and a control group. The effect of methylation of a given CpG site depends on the genomic context13. For instance, hypermethylation of CpG sites in a gene body is commonly associated with higher gene expression14.
Genomic studies have revealed key susceptibility genes for COVID-1915,16,17,18, the most prominent of which is angiotensin-converting enzyme 2 (ACE2)19. Epigenetic investigations add another layer of omics analysis to the study of SARS-CoV-2 and reveal that methylation patterns are predictive of disease severity20,21,22. Three major considerations in deciphering the link between epigenetic variation and infection are how the host responds to viral exposure, how the virus exploits the host’s epigenome to establish infection, and how innate susceptibility differs between individuals. It remains unknown whether the DNAm differences reported thus far in COVID-19 reflect differences in susceptibility, temporary immune modulation, or more permanent changes.
To date, none of the published studies on long-COVID have investigated DNA methylation. Our aim was therefore to explore epigenetic signatures that are correlated with the severity of COVID-19 and the onset of long-COVID. To this end, we conducted a series of EWASs where we compared DNAm profiles in the peripheral blood of 109 Norwegians, eight to twelve weeks after SARS-CoV-2 infection, with the corresponding profiles of 73 controls.
Results
Characteristics of the sample population
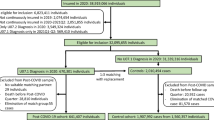
Figure 1 and Table 1 provide details of the sample selection and the study population according to COVID-19 status, sex, mean age, smoking history, presence of chronic diseases, and the average number of days from the time the RT-PCR test was performed to (i) the time of enrolment (hereafter referred to as ‘baseline’), (ii) the blood draw, and (iii) the follow-up questionnaire. Note that the healthy controls did not undergo an RT-PCR test. Among the symptomatic controls with a negative RT-PCR test, four had a positive serology test and were therefore considered false negatives. As these four cases had reported both fever and dyspnea, they were reclassified as belonging to the severe COVID-19 category. The remaining 32 symptomatic controls with a negative RT-PCR test had a confirmatory negative serology test and were thus grouped together with the 41 healthy controls in the control category (n = 73).

Overview of the sampling scheme. 184 of the 200 participants who agreed to a blood draw showed up for their appointment and completed the baseline questionnaire. Two participants lacked data and were excluded from the study, bringing the final number of participants to 182. Four participants with a negative SARS-CoV-2 RT-PCR test and who reported symptoms of severe COVID-19 had a positive serology test. They were considered false negatives and reclassified as severe COVID-19. Ten participants were treated as ‘lost to follow-up’, as they did not return the three-month follow-up questionnaire despite reminders. The control group was not included in the long-COVID EWAS analysis.
36% to 42% of the participants reported smoking in each of the three main categories. Additionally, 15% of the mild COVID-19 cases had at least one chronic disease; the corresponding proportion was 36% among the severe COVID-19 cases and 38% among the controls. At three months of follow-up, 52% of the severe COVID-19 cases reported that their health was worse than a year ago; the corresponding proportion was 19% among the mild cases, 9% among the symptomatic controls, and none among the healthy controls. Among the severe cases, 31% reported fatigue and 23% dyspnea three months after the infection.
Comparison of DNAm profiles
We adopted the analytic strategy outlined in Fig. 2. Specifically, data from males and females were analyzed together for autosomal probes (Figs. 3 and 4, and Supplementary Fig. 1), and separately for sex-chromosome probes (Figs. 5 and 6, and Supplementary Fig. 2). The sex-stratified analyses were motivated by the presence of distinct sex differences in the distribution of mean methylation values on the sex chromosomes (Supplementary Fig. 3). Our primary objective was to compare the DNAm profiles of the following categories of study participants: 1) COVID-19 positive (n = 109) versus COVID-19 negative (n = 73), 2) severe COVID-19 (n = 61) versus mild COVID-19 (n = 48), and 3) long-COVID (n = 41) versus remission (n = 63) (see Fig. 2 and “Methods” for details). We adjusted for the following variables in each EWAS: age, sex, smoking pack-years (estimated from the DNAm data), physical fitness, and estimates of white blood cell counts, i.e., CD8+ naïve and exhausted cytotoxic T cells, CD4+ naïve cells, NK cells, B cells, monocytes, and granulocytes.

Overview of the analytic pipeline. An EWAS was performed for each of the following comparisons: (i) COVID-19 positive vs COVID-19 negative, (ii) severe COVID-19 vs mild COVID-19, and (iii) long-COVID vs the remission group (labeled ‘Remission’ in the figure). Additional comparisons are provided in Supplementary Figs. 8 and 9. We applied the R package limma63 on functional normalized M-values in each EWAS. In total, we conducted nine separate EWASs.

Manhattan plots for the EWASs. The Manhattan plots display the association statistics at each of the 776,892 autosomal probes between (a) COVID-19 positive (n = 109) and COVID-19 negative (n = 73), (b) severe COVID-19 (n = 61) and mild COVID-19 (n = 48), and (c) long-COVID (n = 41) and remission (n = 63). The dotted horizontal line is the Bonferroni threshold (0.05/776,892 CpG sites), and the labeled dots are the significant CpGs at FDR < 0.05.

Volcano plots for the pooled sample of males and females in the analysis of autosomal probes. The plots display the estimated beta coefficients against −log10 of the P-values. (a) COVID-19 positive (n = 109) versus COVID-19 negative (n = 73), (b) severe COVID-19 (n = 61) versus mild COVID-19 (n = 48), and (c) long-COVID (n = 41) versus remission (n = 63). The dotted horizontal line refers to the Bonferroni threshold (0.05/776,892 CpG sites), and the orange-colored dots are the significant CpGs at FDR < 0.05.

