
Abstract
Despite recent progress in acute lymphoblastic leukemia (ALL) therapies, a significant subset of adult and pediatric ALL patients has a dismal prognosis. Better understanding of leukemogenesis and recognition of germline genetic changes may provide new tools for treating patients. Given that hematopoietic stem cell transplantation, often from a family member, is a major form of treatment in ALL, acknowledging the possibility of hereditary predisposition is of special importance. Reports of comprehensive germline analyses performed in adult ALL patients are scarce. Aiming at fulfilling this gap of knowledge, we investigated variants in 93 genes predisposing to hematologic malignancies and 70 other cancer-predisposing genes from exome data obtained from 61 adult and 87 pediatric ALL patients. Our results show that pathogenic (P) or likely pathogenic (LP) germline variants in genes associated with predisposition to ALL or other cancers are prevalent in ALL patients: 8% of adults and 11% of children. Comparison of P/LP germline variants in patients to population-matched controls (gnomAD Finns) revealed a 2.6-fold enrichment in ALL cases (CI 95% 1.5–4.2, p = 0.00071). Acknowledging inherited factors is crucial, especially when considering hematopoietic stem cell transplantation and planning post-therapy follow-up. Harmful germline variants may also predispose patients to excessive toxicity potentially compromising the outcome. We propose integrating germline genetics into precise ALL patient care and providing families genetic counseling.
Similar content being viewed by others

Introduction
Acute lymphoblastic leukemia (ALL) affects individuals of all ages and is the most common pediatric cancer. Although ALL is aggressive and quickly progressing, it usually reacts well to initial treatment1. However, ten to twenty percent of pediatric and 30–60% of adult patients with ALL relapse and high-risk disease remains a challenge2,3. Intensification of ALL therapy by performing allogeneic hematopoietic stem cell transplantation (HSCT) from a family member or registry donor is currently the first option of treatment. Also new treatment modalities, such as targeted antibodies and CAR-T cell therapy are entering the clinics2,4.
The studies in solid malignancies have paved the way for integration of germline genetic information into the care of cancer patients. The identification of inherited genetic variants may affect the choice of treatment in multiple ways and enable early intervention. However, in hematological malignancies, the recognition of these factors has only recently taken root in clinical practice; Constitutional genetic changes are now acknowledged in acute myeloid leukemias (AML) and included in the newest WHO classification of AML5,6. Regarding ALL, data and practices on germline alterations predisposing to the disease are based mostly on pediatric, family, and genome-wide association studies (GWAS), and only one report presenting exome analysis on adult ALL patients (n = 28) has been published7.
Two types of ALL susceptibility have been reported: rare germline mutations with high-penetrance predisposition, such as IKZF18, PAX59,10, and ETV611,12, and common variants with low-penetrance predisposition identified in GWASs13,14 (e.g. ARID5B15, GATA316, and ERG17). Furthermore, in epidemiological studies including also non-pediatric ALL patients, a higher risk of leukemia has been observed in family members, which speaks for shared genetic factors behind the disease18,19.
Here, we have evaluated the extent and distribution of pathogenic (P) and likely pathogenic (LP) germline variation in known leukemia and solid malignancy predisposing genes by performing a comprehensive exome analysis in both adult and pediatric ALL patients.
Methods
Patients
We examined whole exome sequencing (WES) data on freshly collected or biobanked samples of adult (n = 61, aged 16–70) and pediatric (n = 87, aged 0–16) patients with ALL (Finnish Hematological Registry and Biobank – FHRB, and clinical repositories). Adults were diagnosed in the Helsinki University Hospital during the years 2008–2019. WES of adult patients was performed on skin samples except for one remission blood sample. The pediatric samples were collected from ALL patients treated with allogeneic HSCT according to the treatment protocols1,20,21 in the Helsinki University Hospital pediatric unit during 1998–2019. WES in these cases was performed on blood samples taken at the time of HLA typing for HSCT. Specimen preparation and WES are described in detail in Supplementary methods. We also had access to all patients’ clinical information and results of laboratory tests performed during treatment. Patient characteristics are summarized in Table 1 and detailed information on patients is listed in Supplementary Table S1.
