
Abstract
Stenotrophomonas maltophilia is a ubiquitous environmental bacterium capable of causing disease in humans. Antibiotics are largely ineffective against this pathogen due to numerous chromosomally encoded antibiotic resistance mechanisms. An alternative treatment option is phage therapy, the use of bacteriophages to selectively kill target bacteria that are causing infection. To this aim, we isolated the Siphoviridae bacteriophage AXL1 (vB_SmaS-AXL_1) from soil and herein describe its characterization. Host range analysis on a panel of 30 clinical S. maltophilia strains reveals a moderate tropism that includes cross-species infection of Xanthomonas, with AXL1 using the type IV pilus as its host surface receptor for infection. Complete genome sequencing and analysis revealed a 63,962 bp genome encoding 83 putative proteins. Comparative genomics place AXL1 in the genus Pamexvirus, along with seven other phages that infect one of Stenotrophomonas, Pseudomonas or Xanthomonas species. Functional genomic analyses identified an AXL1-encoded dihydrofolate reductase enzyme that provides additional resistance to the antibiotic combination trimethoprim–sulfamethoxazole, the current recommended treatment option for S. maltophilia infections. This research characterizes the sixth type IV pilus-binding phage of S. maltophilia and is an example of phage-encoded antibiotic resistance.
Similar content being viewed by others


Introduction
The genus Stenotrophomonas is a group of over 20 species of phenotypically and genetically diverse Gram-negative, aerobic bacteria found in soil and on plants1,2. Unlike related environmental bacteria of the family Xanthomonadaceae belonging to the genera Xanthomonas and Xylella, bacteria of the Stenotrophomonas genus are not phytopathogenic and instead can secrete compounds to promote plant growth and metabolize numerous damaging organic compounds, such as xenobiotics and phenolics3. Despite this promising potential application for bioremediation, the most prominent bacterial species in this genus, S. maltophilia, is emerging as a nosocomial and community-acquired pathogen. Most commonly associated with respiratory tract infections that are easily transmitted by cough-generated aerosols, S. maltophilia also causes severe bacteremia, wound and soft tissue infections, urinary tract infections, meningitis, endocarditis, pneumonia, osteomyelitis, endophthalmitis, catheter-related bacteremia/septicemia, and acute exacerbations in patients with cystic fibrosis2,4. Prevalence of S. maltophilia infections is rising worldwide largely due to its high innate antibiotic resistance2. This resistance stems from numerous chromosomally encoded multidrug efflux pumps, β-lactamases, and other antibiotic modifying enzymes, ultimately making most frontline antibiotics ineffective at treating S. maltophilia bacterial infections. The current recommended antibiotic treatment option for S. maltophilia infections is trimethoprim–sulfamethoxazole, however resistance to this combination is rising and its use can be problematic due to sulfonamide allergies in patients or drug cross-reactivity2,5. Inhibition of efflux pumps and quorum sensing are promising therapeutic strategies, however well-known inhibitors are ineffective in S. maltophilia6.
To combat the antimicrobial resistance crisis in treating infections caused by S. maltophilia and other highly resistant pathogens, phage therapy, the clinical application of bacterial viruses known as bacteriophages, is being explored2. There is vast genetic diversity found between phages infecting S. maltophilia based upon numerous phages characterized in the last decade2. The recent increase in S. maltophilia phage genomes deposited to NCBI, with 27 genomes released in the last two years, brings the total to 57 described S. maltophilia phages and includes six characterized phages lacking genome sequences in the literature. This highlights the growing interest in phage therapy against S. maltophilia and suggests that we are only just beginning to understand the diversity of S. maltophilia phages.
The evaluation of phages for their possible use in phage therapy requires extensive physical and genomic characterization. In the current study, we describe the isolation and characterization of S. maltophilia phage AXL1. We show that AXL1 is the sixth S. maltophilia phage identified to bind the type IV pilus as its cellular surface receptor. Genomic sequencing places AXL1 in the genus Pamexvirus that contains the previously isolated S. maltophilia phage DLP47. Although a putatively virulent phage, AXL1 encodes a functional dihydrofolate reductase enzyme that is active in its native host, increasing resistance to the antibiotic trimethoprim in the presence of sulfamethoxazole.