Manhattan plots for the sex-stratified EWASs of probes on the sex chromosomes. The plots display the association statistics at each of the 17,183 X-chromosome probes and 51 Y-chromosome probes (indicated by blue dots). The upper panels (a) to (c) are for males only (including the Y-chromosome probes), and the lower panels (d) to (f) are for females only (X-chromosome probes only). Panels (a) and (d) display the result of the comparison between COVID-19 positive and COVID-19 negative. Panels (b) and (e) show the results of the comparison between severe COVID-19 and mild COVID-19. Panels (c) and (f) show the results of the comparison between long-COVID and remission. The dotted line indicates the Bonferroni threshold (0.05/17,183 CpG sites).

Volcano plots for the sex-stratified analyses targeting sex-chromosome probes. The plots display the estimated beta coefficients against -log10 of the P-values. Panels (a) to (c) are for males only and panels (d) to (f) are for females only. Panels (a) and (d) show the results of the COVID-19 positive (n = 109) versus COVID-19 negative (n = 73) comparison. Panels (b) and (e) show the results of the severe COVID-19 (n = 61) versus mild COVID-19 (n = 48) comparison. Panels (c) and (f) show the results of the long-COVID (n = 41) versus remission (n = 63) comparison. The dotted horizontal line indicates the Bonferroni threshold (0.05/17,183 CpG sites for males and 0.05/17,128 CpG sites for females).
The severe COVID-19 group differed significantly from the other groups in terms of cell-type composition. Notably, the B-lymphocyte proportion was greater in the severe COVID-19 group than in the other groups. Additionally, this group differed with regards to the composition of CD8+ T cells, granulocytes, and NK cells (Supplementary Figs. 4 and 5). There were no significant differences in cell type composition between the long-COVID group and the remission group. A comparison of epigenetic age acceleration (EAA) between the groups is presented in Supplementary Fig. 6. We did not find any significant differences in EAA between the groups when we used the DNAmAge clock.
Pooled analyses (males and females combined)
The methylation levels at three CpGs differed significantly (false discovery rate (FDR) < 0.05) in the comparison between COVID-19 positive (n = 109) and COVID-19 negative (n = 73) (Fig. 3). All the CpGs were hypomethylated. The first CpG, cg22399236, is located on chromosome 18 and is not close to any known gene within at least 30 kb, according to a search in the UCSC Genome Browser using GRCh37/hg19. The second CpG, cg03607951, is in the gene ‘interferon induced protein 44 like’ (IFI44L) on chromosome 1p31.1. Groupwise comparisons of differences in the methylation of this CpG is presented in Supplementary Fig. 7. The third CpG, cg09829636, is in the gene ‘ankyrin repeat domain 9’ (ANKRD9) on chromosome 14q32.31. Among the 109 individuals with information on severity of COVID-19 symptoms, we compared the DNAm levels between 61 cases with severe symptoms and 48 cases with mild symptoms. No statistically significant methylation differences were detected. Further, we compared the DNAm levels between 41 cases classified as having long-COVID and 63 cases in remission. No statistically significant methylation differences were detected.
To visualize the local correlation structure of the three significant CpGs, we generated regional co-methylation plots for each CpG using the coMET R package. cg03607951 and cg09829636 showed a moderate degree of correlation with neighboring CpGs (red patches in the heat map in Fig. 7), whereas cg22399236 showed weak correlations.

Visualization of the three differentially methylated CpGs identified in the COVID-19 positive versus COVID-19 negative comparison. (a) cg22399236 (chr18:20651637) (b) cg03607951 (chr1:79085586) in IFI44L (c) cg09829636 (chr14:102976856) in ANKRD9. The correlation map underneath each plot represents pairwise correlations between any two probes (red for high correlation and blue for low correlation). The plot was generated using the coMET R package71.
In addition, we queried the online mQTL database23 to investigate whether methylation of these CpGs is influenced by common SNPs; in other words, we searched for methylation quantitative trait loci (mQTLs) associated with these CpGs. Both cg03607951 and cg09829636 were associated with several trans-acting mQTLs, but the search output did not show any entries for cg22399236 (Supplementary Table 1).
Sex-stratified analyses
As males and females show distinctively different mean methylation values on the sex chromosomes (Supplementary Fig. 3), we performed sex-stratified analyses for probes on the sex chromosomes. No significant differences (FDR < 0.05) in methylation values of CpGs were detected in any of the analyses. However, it may be worth noting that the methylation levels at two CpGs on the X chromosome were slightly lower in the COVID-19 positive males than the COVID-19 negative males (Fig. 6). These were cg08118341 (P = 8.96e−06, chrX:48931823) near the gene ‘PRA1 domain family member 2’ (PRAF2), and cg24340926 (P = 1.19e−05, chrX:129305036) near the gene ‘RAB33A, member RAS oncogene family’ (RAB33A).