Ethics declarations
This study has been approved by Helsinki University Hospital ethics review committee (adults #206/13/03/03/2016 and #303/13/03/01/2011; pediatric patients HUS/114/2018, HUS/284/2019 and V/3235/2019). All samples and data were derived either after written informed consent (living individuals), or authorization by the ethics committee (deceased patients). All methods were conducted in accordance with the relevant guidelines and regulations.
Analyses of the germline variants
We constructed a list of 93 genes (Supplementary Table S2) known to predispose to ALL or other hematological malignancies based on literature22,23. Additionally, a list of 70 genes reported with germline predisposition to cancer (Supplementary Table S3) was compiled based on COSMIC cancer gene census24 (version 94, Tier 1, https://cancer.sanger.ac.uk/census) and NHGRI Clinical Genomics Database (https://research.nhgri.nih.gov/CGD/, accessed 05/03/2021). Variant analysis (workflow presented in Supplementary Figure S1) was done using BasePlayer25, a tool for analyzing and visualizing next-generation sequencing data. Only non-synonymous and splice site variants in protein-coding transcripts (except for TERC, which is a ncRNA, ANKRD26 UTR variants and GATA2 synonymous variants) with a minor allele frequency (MAF) of < 0.01 in both the gnomAD non-cancer whole database and gnomAD non-cancer Finns (version 2.1)26 were considered. Variants filtered in gnomAD due to poor quality were discarded. A 1000 genomes mappability pilot mask track27 and quality measures—genotype quality (GQ) > 20, QUAL > 20, site total coverage > 6 reads, and allelic fraction ≥ 20%—were used to filter out poor-quality variants. All variants were also visually confirmed using BasePlayer: at least two unique reads in both directions with the alternate allele were required and dubious variants in repetitive sequence regions were discarded. For low-coverage variants, also the somatic exome (blood or bone marrow) of the same patient was used to verify the variant, if available. The median coverage was 64 × and 232 × for adult and pediatric samples, respectively.
The classification of the germline variants (benign, likely benign, variant of uncertain significance (VUS), likely pathogenic (LP), or pathogenic (P)), according to the American College of Medical Genetics (ACMG) guidelines28, was done first with Intervar29 (version 2.0.2). Thereafter, to confirm the findings, the P/LP/VUS variants were classified with Varsome30 (ACMG version 9.5.0, https://varsome.com/) since differences in interpretation exist due to various databases and prediction algorithms used. Our own conclusive judgment of pathogenicity for the variants (used throughout the manuscript) was based on careful consideration of all the rules and evidence from InterVar, Varsome and literature. Predictions by both tools are reported in the Table 2 and Supplementary Table S4. The oncoplot in Fig. 1a was made using ComplexHeatmap package (version 2.10.0)31 and the boxplot in Fig. 1b was made using ggplot2 (version 3.3.5, https://ggplot2.tidyverse.org)32 in R (version 4.1.2, https://www.r-project.org/)33. As knowledge on the actionable germline variants is still evolving, we also report variants of uncertain significance, if P/LP variants were identified in the study patients in the respective genes by either of the used prediction tools. The VUS were not included in the calculations of the reported frequencies of (P/LP) variants. Exceptions to the above-mentioned interpretation rules were made with BLM, due to identification of surprisingly high number (7) of the same VUS. Also, a VUS in TERT is reported because it has been shown to correlate with short telomeres34 (Supplementary Table S4).