Results
AXL1 phage physical characteristics
Bacteriophage AXL1 (vB_SmaS-AXL_1) was isolated from potting soil following enrichment with the clinical S. maltophilia strain D1585. AXL1 produces two sizes of clear plaques with diffuse borders averaging 1.67 ± 0.17 mm and 0.73 ± 0.12 mm in diameter after overnight incubation on its isolation host, D1585 (Fig. 1a). This plaque polymorphism is persistent upon picking and propagating individual plaques of each size, a phenotype that has been observed in S. maltophilia phage IME138. The use of chloroform during phage propagation to high titre indicates AXL1 is stable in the presence of this organic compound. Transmission electron microscopy (TEM) permits classification of AXL1 as a Siphoviridae phage of the B2 morphotype9 having an icosahedral elongated capsid 81.0 ± 4.7 nm long and 53.2 ± 3.7 nm wide and a long, non-contractile tail averaging 150.3 ± 4.2 nm in length with a unique baseplate structure present at the distal tail region (Fig. 1c).

AXL1 phage morphology. AXL1 plated on (a) D1585 or (b) 213 show different plaque morphologies. Bar equals 5 mm. (c) Transmission electron micrograph of Siphoviridae AXL1 virion shows a Siphoviridae morphology. High titre CsCl purified lysate was stained with 4% uranyl acetate on a copper grid and viewed at ×110,000 magnification with a transmission electron microscope.
Host range analysis on 30 distinct S. maltophilia clinical isolates revealed a moderate tropism, with AXL1 showing evidence of bacterial cell lysis as clearing of the bacterial lawn on 14 strains when spotted at a high titre of 1011 PFU/mL. These strains were further examined for productive phage infection by calculating the AXL1 efficiency of plating (EOP) from plaque formation in serial dilutions of phage lysate compared to the isolation host strain, D158510. Only two additional hosts produced high levels of phage production having EOPs greater than 0.5, while four strains could be classified as having low phage production with EOPs greater than 0.001 (Table 1). Although plaques formed on five of the remaining strains, this occurred at low dilutions, suggesting little to no phage production on these hosts. No plaque formation was observed on strains 102 and 287 in the dilution series beyond clearing of the bacterial lawn at high titre. Serial passaging of AXL1 on strains with low EOP may train this phage to infect more efficiently11. Of note, AXL1 produced plaques that were significantly smaller in diameter than observed on D1585 on all hosts except D1568, with plaque sizes averaging 0.296 ± 0.047 mm on strain 213 (Fig. 1b); plaques did not increase in size with longer incubation up to 48 h. For ease of plaque enumeration and because strain D1568 is more difficult to grow, strain D1585 was used for all further experiments.
To analyze infection dynamics of AXL1, a one-step growth curve was conducted. Similar to previously characterized S. maltophilia phage AXL312, AXL1 exhibits a long productive cycle having a latent period of approximately 1.5 h and burst size of 58 virions per cell after 5.5 h (Fig. 2a). Inhibition of bacterial growth in liquid culture by AXL1 produced varying levels of growth reduction with changing multiplicities of infection (MOI). At an MOI of 30, bacterial growth began decreasing at 3.5 h (Fig. 2b). This growth reduction was delayed with decreasing MOI, with all phage groups showing resistant outgrowths by 20 h. The higher MOIs tested produce greater levels of resistant growth than lower MOIs, likely due to earlier depletion of sensitive bacterial cells by AXL1 and growth of resistant cells without competition for nutrients. Unexpectedly, AXL1 was ineffective at growth inhibition in liquid culture when the same experiment was conducted at 37 °C and unlike S. maltophilia phage DLP313, in vivo rescue of Galleria mellonella larvae infected with S. maltophilia D1585 was not successful at this temperature (data not shown). Assessment of phage activity on solid media at 37 °C revealed a decreased EOP of 0.002 relative to plaquing ability on D1585 at 30 °C set as an EOP of 1.0 (Table 1). Additional type IV pili-binding phages DLP1 and DLP214 do not exhibit weaker infection at 37 °C on strain D1585 (data not shown). This drop in infection efficiency is not due to temperature instability of AXL1 phage particles, as virions remain active after incubation for 1 h at temperatures ranging from − 20 to 50 °C (Fig. 3).