Location of significant CpGs
We searched for the location of the three significant CpGs using the Ensembl browser24. cg03607951 is located on chromosome 1 (Chr1:79085586–79085635) and cg09829636 on chromosome 14 (Chr14:102976856–102976905). cg03607951 and cg09829636 are both located in gene promoter regions (of IFI44F and ANKRD9, respectively), which is not surprising given that most of the CpGs on the Illumina 450 K array that were migrated over to the more recent Illumina EPIC array are predominantly located within gene promoters and promoter-flanking regions. Ensembl did not return any output for cg22399236, which is probably because this CpG was not present on the former Illumina 450 K platform on which the Ensembl entries are based. The Infinium MethylationEPIC Manifest File25 indicates that this CpG is located on chromosome 18 (nucleotide position 20651395 based on GRch37) and corresponds to the SNP rs576870425. According to the ‘Functional annotation of the mammalian genome 5’ (FANTOM5) database26 and the Illumina Manifest File for the EPIC array, this CpG overlaps with one regulatory feature, ENSR00001016303, which is an enhancer region. Moreover, the Ensembl Regulatory Build indicates that this CpG is located within a promoter-flanking region. Thus, even though cg22399236 is located in a ‘gene desert’ on chromosome 18, it may have regulatory (enhancer) functions in genes located nearby.
Differentially methylated regions
We identified 168 differentially methylated regions (DMRs) from the nine sets of comparisons (Supplementary File 3). An example of a significant DMR with a low P-value was the genomic region chr6:33245490–33246043, containing 19 CpG sites and located near the gene ‘beta-1,3-galactosyltransferase 4’ (B3GALT4). This DMR was less methylated in the COVID-19 positive group (males and females combined) than the COVID-19 negative group (P = 1.24e−15, Bonferroni-corrected P = 1.04e−09). Another example of a significant DMR was the region on chr3:21792248–21792991, containing nine CpGs and located near ‘Zinc Finger Protein 385D’ (ZNF385D). This DMR was differentially methylated in the severe COVID-19 group compared to the mild COVID-19 group (P = 4.82e−14, Bonferroni-corrected P = 4.00e−14). Lastly, two highly significant DMRs were detected in the long-COVID versus remission comparison (chr6:32121225–32121555 near ‘palmitoyl-protein thioesterase 2’ (PPT2) and chr14:63671314–63671737 near ‘ras homolog family member J’ (RHOJ)).
Gene-set enrichment analysis
Table 2 shows the summary statistics for the top 20 gene ontology (GO) enrichment results. Although we did not detect any statistically significant (FDR < 0.05) gene-set enrichment in the GO or “Kyoto Encyclopedia of Genes and Genomes” (KEGG) pathways, the results of the top 20 GO pathways were enriched for several terms related to defense against viral infection. These enrichments were all related to cg03607951 in IFI44L. By contrast, the results of the KEGG analysis did not identify any pathways.
Discussion
The primary objective of this study was to determine whether the DNAm profiles of different groups of COVID-19 patients (severe and mild) differed from those of control individuals eight to twelve weeks after infection with SARS-CoV-2. A secondary objective was to investigate whether the DNAm profiles of individuals with long-COVID differed from those in remission. Overall, only the comparison between the COVID-19 positive and COVID-19 negative individuals revealed differentially methylated CpGs at FDR < 0.05 (specifically, cg22399236, cg03607951, and cg09829636). The analyses of COVID-19 severity and long-COVID did not identify any CpGs with significantly different methylation levels.
The comparison between the COVID-19 positive and COVID-19 negative individuals revealed three differentially methylated CpGs at FDR < 0.05 (cg22399236, cg03607951, and cg09829636). A search of these CpGs in the EWAS atlas27 showed multiple entries linking hypomethylation of cg03607951 in IFI44L to systemic lupus erythematosus (SLE)28, primary Sjögren’s syndrome29, mixed connective tissue disease30, and multiple other autoimmune disorders31. By contrast, there were no entries for cg22399236 and cg09829636 in the EWAS atlas. Further, a search in the online mQTL database showed several trans mQTLs associated with cg03607951 and cg09829636, but no cis or trans mQTLs associated with cg22399236. Trans mQTLs are known to be more polygenic than cis mQTLs and to explain less of the trait variance than cis mQTLs32. More studies are needed to elucidate how these trans mQTLs affect methylation levels at the two CpGs in relation to COVID-19.
IFI44L plays an important role in interferon-induced innate viral response and protection against disease. It has targeted antiviral specificity to several viral species33. Such a function is not surprising given that IFI44L is a paralog of IFI44, a key gene involved in the induction of type I and type III interferon signaling34,35. However, a recent study showed that inhibition of IFI44L impairs the replication of several viruses, while the expression of IFI44L impairs antiviral response34. Based on these findings, it is interesting to note that a recent transcriptomics study concluded that asymptomatic SARS-CoV-2 infection might be due to decreased expression of six genes, one of which was IFI44L36. Another transcriptomics study of myocardial tissue from SARS-CoV-2-positive autopsy cases revealed an upregulation of two interferon-related genes (IFIT3 and IFI44L) among those with cardiac involvement37. Transcriptome data from human bronchial epithelial cells during SARS-CoV-2 infection have also identified IFI44L as one of the top genes upregulated in severe COVID-1938,39.
Collectively, the above studies indicate that downregulation of IFI44L early in the disease course may be a beneficial physiologic response to control SARS-CoV-2 infection and that it may be associated with a positive outcome. We did not find any methylation differences between severe and mild COVID-19 three months after the infection. However, this may be because both the mild and severe group in our study consisted of outpatients who all survived. Although nonsignificant, the severe COVID-19 group in our study did show a higher expression of IFI44L.