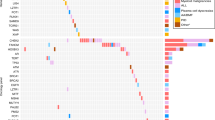
(a) The distribution of all pathogenic and likely pathogenic variants and variants of uncertain significance (VUS) in the ALL patients. The frequency and number of patients harboring a variant in each gene are shown on the right-hand panel. B-ALL, Ph-negative B-ALL; Ph-ALL, Ph-positive B-ALL. (b) Enrichment of pathogenic variants in ALL patients compared to healthy controls (gnomAD Finns). Variants in genes with known predisposition to hematological malignancies (n = 93) and other known cancer-predisposition genes (n = 70) are shown separately and together for both age groups and overall. OR, odds ratio; CI, confidence interval.
Determining the germline/somatic origin of the pediatric patients’ variants
The exome sequenced blood samples from pediatric patients were obtained at different phases of ALL with a varying degree of blastemia. Germline variants’ variant allele fraction (VAF) should be close to 0.5 for heterozygous variants and 1.0 for homozygous variants. We classified the variants as somatic or germline manually by considering the blast percentage, VAF and karyotype in each sample. To support the interpretation of the origin of the variant, we also verified if the variant was reported in the gnomAD database, ClinVar35 (germline variants) (https://www.ncbi.nlm.nih.gov/clinvar/), or in cBioPortal36,37 (https://www.cbioportal.org/) (somatic variants) (Supplementary Table S5). Variants that have been reported in normal population and never as somatic, are likely germline, and vice versa. This is even more pronounced in Finland, where certain germline variants are enriched and therefore these variants are even more likely to be of germline origin (Supplementary Table S5). Only variants which were evidently of germline origin were included in the analyses. If the judgement was uncertain, the variant was not interpreted as of germline origin. All variants in patients with ≥ 50% blasts are reported in Supplementary Table S5, along with justification of germline origin of the variant.
Enrichment of pathogenic or likely pathogenic variants
To define the potential enrichment of rare (MAF < 0.01) P/LP variants in the studied genes, we compared the frequency of all rare P/LP variants in these gene sets with respect to ACMG classification in adult and pediatric ALL patients and in gnomAD non-cancer Finns26 v.2.1 (n = 21,632 alleles). We also compared the frequency of all P/LP variants in each studied gene separately (with at least one P/LP variant) in patients and gnomAD non-cancer Finns. The differences were evaluated using 2-sided Fisher exact tests. Benjamini–Hochberg procedure was used to adjust the p-values for multiple testing. In genes with multiple truncating variants that were determined as VUS, we compared the frequencies of truncating variants only. As an exception for other analyses in this study, we used Intervar as the only prediction tool here since it is open source and feasible to be integrated into pipeline to classify large amounts of variants.
Results
Frequency and description of the identified germline variants in ALL patients
Overall, by using stringent classification criteria 11/61 (18%) of adults and 21/87 (24%) of children had heterozygous P/LP variants in the genes analyzed (Fig. 1a, Supplementary Table S4). If we excluded the heterozygous variants in genes where the acknowledged mode of heritance is autosomal recessive, the respective numbers were 5/61 (8%) in adult and 10/87 (11%) in pediatric patients (Table 2). Most of these were previously reported P variants with a known cancer risk—in 5/61 (8%) adults and 8/87 (9%) pediatric patients. All the identified P/LP/VUS are demonstrated in the oncoplot (Fig. 1a), and the classification criteria in the Supplementary Table S4.
We discovered one P variant in high-risk (not lineage-restricted) leukemia predisposition genes: RUNX1 p.Arg204Gln in an adult patient. A truncating variant in SAMD9 p.Ser844ValfsTer10 was found in a pediatric patient. We classified it as a VUS since functional information and segregation analysis on this variant were not available, and most of the pathogenic variants in SAMD9 are missense variants. However, a few rare loss-of-function variants in SAMD9 have also been identified in myelodysplastic syndrome patients38,39. Most malignancies caused by germline RUNX1 and SAMD9 variants are myeloid, but also predisposition to lymphoid malignancies has been reported40,41. Regarding genes causing syndromes with a risk of ALL, we found two P/LP variants in Li-Fraumeni and Noonan syndrome -causing genes: one P variant in TP53 (p.Gly245Ser) and one in LZTR1 (c.2407-1G > A) (Fig. 1a, Table 2).