Infection dynamics of AXL1 on S. maltophilia strain D1585. (a) A one step growth curve of AXL1 at an MOI of 1 shows a latent period of 1.5 h and burst size of approximately 58 virions per cell. (b) Liquid bacterial growth reduction curves were conducted at multiple MOIs over 48 h at 30 °C. Data from three biological replicates are plotted as mean ± SEM. Where error bars are smaller than the size of the symbol, they are not visible.

Temperature stability of AXL1 virions. AXL1 phage lysate diluted to 107 PFU/mL was incubated at temperatures ranging from − 20 °C to 90 °C for 1 h, followed by serial dilution and calculation of titre by spot assay on S. maltophilia D1585 after overnight incubation at 30 °C. Bars represent mean titres from three replicates and error bars show standard deviation.
Phage receptor analysis
Several phages previously isolated on S. maltophilia D1585 were shown to use the type IV pilus as their cell surface receptors7,12,13,14. Assessment of AXL1 infection on a previously constructed D1585 ΔpilA1 mutant12,14 lacking the major pilin subunit showed results consistent with the previous phages; mutants lacking a type IV pilus are resistant to AXL1 as evidenced by a lack of plaque formation or clearing of the bacterial lawn compared to infection of wildtype cells (Fig. 4a). Genetic complementation of pilA1 restored AXL1 infection efficiency to wildtype levels. The type IV pilus is a surface expressed virulence factor found on many bacterial pathogens that is used for adhering to biotic and abiotic surfaces, contributes to the formation of biofilms, and is the sole protein structure responsible for a form of surface translocation known as twitching motility15,16. Through extension and retraction of the pilus, which is controlled by intracellular ATPases PilB and PilT respectively, combined with adherence to a surface, a bacterium may travel across the surface via twitching motility. Deletion of either ATPase-encoding genes results in a non-function type IV pilus, and in the case of a ΔpilT mutant, hyperpiliated cells with numerous pili expressed at the pole of the cell in bundles (Supplementary Fig. S1). In addition to loss of twitching motility, deletion of pilT in D158513 prevents infection by AXL1 phage and complementation restores phage infection to wildtype levels (Fig. 4a). These results are consistent with previously characterized S. maltophilia phages isolated from soil samples7,12,13,14. Although AXL1 was unable to infect at high efficiency at 37 °C and this was not due to temperature instability of the phage particles, decreased host receptor expression was also not involved. The area of the twitching motility zone of host strain D1585 did not change under different temperature conditions. Interestingly, in one S. maltophilia strain 213, twitching motility did increase to produce a zone of 122.2 ± 18.4 mm2 at 37 °C, up from 64.7 ± 12.1 mm2 in diameter at 30 °C.

S. maltophilia bacteriophage AXL1 requires a functional type IV pili for infection. (a) Wildtype (WT) S. maltophilia strain D1585 is susceptible to AXL1. Deletion of the major pilin subunit encoded by pilA1, or the retraction ATPase encoded by pilT, abolishes infection by AXL1. Complementation restores phage infection to wildtype levels. (b) Exogenous expression of the D1585 pilA1 gene in phage-resistant hosts, S. maltophilia D1571 and P. aeruginosa PA01 pilA−, permits AXL1 infection. Images are representative of three biological replicates, each with three technical replicates.