The observed hypomethylation of IFI44L three months after infection in our study could reflect that it takes time to reverse the immune responses induced by infection. However, it could also reflect a physiological response to control the infection, as complete viral clearance may take months to achieve in some individuals40,41. Another explanation is that it could indicate a possible link between COVID-19 and systemic autoimmune diseases. Notably, Zhao et al.42 proposed that hypomethylation of the IFI44L promoter might be a reliable biomarker for the diagnosis of SLE. Furthermore, SARS-CoV-2 has been shown to induce autoantibodies (e.g., antiphospholipid antibodies and transient lupus anticoagulant43,44) and trigger autoimmune responses such as hemolytic anemia, thrombocytopenia, and myocarditis44. There are recent reports of SARS-CoV-2 infection preceding various autoimmune diseases, including SLE45,46. Therefore, longitudinal data allowing an evaluation of the duration of the methylation changes post infection might enhance our understanding of possible long-term effects.
Besides cg03607951 in IFI44L, the comparison between COVID-19 positive and COVID-19 negative individuals identified a statistically significant CpG (cg09829636) in ‘ankyrin repeat domain 9’ (ANKRD9). ANKRD9 is a highly conserved protein that modulates the intracellular properties of the cytosolic enzyme inosine monophosphate dehydrogenase 2 (IMPDH2) and facilitates its degradation47. The link between ANKRD9 and IMPDH2 is compelling in light of the role of IMPDHs as therapeutic targets for COVID-19. Specifically, the IMPDH inhibitor, Ribavirin (aka tribavirin), is an antiviral medication used for the treatment of COVID-1948. Thus, hypomethylation of ANKRD9 in COVID-19 patients might reflect a physiologic response to control the infection.
The top GO pathways identified in our analyses, although nonsignificant at FDR < 0.05, suggest that IFI44L has a central role in viral response. Indeed, cg03607951 in IFI44L seemed to be the sole contributor of the three significant CpGs in the GO pathways related to viral response. Our top GO pathway (GO:0051607) “Defense response to virus” has also been reported in two other transcriptomic studies of COVID-19-infected humans and mammals49,50.
Our analysis comparing the long-COVID group to the remission group did not identify any significant methylation differences. The lack of significant findings may be real or reflect the small sample size available for this comparison. The proportion of patients evaluated to have long-term symptoms was similar to the numbers reported in previous studies (38% vs 33% and 40%)1,2,51.
To our knowledge, the current study is the only EWAS of post-COVID-19 and long-COVID patients. Other EWASs of COVID-19 in the literature include those by Castro de Moura et al.20, Balnis et al.22, Zhou et al.52 and a multi-omics study by Bernardes et al.53. These studies examined DNAm changes early in the disease course, i.e., during the ongoing infection when the immune system is highly activated and found distinct patterns correlating with disease severity. Castro de Moura et al. included data on mild and severe COVID-19 cases without underlying conditions, and their DNAm data were also generated on the Illumina EPIC platform. Consistent with our findings, Castro de Moura et al. also found hypomethylation of IFI44L among their top ten genes associated with severe COVID-19 early in the disease course. They concluded that the methylation changes were likely due to innate susceptibility rather than changes induced by the virus itself. Apart from IFI44L, we had no other overlapping findings with the study by Castro de Moura et al. This may be due to differences in study populations (we only had seven patients in need of ventilator support). It could also be that the methylation signals at the other loci were transient and were reversed three months after infection.
Balnis et al.22 also had data on COVID-19 positive and COVID-19 negative individuals, and like the above-mentioned study by Castro de Moura et al.20, their DNAm data were also generated on the Illumina EPIC platform. Despite these similarities, Balnis et al. did not detect any global mean methylation differences between the two groups of participants. Again, this could be due to differences in the study populations (the controls in Balnis et al. were an intensive care population). The authors did, however, detect DMRs in another interferon-induced gene, ‘interferon alpha inducible protein 27’ (IFI27), as well as in ‘2′-5′-oligoadenylate synthetase 2’ (OAS2) which is a member of the 2–5 A synthetase family known to be involved in the innate immune response to viral infection. The results of their gene ontology and pathway enrichment analysis were also congruent with ours, pointing to host-defense responses and terms such as ‘response to type I interferon’ and ‘response to virus’, among others.
Despite limited sample sizes (n = 21 and n = 13), the studies by Zhou et al.52 and Bernardes et al.53 reported significant methylation changes between mild and severe COVID-19. Zhou et al. reported the downregulation of four genes, in particular “G Protein Subunit Gamma 7” (GNG7) and “Guanine nucleotide binding protein” (GNAS), among patients with severe disease. Likewise, the longitudinally combined transcriptomic and methylation analysis by Bernardes et al. revealed the downregulation of GNG7 and GNAS2 in severe COVID-19. However, neither study detected methylation changes related to IFI44L or ANKRD9. The epigenome-wide investigations described above collectively point to innate response to viral infection via interferon-inducible proteins as a possible mechanism for COVID-19 disease progression. In support, our gene-enrichment pathway analysis revealed enrichment of GO terms that were specific for defense responses to viral infection.
Although we did not collect peripheral blood mononuclear cells (PBMCs), Su et al. did not find any link to IFI44L when examining RNA expression in samples taken three months after SARS-CoV-2 infection in subjects with long-COVID54.
Although there have been reports of EAA in severe COVID-1921, we did not find any differences in EAA in our study. Interestingly, Cao et al.55 found a dynamic increasing EAA in the initial phases of COVID-19, while this increase was partly reversed in the convalescent phase, indicating that the infection might accelerate epigenetic aging. Another study by Mongelli et al.56 reported increased EAA in younger but not older COVID-19 survivors. More studies are needed to determine the association between EAA and SARS-CoV-2 infection.
Strengths and limitations of the study
Only a few studies have reported blood-based DNAm in COVID-19, and, to our knowledge, there are no prior reports comparing differentially methylated CpGs three months after SARS-CoV-2 infection. Our study also includes data from confirmatory serology tests performed on all participants, including those with long-COVID, which helps to minimize false positives/negatives. Our study is also based on prospective follow-up questionnaires with high response rates, enabling detailed longitudinal assessments.