We also detected six heterozygous truncating P/LP variants and two VUS missense variants in DNAJC21, in which autosomal recessively inherited mutations have been reported to cause Shwachman-Diamond-like disease42.
Seven patients had P/LP variants in colorectal cancer risk -associated genes MUTYH and PMS2. Four of them had a single P variant in MUTYH – p.Arg19Ter (n = 1) and hotspot variants p.Gly396Asp (n = 2), and p.Tyr179Cys (n = 1); while one pediatric patient was compound heterozygous for the hotspot variants p.Gly396Asp and p.Tyr179Cys. In PMS2 we identified two pathogenic heterozygous variants (p.Tyr255Ter and p.Glu109GlyfsTer30) in one adult and one pediatric patient, respectively. There was no known family history of colorectal cancer in these patients.
We also identified heterozygous P/LP variants in genes predisposing to other solid tumors: BRCA1 (n = 1), BRIP1 (n = 1) and CHEK2 (n = 6), that are breast cancer –predisposing genes, RET (n = 1), which predisposes to multiple endocrine neoplasia, and SDHB (n = 1) and SDHC (n = 1) that are known to predispose familial paraganglioma- pheocromocytoma (Table 2, Fig. 1a).
The comparison of the frequency of harmful germline variants with population-matched controls
We compared the frequency of P/LP germline predisposition variants in ALL patients to population-matched (Finns) GnomAD non-cancer control data. We found a 2.6–fold (CI 95% 1.5–4.2, p = 0.00071) enrichment of variants in the adult and pediatric patient sets combined (Fig. 1b, Supplementary Table S6). The enrichment of variants was pronounced in traditional cancer predisposition genes in adults, and in hematological malignancy genes in children, 4.2- and 2.5-fold, respectively (Fig. 1b, Supplementary Table S6).
Statistically, the most significant differences in the frequency of P/LP germline variants between patients and population-matched non-cancer controls were detected in PMS2, DNAJC21 (truncating variants only), and MUTYH (OR ranging from 4.1 to 48.9) (Supplementary Table S7). P/LP variants in colorectal cancer risk—associated genes were enriched in our ALL patients compared to non-cancer Finns (PMS2 OR 48.91, 95% CI 4.08–434.55; p = 0.0018, MUTYH OR 5.12, 95% CI 1.82–11.74, p = 0.0016). Despite the recessive inheritance of DNAJC21, enrichment of truncating heterozygous variants was seen in the study patients compared to population-matched (gnomAD non-cancer Finns) controls (OR 4.07, 95% CI 1.28–9.95, p = 0.00976). All significant (p < 0.05) results, except CHEK2, survived correction for multiple testing (FDR < 10%) (Supplementary Table S7).
Discussion
In this study we present data on germline alterations in pediatric and adult ALL patients. Previous reports on this matter and especially in adult cases are scarce. In the analyses, we took advantage of having a large population-specific reference dataset in Finland to assess the variant enrichment in the patients compared to population-matched non-cancer controls. This is, to our knowledge, the largest assessment of predisposition variants in adult ALL patients performed to date, which is important given the inferior survival of adult ALL patients. Altogether, we identified likely harmful germline variants in 8% of adult and 15% of pediatric ALL patients. These rates are parallel to results attained from studies on myeloid malignancies6,43. A previous study on childhood cancers found germline predisposition in childhood ALL in ~ 4% of the patients44. In our pediatric series the frequency was more than three-fold. However, as Zhang et al. discuss, their results may be underestimates. Furthermore, our study included only children with high-risk ALL who had undergone allogeneic HSCT, in distinction to Zhang et al. report. A study on Korean ALL patients (65 pediatric, 28 adult cases) found only 1.1% carrying pathogenic variants in ALL-predisposing syndrome genes and one multi-exon deletion7. Interestingly, both pathogenic variants were discovered in adult patients (> 16yrs) (2/28). Evidently, additional studies and larger material are needed, especially on adult ALL, to further define the role of germline predisposition in ALL.