Although a common virulence factor with highly conserved machinery, type IV pili major pilin proteins are highly variable between species and even strains, allowing for the evasion of host immune responses17 and phage resistance18. To further assess phage recognition of the type IV pilus, the D1585 pilA1 gene was expressed in a S. maltophilia host strain, D1571, that is not susceptible to AXL1 and infection by AXL1 was examined. Expression of the exogenous D1585 pilA1 gene allows phage infection, with plaque formation occurring at 106 PFU/mL compared to lack of infection in the empty vector control (Fig. 4b). Additionally, cross-genera expression of the D1585 pilA1 gene in a Pseudomonas aeruginosa PA01 pilA− mutant permits binding of AXL1 virions and cell lysis as shown by infection as compared to the empty vector control, albeit at low efficiency. This is reminiscent of S. maltophilia DLP2, a phage capable of cross-taxonomic order infection, that is only capable of infecting strain PA01 with the expression of the D1585 pilin14.
Together, these results identify the type IV pilus as the host cell surface receptor for AXL1, interacting directly with the PilA1 pilin subunit and requiring a functional pilus capable of retraction for successful infection. AXL1 is the sixth S. maltophilia phage experimentally determined to bind the type IV pilus, all of which are Siphoviridae phages isolated from soil samples7,12,13,14,19. While the majority of the S. maltophilia strains in our collection do not have fully sequenced genomes, the pilA1 genes could be identified in partial genome sequencing or from Sanger sequencing of PCR products using primers14 designed against the D1585 pilA1 gene for six strains. Comparison of the amino acid sequences revealed pilins with 98.55% identity between the main host D1585 and 213, a strain with the highest AXL1 productivity and infection efficiency (Table 1). Strains with low efficiency of plating, 280 and 287, have lower percent identity scores with D1585 PilA1 of 61.87% and 66.91%, respectively. The final two strains with sequenced PilA1, D1571 and ATCC13637, are not susceptible to AXL1, and not unsurprisingly, have pilin percent identity scores with the D1585 PilA1 sequence in the 40s; most of this identity is found in the conserved N-terminal region of the pilin that is required for pilus assembly.
Genomic characterization
The AXL1 genome assembled into a single contig 63,692 bp in length (Fig. 5) with a GC content of 67.3% that is similar to the host S. maltophilia GC content. BLASTn analysis of AXL1 showed high relatedness to phages in the genus Pamexvirus, most closely aligning to Xanthomonas phage Bosa20 with 91.01% identity over 97% of the genome. Among this genus, in order of percent identity to AXL1, are Xanthomonas phage Xp1221, Stenotrophomonas phage DLP47, Xanthomonas phage Xoo-sp222, and the Pseudomonas phages AAT-123, PaMx28, and PaMx7424. Despite shared nucleotide sequence identity with three P. aeruginosa phages and previous observation of cross-taxonomic order infectivity of S. maltophilia phages DLP1 and DLP219, AXL1 was incapable of infecting 21 P. aeruginosa strains tested. To the contrary, examination of AXL1 infectivity of strains of four Xanthomonas species revealed infection of X. axonopodis pv. vasculorum FB570 at an EOP of 0.03 as compared to infection of S. maltophilia D1585. No phage lysis was observed on the X. oryzae host strain of related phage Xp12 or X. translucens pv. translucens ATCC 19319 and X. campestris ATCC 33440.

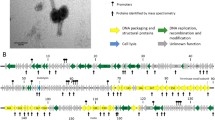
Circularized genomic map of AXL1. The scale (in bp) is shown on the outer periphery. Assigned putative functions for each of the 83 predicted open reading frames are classified as follows: lysis (red), DNA replication and repair (blue), DNA packaging (teal), virion morphogenesis (green), hypothetical (grey), moron (purple). Regulatory elements are promoters (pink) and terminators (black). No tRNA genes were identified. AXL1 has a GC content of 67.3%. Image created using Geneious Prime25.
Comparative genomic analysis of AXL1 with the seven members of the Pamexvirus genus shows high relatedness to Xanthomonas phages Bosa and Xp12, as well as the Stenotrophomonas phage DLP4, whereas Xanthomonas phage Xoo-sp2 shares a high percent identity with only half of the morphogenesis proteins (Fig. 6). The three Pseudomonas phages show greater amino acid sequence identity with each other than with the Stenotrophomonas and Xanthomonas phages. This comparison supports the classification of AXL1 as the eighth member of the Pamexvirus genus.