Our study also has a few limitations. The evaluation of disease severity was based on self-reported symptoms, and not on an objective assessment. It is difficult to gauge the extent to which recall bias and other types of misclassifications might have influenced the results presented here. The evaluation of long-COVID using the RAND 36-item health survey questionnaire is known to have high sensitivity; however, specificity may be low. Notably, 12% of the Norwegian population reported that their health had, in general, worsened compared to the preceding year. At the same time, the population included in the current study was apparently healthier than the general population. For instance, in the first wave of the pandemic, the COVID-19 positive individuals reported higher physical fitness and socioeconomic status, which is also reflected in our control group where only 4% of the participants reported worse health compared to a year before. Additionally, the evaluation of long-COVID versus remission was likely affected by the small sample size.
In addition, we were unable to explore the expression of IFI44L and ANRKD9 in PBMCs, as extracting these cells from whole blood after freezing is not feasible. The study setup we had at the time of recruitment did not allow for repeated measurements on the same patients both in the acute phase of infection and after three months, which would have provided relevant longitudinal information rather than a single “snapshot” of disease progression.
In conclusion, our study adds to the growing knowledgebase regarding epigenetic contributions to COVID-19. It confirmed previously reported associations with IFI44L and the involvement of interferon-responsive genes in the underlying pathophysiology of COVID-19 and showed that such signals can be identified months after the infection. We identified a novel link to ANKRD9, which is noteworthy given that IMPDH inhibitors are used in the treatment of COVID-19. This and the other genes identified here would need to be replicated in other EWASs before being dismissed as false positives. We have thus provided all the results of our main EWASs, so that other researchers would be able to easily compare their results to ours.
Methods
Study design
The study was designed as a retrospective case–control study nested within the Norwegian Corona Cohort Study (ClinicalTrials.gov Identifier: NCT04320732), which is an ongoing prospective observational study established in March 2020 during the first wave of the COVID-19 pandemic. The cohort consists of two subgroups: (i) adults with a conclusive SARS-CoV-2 RT-PCR test (n = 23,948) invited through four laboratories in the greater Oslo area (Oslo University Hospital, Akershus University Hospital, Vestre Viken hospital, and Fürst Medical Laboratory), and (ii) adults signing up to the study through a media campaign (n = 92,730). All the participants completed an online baseline questionnaire upon enrollment and were invited to follow-up questionnaires at three and six months into the study. Whole blood and serum samples from 110 confirmed COVID-19 cases and 74 controls were retrospectively collected on the 27th and 28th of May 2020 at the Oslo University Hospital.
Inclusion/exclusion criteria
Participants who were enrolled in the Norwegian Corona Cohort Study and who lived in the greater Oslo area were invited to participate in the current study. They were categorized into three groups based on the following criteria:
-
Severe COVID-19 Participants with a positive SARS-CoV-2 RT-PCR test who had either been hospitalized because of COVID-19 or reported all of the following symptoms: fever > 38 °C, dyspnea, cough, and fatigue.
-
Mild COVID-19 Participants with a positive SARS-CoV-2 RT-PCR test not requiring hospitalization and who reported neither fever > 38 °C nor dyspnea.
-
Controls This group consisted of both symptomatic and healthy controls. The symptomatic control group, designed similarly to the severe COVID-19 group, were those who had a negative SARS-CoV-2 RT-PCR test and who reported all of the following symptoms at the time of testing: fever > 38 °C, dyspnea, cough, and fatigue. The healthy control group reported none of the following symptoms during the three weeks preceding inclusion into the study: temperature > 38 °C, dyspnea, cough, fatigue, altered sense of smell and taste, sore throat, nasal symptoms, or headache, and no infections during the past six months. For this reason, the healthy controls had not undergone any SARS-CoV-2 RT-PCR testing.
Eligible participants from each group were randomly invited to donate a blood sample at the Oslo University Hospital. Of those who consented, a list of 318 randomly selected potential participants was prepared. The study staff called and invited participants into the substudy until approximately 200 appointments were made. To minimize sampling bias, all the participants were given the opportunity to choose their own time slot for the blood draw.
Data collection
The evaluation of disease severity was based on self-reported information from the baseline questionnaire, which covered previous medical history, symptoms, disease duration, hospitalization, and remission. SARS-CoV-2 RT-PCR test results were obtained from the following four laboratories in the greater Oslo area: Fürst Medical laboratory, Oslo University Hospital, Akershus University Hospital, and Vestre Viken Hospital. A SARS-CoV-2 serology test was performed on all participants.
Evaluation of long-COVID was performed through an electronic follow-up questionnaire distributed three months after the blood draw. Reminders were sent to non-responders via email and SMS. Long-COVID was defined as a worsening in self-reported health from a year ago, assessed by a single-item, five-level question from the RAND 36-item health survey questionnaire57. This definition is considered to have high sensitivity for long-COVID. However, 12% of Norwegians reported a worsening in health in a general population survey based on this questionnaire58. Therefore, the specificity in the study population is estimated to be approximately 70%.
Sampling scheme
Figure 1 provides a schematic overview of the sampling scheme used to assign the participants into distinct categories (i.e., severe COVID-19, mild COVID-19 and controls, and long-COVID and remission) according to the inclusion/exclusion criteria. Of the approximately 200 individuals who consented to a blood draw, 184 showed up for their appointment and completed the baseline questionnaire. Two of the participants failed the data file transfer, bringing the final number of participants included in the current analyses to 182. Of these, 48 were categorized as mild COVID-19, 61 as severe COVID-19, and 73 as controls. Based on the participants’ answers in the follow-up questionnaire three months after inclusion, we were able to reclassify four subjects from the mild and severe COVID-19 categories as belonging to the long-COVID category.