When focusing on the variants we discovered, we didn’t find any variants in genes predisposing solely to ALL. Nevertheless, we identified a rare harmful variant in RUNX1, first recognized in AML. This RUNX1 variant seen in patient 2167 has been reported in a family with AML and T-ALL40. Also, our patient suffered from T-cell ALL, the rarer lineage but typical for germline RUNX1 mutation-linked lymphoblastic leukemia40,41. Interestingly, the patient’s family history of cancer did not suggest a high-penetrance germline mutation.
In children, two rare harmful variants were identified in genes causing syndromes with ALL predisposition: one in TP53 and one in LZTR1. Germline TP53 mutations cause Li-Fraumeni syndrome, which is characterized by a high risk for malignancies, including ALL, in particular hypodiploid ALL45. In our patient with the germline TP53 mutation, ALL (not hypodiploid) was the first presentation of Li-Fraumeni syndrome (at the age 2). An increased risk for leukemias in general, but also in ALL, has been described in Noonan syndrome patients with different germline mutations causing the disease46,47. Patients with Noonan syndrome may have some characteristic features, such as cardiac abnormalities and facial features, but there were no indications of these in our patients. Noonan syndrome -causing LZTR1 variants have been reported in the literature both in homozygous and heterozygous forms, depending on the variant48,49. The LP LZTR1 splice site variant (c.2407-1G > A) identified in our study has not been reported before, but a variant in the adjacent base (c.2407-2A > G) has been reported in Noonan syndrome patients (homozygous and one heterozygote in ClinVar) and in schwannomatosis patients49,50. This suggests that the variant is damaging.
Homozygous or compound heterozygous mutations in DNAJC21 have been reported to cause Shwachman-Diamond -like hematological phenotype42. We identified several heterozygous variants in our data, which is statistically more than observed in the control set. The implication of these DNAJC21 variants in leukemogenesis needs to be validated in other patient sets and functional experiments before further interpretation.
In addition to the genes known to predispose to hematological malignancies, patients harbored several interesting variants in genes primarily associated with an increased risk for solid tumors (MUTYH, PMS2, and CHEK2). Also, for some genes the mode of inheritance is not unequivocal. MUTYH is involved in base excision repair and biallelic germline mutations in it cause MUTYH-associated polyposis. Although MUTYH is considered an AR predisposition gene, several studies also suggest a modestly increased cancer risk for heterozygous mutation carriers51,52,53. Specifically, the hotspot variant p.Gly396Asp seen in our patients, has been suggested to result in increased risk for some cancers, such as small intestinal neuroendocrine tumors and a modest increase in breast cancer risk as monoallelic52,53. Germline PMS2 mutations cause hereditary nonpolyposis colorectal cancer (AD) and constitutional mismatch repair deficiency syndrome (AR).The CHEK2 variant c.1100delC is not a very rare variant in Finnish population, but it is a known pathogenic variant with a modestly increased breast cancer risk54. Also, predisposition to other solid malignancies has been reported55, given the universal role of CHEK2 in DNA repair.
In Finland we have the advantage of having a large population-specific reference dataset (gnomAD Finns26 v.2.1, n = 21,632 alleles). This enables more precise assessment of variant enrichment in the patients. In general, we found an enrichment of P/LP variants in cancer-predisposing genes in the ALL patients compared to population-matched control data. However, our results are transferable, as the prevalence of ALL in Finland is comparable with other developed countries, according to the published registry data56.