Comparative genome alignment of AXL1 and phages of the Pamexvirus genus. A linear representation of AXL1 (bold) and seven related phage genomes shows high amino acid sequence identity following analysis with Clinker26. Arrows represent phage coding sequences coloured to indicate homologous groups and are linked by grey regions, with shading representing percentage amino acid identity. Genome accession numbers are in parentheses for each phage.
Restriction fragment length polymorphism (RFLP) analysis of the AXL1 genome using 31 restriction enzymes with recognition sequences present in the genome revealed digestion by only five enzymes: AseI, Eam1105I, KpnI, TasI, and Tru1I (Supplementary Fig. S2). A similar result has been observed for Xanthomonas phage Xp12, known to contain 5-methylcytosine in place of all cytosines in its genome21; as AseI, TasI, and Tru1I contain only A/T bases in their recognition sequences, digestion is not impaired by this base modification. Compared to the expected digestion patterns, Eam1105I and KpnI partially digest the AXL1 genome. For the Pamexvirus phages with RFLP analysis, resistance to digestion with some restriction enzymes was also observed for DLP47 and PaMx28 and PaMx7424.
AXL1 is predicted to encode 83 open reading frames (ORFs) (Fig. 5, Supplementary Table S1) producing a coding density of approximately 93%. The majority of start codons are ATG (71 out of 83), with fewer GTG and TTG start codons present in 10 and two ORFs, respectively. The stop codon TGA is found in 58 ORFs, with TAA second most abundant in 20 ORFs, and TAG in only five. No standard tRNA genes were detected. Functional predictions based on BLASTp analysis produced significant hits for all 83 putative proteins, however putative functions beyond hypothetical could be assigned for only 37 proteins. The majority of top hits were to Stenotrophomonas phage DLP4, having 30 unique hits, and Xanthomonas phage Bosa, having 17, with an additional 28 proteins identical between the two phages as noted by the asterisks in Supplementary Table S1. Seven of the remaining proteins shared the highest percent identity with Xanthomonas phage Xp12 and only gp5 hit to Xanthomonas phage Xoo-sp2. The assigned functions of these proteins place them in distinct modules consisting of those related to DNA replication and repair (blue) or DNA packaging (teal) on the positive and negative strands, virion morphogenesis (green) and lysis (red) on the positive strand, and a small operon of unknown function (purple) on the negative strand containing a gene encoding a YbiA homolog (Fig. 5). The genome sequence of AXL1 with putative annotations has been deposited in Genbank under the accession number OL674541.
DNA replication, repair, and packaging module
There are at least 14 genes encoded in two bi-directional gene clusters in the AXL1 genome involved in DNA replication, processing of nucleotides, and genome packaging (Fig. 5, Supplementary Table S1). Gene products that could be assigned putative enzymatic functions within the positive stranded region AXL1_1 to AXL1_14 include terminase small subunit (gp1), terminase large subunit (gp6, TerL), ribonucleotide reductase of class 1a (aerobic) alpha (gp9) and beta (gp10) subunits, 3’-phosphatase/5’-polynucleotide kinase (gp13), and DNA ligase (gp14), as well as bifunctional DNA primase/polymerase (gp83). The class 1a ribonucleotide reductase (RNR) complex composed of gp9 and gp10 contributes to DNA synthesis by catalyzing the reduction of ribonucleosides (NDPs) into their corresponding deoxyribonucleosides (dNDPs)27.