Laboratory methods
The serum tubes were centrifuged, aliquoted, and frozen within four hours of the blood draw. The EDTA tubes with whole-blood samples were kept on ice, aliquoted, and frozen at − 80 °C within two hours of the blood draw. Confirmatory serology was performed based on the detection of anti-SARS-CoV-2 antibody against nucleocapsid, as measured by the Roche Cobas e601 module (Roche Diagnostics GmbH, Mannheim, Germany).
DNAm measurement
DNA was extracted from 200 µl of EDTA-anticoagulated whole blood using the QIAsymphony DSP DNA Mini Kit (QIAGEN, catalog number 937236) at the Oslo University Hospital. The tubes were initially placed in random order but were not formally randomized before bisulfite conversion using the Zymo EZ-96DNA Methylation-Lightning MagPrep kit (Zymo Research, Irvine, USA). DNAm was measured using the Illumina Infinium MethylationEPIC BeadChip (Illumina, San Diego, USA) at Life & Brain GmbH, Bonn, Germany.
The raw signal intensity data were extracted from the IDAT files using a standard pipeline powered by the R packages minfi and DMRcate59. We applied the multiple sample/probe exclusion criteria prior to background correction and normalization. The detectionP function was used to exclude samples with a mean detection p-value greater than 0.01 and probes with a detection p-value greater than 0.01. Cross-reactive probes and probes within two base-pairs from a single-nucleotide polymorphism (SNP) with a minor allele frequency (MAF) greater than 0.05 were removed using the rmSNPandCH function in DMRcate. In addition, cross-hybridizing probes specific to the EPIC array, as previously reported by McCartney and co-workers60, were also excluded. In addition, the output of the minfi qcReport and plotQC were visually inspected for inconsistencies. Finally, the data were background corrected and normalized using the default settings of the preprocessFunnorm function in minfi.
Following the above QC steps, DNAm data on 182 individuals (109 individuals in the COVID-19 group and 73 in the non-COVID-19 group) and 794,075 probes remained for the current EWASs.
Statistical analyses
EWAS
We stratified the methylation data into the following three subsets: (1) the combined sample of males and females and 776,892 autosomal probes, (2) males only and 17,183 sex chromosome probes (17,128 X-linked and 55 Y-linked), and (3) females only and 17,128 X-linked probes. In each subset, we compared the mean methylation levels, i.e., the M-values61, in the following group comparisons: (a) COVID-19 positive (n = 109) versus COVID-19 negative (n = 73), (b) severe COVID-19 (n = 61) versus mild COVID-19 (n = 48), and (c) long-COVID (n = 41) versus remission (n = 63). This analytic strategy is outlined in Fig. 2. The results from additional group comparisons can be found in Supplementary Figs. 8 and 9.
We fit linear regressions of the M-values on the COVID-19 variables, with adjustment for age, sex, imputed smoking pack-years, physical fitness, imputed white blood cell counts (CD8 + naïve and exhausted cytotoxic T cell, CD4 + naïve cells, natural killer cells, B cells, monocytes, and granulocytes), and plate. The imputed smoking pack-years and white blood cell counts were derived using Horvath's online calculator62. Next, we derived empirical Bayes moderated t-statistics and the corresponding P-values using the limma R package63.
The entire EWAS summary statistics can be found in Supplementary Files 1 and 2, respectively. All analyses were performed in the statistical programming language R, version 4.0.5.
Additional post-processing analyses
To identify differentially methylated regions (DMRs), we applied the dmrff function from the dmrff R package64 to the EWAS summary statistics. We chose dmrff because it was reported to be the most powerful method in a comparison involving four other popular methods for DMR detection (DMRcate, comb-p, seqlm, and GlobalP)65. The maximum distance between consecutive probes was set to 500 base-pairs (the default value). We defined a DMR as being statistically significant if it had a Bonferroni-corrected P-value less than 0.05.
Next, we performed a gene-set enrichment analysis of the significant CpGs detected by the EWASs to test for potential enrichment in biological pathways. We used the gometh function implemented in the missMethyl R package66, which queries Gene Ontology (GO) categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
We used the Illumina Infinium MethylationEPIC manifest file (v1.0 B5)25 to define and analyze the target genes. This file contains detailed information on whether a given CpG is located within specific regions of interest (e.g., gene-promoter region, promoter-flanking region, gene-body, CpG island, shelf, shore, and open sea), and whether the CpG is associated with specific regulatory features, such as DNase hypersensitive regions, chromatin regions, and enhancers (as defined by FANTOM5 annotations), etc.
Cell-type composition was estimated using the function estimateCellCounts in the minfi R package67.
Epigenetic age acceleration
Epigenetic age was estimated using the DNA methylation based age predictors DNAmAge68 and Hannum69 using the University of California Los Angeles (UCLA) web-based service70 that adjusts for blood cell composition. We used the (linear regression) residuals between estimated epigenetic age and true chronological age as the measurement of epigenetic age acceleration (EAA), as recommended by the authors of these epigenetic clocks. A positive EAA indicates that the epigenetic age is higher than the chronological age.
Ethics
The study was approved by the Regional Committees for Medical and Health Research Ethics (REK) in Norway (Reference Number 2021/8504) and conducted in accordance with the Declaration of Helsinki. All participants in the Norwegian Corona Cohort Study provided written informed consent.