Since we took into account only P/LP variants (and excluded VUS) and the gene lists comprised only of currently known predisposition genes, this result presents the minimum number of rare germline predisposition variants. We identified P/LP germline variants also in adult ALL patients, and therefore, germline predisposition should not be ignored in ALL in any age groups. Our patients are, however, more high-risk than average, but it is not known if the predisposition to ALL is related to the severity of the disease. To our knowledge our exome data set in adult ALL patients is unique and even for pediatric patients only few estimates on the frequency of germline predisposition to ALL have been reported.
The difference in the frequency between patients and controls was more pronounced in hematological genes in children and in cancer genes in adults. The explanation for this may be explained by the fact that the list of hematological malignancy genes consists of high-penetrance ALL predisposition factors found originally in pediatric patient sets and leukemia families. Furthermore, those harboring high-penetrance predisposition mutations for ALL are more likely to have the disease in childhood. However, more studies are needed to estimate more precisely the gene/variant -specific risk. The clustering of the solid malignancy predisposition gene variants in the adult patient set, instead, may indicate their contribution to leukemogenesis later in life. Identifying carriers of general cancer predisposition gene defects—often involved in DNA repair—should not be ignored. This is especially reflected in the setting of allogeneic HSCT given to 20–50% of adult and 5–10% of pediatric ALL patients. The procedure comprises of multiple steps where germline defects may interfere donor selection from family-members, intensity of conditioning therapy, and need for individualized surveillance plan regarding risks for second or secondary malignancies or late toxicities.
This study was not aimed at finding causal connection between variants and ALL, but to analyze the frequency of P/LP variants in genes possibly contributing to ALL pathogenesis and their enrichment in ALL patients. Identification of harmful rare germline variants may, however, contribute to the selection of optimal therapy (HSCT from siblings/donor registry, possible new treatments). It may also affect the surveillance plan of the patients regarding the risk of secondary or second cancers, and genetic counseling57.
Certainly, some ALL-predisposing genes remain unveiled. The rarity of ALL in adults makes it more difficult to find new adult-specific genes with ALL predisposition without exceptional families. We found some P/LP variants in genes with high penetrance for hematological malignancies among our study patients despite not focusing on families with multiple ALL cases. Family history is only indicative, but not definitive of germline predisposition and mutations can also arise de novo.
Conclusions
A proportion of ALL patients of all ages, carries P/LP germline variants in cancer-predisposing genes even without clear indications for germline testing (such as personal or family history of leukemia or other cancer). In contrast to pediatric ALL patients, the possibility of inherited predisposition in adults has been oversighted in the clinical practice. Acknowledging inherited factors is crucial, especially when considering HSCT donors and planning post-therapy follow-up. Harmful germline variants may also predispose patients to excessive toxicity potentially compromising the outcome. Furthermore, recognition of the role of germline factors in leukemogenesis may also provide new tools for therapy development. Analogous to AML, we propose integrating germline genetics into precise ALL patient care and providing genetic counseling to affected families.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available due to privacy and ethical restrictions but are available from the corresponding author upon reasonable request.
References
Toft, N. et al. Results of NOPHO ALL2008 treatment for patients aged 1–45 years with acute lymphoblastic leukemia. Leukemia 32, 606–615 (2018).
Hunger, S. P. & Raetz, E. A. How I treat relapsed acute lymphoblastic leukemia in the pediatric population. Blood 136, 1803–1812 (2020).
Roberts, K. G. Genetics and prognosis of ALL in children vs adults. Hematology 2018, 137–145 (2018).
Malard, F. & Mohty, M. Acute lymphoblastic leukaemia. Lancet 395, 1146–1162 (2020).
Arber, D. A. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2406 (2016).
Wartiovaara-Kautto, U. et al. Germline alterations in a consecutive series of acute myeloid leukemia. Leukemia 32, 2282–2285 (2018).
Shin, S.-Y. et al. Recurrent somatic mutations and low germline predisposition mutations in Korean ALL patients. Sci. Rep. 11, 8893 (2021).
Churchman, M. L. et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell 33, 937-948.e8 (2018).
Auer, F. et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 28, 1136–1138 (2014).