On the negative strand within AXL1_53 to AXL1_81, putative functional annotations include dihydrofolate reductase (gp61), dCMP deaminase (gp62), thymidylate synthase (gp63), nucleotide pyrophosphohydrolase (gp64), DNA helicase (gp69), DNA polymerase I (gp70), and Cas4 nuclease (gp79). All of these proteins also have conserved domains associated with their assigned functions, except for the small and large terminase subunits (Table 2). Similar to Stenotrophomonas phages DLP4 and AXL3 are the enzymes involved in the thymidylate synthesis pathway, gp62 and gp63, that function to convert deoxycytidylate (dCMP) into deoxyuridine monophosphate (dUMP), and dUMP into deoxythymidine monophosphate (dTMP), respectively28,29. The reductive methylation of dUMP into dTMP relies on the cofactor 5,10-methylenetetrahydrofolate that is converted into dihydrofolate in the process. This cofactor can be regenerated by the function of dihydrofolate reductase, gp61 in AXL1, that reduces dihydrofolate into tetrahydrofolate, which is subsequently processed by serine hydroxymethyltransferase, not encoded by AXL1, into the cofactor for use by thymidylate synthase30.
Two additional proteins of interest are gp56 that contains a HIRAN domain predicted to bind DNA, helping to resolve stalled replication forks or recognize regions of DNA damage31, and gp65, a protein found only in the related Xanthomonas phage Xp12, containing an ENDO3c domain with an intact minor groove reading motif and helix-hairpin-helix signature motif. This domain is found in endonuclease III enzymes that act in the site-specific repair of DNA damage. However, conserved domain search32 results showed the absence of the canonical substrate binding pocket and active site motifs, and COFACTOR33 and COACH34 analysis based on I-TASSER35 structural prediction produced an aspartate at position 141 as the putative catalytic site with a confidence score of only 0.33 based on structural similarity with the Escherichia coli endonuclease III. Additionally, hypothetical proteins gp71, gp74, and gp77 contain conserved domains of unknown function.
Structural proteins within the virion morphogenesis module
The virion morphogenesis module includes 26 genes spanning AXL1_15 to AXL1_47 on the positive strand, however this module is interrupted by an operon containing six genes, AXL1_19 to AXL1_25, in the reverse orientation that is described below. The gene products for 20 of the 26 genes could be assigned function based on BLASTp comparisons (Fig. 5, Supplementary Table S1). Proteins involved in capsid assembly and packaging include portal protein (gp15), minor head protein (gp16), and major head protein (gp26). 17 proteins identified as structural proteins in phage assembly and tail morphogenesis include seven virion structural proteins (gp18, gp29-31, gp38, gp39, gp41), tail terminator protein (gp32), major tail structural protein (gp33), tape measure protein (gp36), tail fiber protein (gp37), FAD/FMN-containing dehydrogenase (gp40), central tail hub protein (gp43), three tail assembly proteins (gp44-46), and tail fiber protein (gp47). Proteomic analysis of CsCl-purified AXL1 virions by HPLC–MS confirmed 17 of the above proteins as virion-associated, in addition to the hypothetical protein gp27; below the limit of detection were gp18, gp41, and gp45 (Table 3). The most abundant virion protein identified, and the only band evident in SDS-PAGE, is the major head protein, gp26, an estimated 32.5 kDa protein. Interestingly, a single S. maltophilia protein, bacterioferritin, was identified in the AXL1 sample by the presence of two peptides after analysis against D1585 proteins (Table 3).
In general, the largest gene in Siphoviridae phage genomes encodes the tape measure protein36, which determines the length of the phage tail. In AXL1 however, this gene is second in length to AXL1_16 encoding the putative minor head protein. A conserved domain search of this protein revealed a Phage_Mu_F domain at the N-terminal end of the protein that is commonly found in head morphogenesis proteins of phages, as well as an unexpected VIP2 domain at the C-terminal end that is found in actin-ADP-ribosylating toxins such as Clostridium botulinum C2 toxin and C. difficile toxin37 (Table 2). The function of this protein in AXL1 infection is unknown, however polyvalent proteins containing MuF and VIP2 domains have been identified in phages infecting Microbacterium38 and are overrepresented in prophages of Firmicutes in the gut microbiota39. In E. coli phage T4, the ADP-ribosylating protein Alt is packaged in the phage capsid and injected into the bacterial cell with the phage genome where Alt ADP-ribosylates the host RNA polymerase to commandeer it for transcription of viral genes40. A search of the Virulence Factor Database (VFDB)41 also showed similarity of the C-terminus of AXL1 gp16 with T3SS and T4SS effector proteins with ADP ribosyltransferase activity. In the Pamexvirus phages, only Pseudomonas phage PaMx74 lacks this VIP2 domain fused to its minor head protein (Fig. 6). The enzymatic function of this fusion protein in AXL1 virion morphogenesis or during phage replication to alter phage gene expression is unknown.