Data availability
To enhance data sharing and enable other researchers to compare their results with ours, we have provided the entire EWAS summary statistics for the joint analysis of males and females on 776,892 autosomal probes in Supplementary File 1. The corresponding EWAS summary statistics for the sex-stratified analyses of probes on the sex chromosomes (males and female separately on 17,183 sex-chromosome probes) are provided in Supplementary File 2. Due to written consent and ethical issues, the datasets in the current study are not publicly available. However, researchers may obtain a de-identified dataset upon reasonable request to the study authors and after approval from the study board. Data requests may be subjected to further review by the national register authority and the national ethics committee.
References
Søraas, A. et al. Self-reported memory problems 8 months after COVID-19 infection. JAMA Netw. Open 4, e2118717 (2021).
Stavem, K., Ghanima, W., Olsen, M. K., Gilboe, H. M. & Einvik, G. Persistent symptoms 1.5–6 months after COVID-19 in non-hospitalised subjects: a population-based cohort study. Thorax 76, 405–407 (2021).
Barizien, N. et al. Clinical characterization of dysautonomia in long COVID-19 patients. Sci. Rep. 11, 14042 (2021).
Ramakrishnan, R. K., Kashour, T., Hamid, Q., Halwani, R. & Tleyjeh, I. M. Unraveling the mystery surrounding post-acute sequelae of COVID-19. Front. Immunol. 12, 686029 (2021).
Scherlinger, M. et al. Refining “long-COVID” by a prospective multimodal evaluation of patients with long-term symptoms attributed to SARS-CoV-2 infection. Infect. Dis. Ther. 10, 1747–1763 (2021).
Peluso, M. J. et al. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 36, 109518 (2021).
Ni, L. et al. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity 52, 971-977.e3 (2020).
Langevin, S. M. & Kelsey, K. T. The fate is not always written in the genes: epigenomics in epidemiologic studies. Environ. Mol. Mutagen. 54, 533–541 (2013).
Lappalainen, T. & Greally, J. M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 18, 441–451 (2017).
Rakyan, V. K., Down, T. A., Balding, D. J. & Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 12, 529–541 (2011).
Greenberg, M. V. C. & Bourchis, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607 (2019).
Saxonov, S., Berg, P. & Brutlag, D. L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 103, 1412–1417 (2006).
Zhu, H., Wang, G. & Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 17, 551–565 (2016).
Anastasiadi, D., Esteve-Codina, A. & Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenet. Chromatin 11, 37 (2018).
COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Zeberg, H. & Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020).
Wei, J. et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 184, 76-91.e13 (2021).
Severe Covid-19 GWAS Group. Genomewide association study of severe covid-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534 (2020).
Choudhary, S., Sreenivasulu, K., Mitra, P., Misra, S. & Sharma, P. Role of genetic variants and gene expression in the susceptibility and severity of COVID-19. Ann. Lab. Med. 41, 129–138 (2021).
de Moura, M. C. et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66, 103339 (2021).
Corley, M. J. et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 110, 21–26 (2021).
Balnis, J. et al. Blood DNA methylation and COVID-19 outcomes. Clin. Epigenet. 13, 118 (2021).
Gaunt, T. R. mQTLdb. Large-Scale Genome-Wide DNA Methylation Analysis of 1,000 Mother-Child Pairs at Serial Time Points Across the Life-Course (ARIES) http://www.mqtldb.org/.
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
The Infinium MethylationEPIC Manifest File. https://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html.
Ramilowski, J. A. et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res. 30, 1060–1072 (2020).
Li, M. et al. EWAS Atlas: A curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 47, D983–D988 (2019).
Zhang, B. et al. A simple and highly efficient method of IFI44L methylation detection for the diagnosis of systemic lupus erythematosus. Clin. Immunol. 221, 108612 (2020).
Imgenberg-Kreuz, J. et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 75, 2029–2036 (2016).
Carnero-Montoro, E. et al. Epigenome-wide comparative study reveals key differences between mixed connective tissue disease and related systemic autoimmune diseases. Front. Immunol. 10, 1880 (2019).
Chen, S. et al. Genome-wide DNA methylation profiles reveal common epigenetic patterns of interferon-related genes in multiple autoimmune diseases. Front. Genet. 10, 223 (2019).
Gaunt, T. R. et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 17, 61 (2016).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
DeDiego, M. L., Martinez-Sobrido, L. & Topham, D. J. Novel functions of IFI44L as a feedback regulator of host antiviral responses. J. Virol. 93, 21 (2019).
DeDiego, M. L., Nogales, A., Martinez-Sobrido, L. & Topham, D. J. Interferon-induced protein 44 interacts with cellular FK506-binding protein 5, negatively regulates host antiviral responses, and supports virus replication. MBio 10, 19 (2019).
Sfikakis, P. P. et al. Blood transcriptomes of anti-SARS-CoV-2 antibody-positive healthy individuals who experienced asymptomatic versus clinical infection. Front. Immunol. 12, 746203 (2021).
Bräuninger, H. et al. Cardiac SARS-CoV-2 infection is associated with pro-inflammatory transcriptomic alterations within the heart. Cardiovasc. Res. 118, 542–555 (2022).
Vishnubalaji, R., Shaath, H. & Alajez, N. M. Protein coding and long noncoding RNA (lncRNA) transcriptional landscape in SARS-CoV-2 infected bronchial epithelial cells highlight a role for interferon and inflammatory response. Genes 11, 760 (2020).