Shah, S. et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat. Genet. 45, 1226–1231 (2013).
Moriyama, T. et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: A systematic genetic study. Lancet Oncol. 16, 1659–1666 (2015).
Topka, S. et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. 11, 1–14 (2015).
Inaba, H. & Mullighan, C. G. Pediatric acute lymphoblastic leukemia. Haematologica 105, 2524–2539 (2020).
Gocho, Y. & Yang, J. J. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood 134, 793–797 (2019).
Trevĩo, L. R. et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat. Genet. 41, 1001–1005 (2009).
Perez-Andreu, V. et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat. Genet. 45, 1494–1498 (2013).
de Smith, A. J. et al. Heritable variation at the chromosome 21 gene ERG is associated with acute lymphoblastic leukemia risk in children with and without down syndrome. Leukemia 33(11), 2746–2751 (2019).
Rönkkö, R. et al. Familial aggregation of early-onset haematological malignancies. Br. J. Haematol. 193, 1134–1141 (2021).
Kharazmi, E. et al. Familial risks for childhood acute lymphocytic leukaemia in Sweden and Finland: Far exceeding the effects of known germline variants. Br. J. Haematol. 159, 585–588 (2012).
Saarinen-Pihkala, U. M. et al. Outcome of children with high-risk acute lymphoblastic leukemia (HR-ALL): Nordic results on an intensive regimen with restricted central nervous system irradiation. Pediatr. Blood Cancer 42, 8–23 (2004).
Oskarsson, T. et al. Relapsed childhood acute lymphoblastic leukemia in the Nordic countries: Prognostic factors, treatment and outcome. Haematologica 101, 68–76 (2016).
Trottier, A. M. & Godley, L. A. Inherited predisposition to haematopoietic malignancies: Overcoming barriers and exploring opportunities. Br. J. Haematol. 194, 663–676 (2021).
Furutani, E. & Shimamura, A. Germline genetic predisposition to hematologic malignancy. J. Clin. Oncol. 35, 1018–1028 (2017).
Sondka, Z. et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018).
Katainen, R. et al. Discovery of potential causative mutations in human coding and noncoding genome with the interactive software BasePlayer. Nat. Protoc. 13, 2580–2600 (2018).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Li, Q. & Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100, 267–280 (2017).
Kopanos, C. et al. VarSome: The human genomic variant search engine. Bioinformatics 35, 1978 (2019).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
R Core Team. R: A Language and Environment for Statistical Computing (2021).
Yamaguchi, H. et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in Aplastic Anemia. N. Engl. J. Med. 352, 1413–1424 (2005).
Landrum, M. J. et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46(D1), D1062–D1067 (2018).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, (2013).
Cerami, E. et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Sahoo, S. S. et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 27, 1806–1817 (2021).
Nagata, Y. et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 132, 2309 (2018).
Brown, A. L. et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 4, 1131–1144 (2020).
Nishimoto, N. et al. T cell acute lymphoblastic leukemia arising from familial platelet disorder. Int. J. Hematol. 92, 194–197 (2010).
D’Amours, G. et al. Refining the phenotype associated with biallelic DNAJC21 mutations. Clin. Genet. 94, 252–258 (2018).
Tawana, K., Brown, A. L. & Churpek, J. E. Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: Current strategies and challenges. Br. J. Haematol. 196(6), 1293–1310 (2021).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015).
Comeaux, E. Q. & Mullighan, C. G. TP53 mutations in hypodiploid acute lymphoblastic leukemia. Cold Spring Harb. Perspect. Med. 7, 026286 (2017).
Cavé, H. et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur. J. Med. Genet. 59, 173–178 (2016).
Chinton, J. et al. Providing more evidence on LZTR1 variants in Noonan syndrome patients. Am. J. Med. Genet. A 182, 409–414 (2020).
Yamamoto, G. L. et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J. Med. Genet. 52, 413–421 (2015).