Within the tail morphogenesis proteins, numerous proteins of interest stand out as potential receptor binding proteins for virion interaction with the type IV pilus. Located between the tape measure protein and lysis module, gp43 to gp47 comprise the distal tail tip proteins and show amino acid sequence identity to known type IV pili binding phages. Specifically, gp44 shared nearly 100% query coverage with type IV pili binding S. maltophilia phages DLP313 and DLP47 and Xylella phages Sano and Salvo42. A conserved domain search revealed a major tropism determinant N-terminal (Mtd_N) domain (Table 2) found in the major tropism determinant (Mtd) protein known to determine receptor binding in Bordetella phage BPP-143. Conserved domain searches also revealed a phage-tail_3 domain in the structural protein gp43; this domain is present in the central tail hub or major baseplate proteins of numerous type IV pili binding phages that infect Stenotrophomonas, Xylella, and Pseudomonas and is hypothesized to play a role in receptor binding12,14. Further research is ongoing to determine the function of these putative receptor binding proteins in host recognition.
YbiA operon
The genes AXL1_19 to AXL1_25 encoded on the negative strand that disrupt the virion morphogenesis module are expressed from a promoter 58 bp upstream of AXL1_25 (Fig. 5, Supplementary Table S1). This operon includes seven genes encoding hypothetical proteins with unknown function and a YbiA homolog (AXL1_19) shown to play a role in swarming motility in E. coli44. A similar protein is present in all members of the Pamexvirus genus except PaMx74 (Fig. 6), and although swarming motility capability has not been confirmed in S. maltophilia, including our host strain D1585, the ybiA gene from Stenotrophomonas phage DLP4 was experimentally determined to complement swarming motility in an E. coli ybiA insertional mutant7. No change in swarming was observed in S. maltophilia D1585 expressing the DLP4 ybiA gene7.
Although originally identified to play a role in swarming motility, recent research describes the NADAR and COG3236 domains found in the structurally related E. coli YbiA protein to putatively function in ADP ribose metabolism based on clustering with related genes45 and was experimentally shown to function as an N-glycosidase in riboflavin biosynthesis46. The absence of putative functions or conserved domains in neighbouring genes in this operon limit predictions for the classification of the AXL1 YbiA-like protein, however structural predictions by I-TASSER analysis produced high structural homology to the E. coli YbiA protein, with a TM-score of 0.902 and 0.913 coverage. Additionally, catalytic residues identified to be essential for hydrolysis of N-glycosidic bonds46 are conserved indicating enzymatic function. The function of this operon in phage replication is unknown; in S. maltophilia, the ybiA-like gene is located between genes encoding a transketolase and acetyl-CoA hydrolase, however in the D1585 host for AXL1, this ybiA gene is absent.