Shaath, H., Vishnubalaji, R., Elkord, E. & Alajez, N. M. Single-cell transcriptome analysis highlights a role for neutrophils and inflammatory macrophages in the pathogenesis of severe COVID-19. Cells 9, 2374 (2020).
Vibholm, L. K. et al. SARS-CoV-2 persistence is associated with antigen-specific CD8 T-cell responses. EBioMedicine 64, 103230 (2021).
Hu, F. et al. A compromised specific humoral immune response against the SARS-CoV-2 receptor-binding domain is related to viral persistence and periodic shedding in the gastrointestinal tract. Cell. Mol. Immunol. 17, 1119–1125 (2020).
Zhao, M. et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann. Rheum. Dis. 75, 1998–2006 (2016).
Zhang, Y. et al. Coagulopathy and antiphospholipid antibodies in patients with covid-19. N. Engl. J. Med. 382, e38 (2020).
Rodríguez, Y. et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J. Autoimmun. 114, 102506 (2020).
Galeotti, C. & Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 16, 413–414 (2020).
MantovaniCardoso, E., Hundal, J., Feterman, D. & Magaldi, J. Concomitant new diagnosis of systemic lupus erythematosus and COVID-19 with possible antiphospholipid syndrome. Just a coincidence? A case report and review of intertwining pathophysiology. Clin. Rheumatol. 39, 2811–2815 (2020).
Hayward, D. et al. ANKRD9 is a metabolically-controlled regulator of IMPDH2 abundance and macro-assembly. J. Biol. Chem. 294, 14454–14466 (2019).
Messina, E. et al. Ribavirin aerosol in the treatment of SARS-CoV-2: A case series. Infect. Dis. Ther. 10, 2791–2804 (2021).
Zhang, F. et al. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med. 13, 64 (2021).
Hachim, M. Y. et al. Interferon-induced transmembrane protein (IFITM3) is upregulated explicitly in SARS-CoV-2 infected lung epithelial cells. Front. Immunol. 11, 1372 (2020).
Davis, H. E. et al. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClin. Med. 38, 101019 (2021).
Zhou, S. et al. An epigenome-wide DNA methylation study of patients with COVID-19. Ann. Hum. Genet. 85, 221–234 (2021).
Bernardes, J. P. et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity 53, 1296-1314.e9 (2020).
Su, Y. et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 185, 881–895 (2022).
Cao, X. et al. Accelerated biological aging in COVID-19 patients. Nat. Commun. 13, 2135 (2022).
Mongelli, A. et al. Evidence for biological age acceleration and telomere shortening in COVID-19 survivors. Int. J. Mol. Sci. 22, 6151 (2021).
Hays, R. D. & Morales, L. S. The RAND-36 measure of health-related quality of life. Ann. Med. 33, 350–357 (2001).
Garratt, A. M. & Stavem, K. Measurement properties and normative data for the Norwegian SF-36: Results from a general population survey. Health Qual. Life Outcomes 15, 51 (2017).
Aryee, M. J. et al. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
McCartney, D. L. et al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 9, 22–24 (2016).
Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 11, 587 (2010).
Horvath, S. DNA Methylation Age Calculator. http://dnamage.genetics.ucla.edu/.
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Suderman, M. et al. dmrff: Identifying differentially methylated regions efficiently with power and control. BioRxiv https://doi.org/10.1101/508556 (2018).
Lent, S. et al. Detecting differentially methylated regions with multiple distinct associations. Epigenomics 13, 451–464 (2021).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2016).
Andrews, S. V., Ladd-Acosta, C., Feinberg, A. P., Hansen, K. D. & Fallin, M. D. “Gap hunting” to characterize clustered probe signals in Illumina methylation array data. Epigenet. Chromatin 9, 56 (2016).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
UCLA. DNA Methylation Age Calculator. http://dnamage.genetics.ucla.edu/.
Martin, T. C., Yet, I., Tsai, P.-C. & Bell, J. T. coMET: visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns. BMC Bioinform. 16, 131 (2015).
Acknowledgements
We thank the nurses at the Department of Orthopedics, Oslo University Hospital, for their help with the blood draws, and the Internal Medicine Research Lab at the same hospital for lending us their lab facilities. We are grateful to all the study participants for their valuable contributions to this study.
Funding
This project was funded by the Research Council of Norway (RCN), Grant Number 313651. The funding body did not play any role in the design of the study, data collection, analysis, interpretation of the results, and the writing of this manuscript. The work of JRH and AJ is supported, in part, by the Research Council of Norway through its Centres of Excellence funding scheme, Project Number 262700.
Author information
Authors and Affiliations
Contributions
K.T.K., A.S., C.L.H., E.R., A.J., Y.L., and J.A.D. designed the study. M.I., C.L.H., A.S. and O.R. recruited the participants and biobanked the samples. Y.L., E.R., A.J., and K.T.K. performed quality control. Y.L. and E.R. performed the statistical analyses. F.L.J. and A.L. performed the serology analysis. K.T.K., C.L.H., A.J., J.R.H., Y.L., J.A.D., and E.R. acquired funding. C.L.H., A.J., Y.L. and E.R. drafted the manuscript. All authors provided critical input, revised the manuscript, and approved the final version.
Corresponding author
Ethics declarations
Competing interests
ER, AS, KTK and CLH are shareholders of the Age Labs company. The remaining authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, Y., Riskedal, E., Kalleberg, K.T. et al. EWAS of post-COVID-19 patients shows methylation differences in the immune-response associated gene, IFI44L, three months after COVID-19 infection. Sci Rep 12, 11478 (2022). https://doi.org/10.1038/s41598-022-15467-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15467-1
- Springer Nature Limited