Johnston, J. J. et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 20, 1175–1185 (2018).
Louvrier, C. et al. Targeted next-generation sequencing for differential diagnosis of neurofibromatosis type 2, schwannomatosis, and meningiomatosis. Neuro Oncol. 20, 917–929 (2018).
Mazzei, F., Viel, A. & Bignami, M. Role of MUTYH in human cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 743–744, 33–43 (2013).
Dumanski, J. P. et al. A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr. Relat. Cancer 24, 427–443 (2017).
Rennert, G. et al. MutYH mutation carriers have increased breast cancer risk. Cancer 118, 1989–1993 (2012).
Schmidt, M. K. et al. Age-And tumor subtype-specific breast cancer risk estimates for CHEK2∗1100delC Carriers. J. Clin. Oncol. 34, 2750–2760 (2016).
Naslund-Koch, C., Nordestgaard, B. G. & Bojesen, S. E. Increased risk for other cancers in addition to breast cancer for CHEK2∗1100delC heterozygotes estimated from the copenhagen general population study. J. Clin. Oncol. 34, 1208–1216 (2016).
Allemani, C. et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 391, 1023–1075 (2018).
Singhal, D. et al. Targeted gene panels identify a high frequency of pathogenic germline variants in patients diagnosed with a hematological malignancy and at least one other independent cancer. Leukemia 35(11), 3245–3256 (2021).
Acknowledgements
We would like to thank Professor Kimmo Porkka for initiating the adult ALL exome sequencing project and supporting our research and clinical geneticist Kirsimari Aaltonen, MD, PhD for reviewing and commenting our manuscript. Lotta Katainen and Jaana Vettenranta are thanked for their extremely precise technical help. The Finnish Hematology Registry and Clinical Biobank provided some of the samples and data used in this project. We would like to thank the Institute for Molecular Medicine Finland (FIMM) and Biomedicum Functional Genomics Unit (FuGU) for the sequencing services and Helsinki Institute of Life Science (HiLIFE) and Biocenter Finland (BF) for research infrastructures.
Funding
This study was funded by grants from the Academy of Finland (EP: 322675, OK: 137680 and 274474), the Sigrid Jusélius Foundation, the Finnish Special Governmental Subsidy for Health Sciences, Research, and Training, the Helsinki University Hospital Comprehensive Cancer Research Funding, the Finnish Funding Agency for Technology and Innovation (TEKES), the Väre Foundation for Pediatric Cancer Research, the iCAN Digital Precision Cancer Medicine, the Swedish Childhood Cancer Foundation, Cancer Foundation Finland, the Foundation for Pediatric Research (Finland), Ane and Signe Gyllenberg Foundation, the Orion Research Foundation, the Ida Montin Foundation, the Finnish Hematology Association, the Biomedicum Helsinki Foundation, the Paulo Foundation, and the Päivikki and Sakari Sohlberg Foundation.
Author information
Authors and Affiliations
Contributions
S.D. performed research and analyzed data; A.L. collected and analyzed data; J.K. analyzed data; L.L. collected and analyzed data, M.A.I.K. collected and analyzed data; M.K. contributed data; C.H. contributed data; K.J. contributed and analyzed data; E.P. designed research, supervised students, and analyzed and interpreted data; U.W.-K. and O.K. designed research, supervised students and interpreted data. S.D., A.L., J.K., E.P., U.W.-K., and O.K. wrote the manuscript. All authors have approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Douglas, S.P.M., Lahtinen, A.K., Koski, J.R. et al. Enrichment of cancer-predisposing germline variants in adult and pediatric patients with acute lymphoblastic leukemia. Sci Rep 12, 10670 (2022). https://doi.org/10.1038/s41598-022-14364-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14364-x
- Springer Nature Limited
This article is cited by
-
Clinically relevant germline variants in allogeneic hematopoietic stem cell transplant recipients
Bone Marrow Transplantation (2023)