Lysis module
Similar to S. maltophilia phage DLP4, the AXL1 lysis module contains five genes, AXL1_48 to AXL1_52, directly downstream of the virion morphogenesis module (Fig. 5, Supplementary Table S1). BLASTp results combined with LipoP 1.0 and TMHMM analyses to identify lipoproteins and transmembrane domains, respectively, provide putative functional roles of these proteins in cell lysis. The first gene in this module encodes a putative holin, gp48, based on the presence of two transmembrane domains47. The insertion of canonical holin proteins in the cytoplasmic membrane create pores upon activation that allow phage endolysin to enter the periplasm and degrade the peptidoglycan47. In AXL1, the endolysin gp49 contains a conserved l-alanyl-d-glutamate peptidase domain identified by CD-Search (Table 2) that cleaves between the l-alanine and d-glutamate residues of the peptidoglycan cell wall. For complete cell lysis to occur, phage spanin proteins form a complex to disrupt the outer membrane48. In AXL1, the i-spanin, gp50, contains a predicted N-terminal transmembrane domain that anchors the protein in the cytoplasmic membrane where it can span the periplasm to interact with the o-spanin localized in the outer membrane. Encoded by a gene overlapping the i-spanin coding sequence, gp51 putatively functions as the o-spanin based on the presence of a predicted lipoprotein signal peptide II cleavage site located between amino acids 32 and 33. The final gene in this module encodes a protein with a single transmembrane domain at the N-terminus, however its role in cell lysis is unknown.
Lifestyle analysis
Based on high sequence identity with the temperate Stenotrophomonas phage DLP4, we sought to isolate AXL1 lysogens to confirm its temperate lifestyle. Using the primary host strain, D1585, phage resistant colonies were isolated and tested for the presence of viral DNA using AXL1 specific primers. AXL1-positive colonies were isolated on few occasions and did not maintain AXL1 as a stable prophage. Attempts to isolate stable lysogens of strain 213 were also unsuccessful. Due to the low lytic phage production observed at 37 °C, we hypothesized that AXL1 may lysogenize more stably at this temperature, as some Burkholderia tropical phages have been shown to have temperature-dependent lifestyles49,50. However, this was not the case for AXL1. It is possible that AXL1 cannot stably integrate into the host genomes tested under lab conditions in rich media. However, the genome of AXL1, as well as all Pamexvirus genomes, lacks any identifiable lysogeny-associated repressor or integrase genes, which is indicative of virulent phages. BLASTn analysis of the AXL1 genome against Gamma proteobacteria (taxid:1236) also produced no significant results with query coverage greater than 3%, suggesting a lack of remnants of AXL1 as prophage elements in bacterial genomes.
AXL1-encoded DHFR contributes to host resistance to trimethoprim
Dihydrofolate reductase is an essential enzyme in folate metabolism required for the synthesis of DNA and bacterial growth and is also the target of the antibiotic trimethoprim in bacteria. Trimethoprim binds to and inhibits the enzymatic activity of dihydrofolate reductase, preventing the conversion of dihydrofolate into tetrahydrofolate, the active form of folic acid that is required for the synthesis of thymidine51. We therefore sought to examine the function of the AXL1-encoded dihydrofolate reductase (gp61, DHFR) in providing resistance to trimethoprim in the S. maltophilia host. Putative DHFR enzymes are encoded by all eight viruses of the Pamexvirus genus, with varying degrees of homology to the AXL1 protein ranging from approximately 91% identity with Xanthomonas phage Xp12 to 44% identity with Pseudomonas phage PaMx28. Despite the known role of DHFR in antibiotic resistance, the possible contribution of these phage-encoded enzymes to host antimicrobial resistance was only examined in S. maltophilia phage DLP4 where we found a significant increase in bacterial resistance to high concentrations of trimethoprim in a D1585::DLP4 lysogen compared to the wildtype bacterial host, as well as in E. coli expressing the DLP4 dhfr gene compared to an empty vector control7. We conducted a similar experiment to test the function of AXL1 dhfr in promoting resistance to trimethoprim in E. coli DH5α; minimum inhibitory concentration (MIC) assays with E. coli carrying the AXL1 dhfr gene on a plasmid resulted in a significant increase in growth in the presence of trimethoprim, with the MIC greater than 256 µg/mL compared to the empty vector control strain having an MIC of less than 1 µg/mL (Table 4). These results indicate that the AXL1 DHFR variant is less vulnerable to inhibition by trimethoprim than the E. coli dihydrofolate reductase protein and may play a role in increasing antibiotic resistance in its native bacterial host.
In S. maltophilia, resistance to trimethoprim is naturally high due to chromosomally encoded efflux pumps SmeDEF, SmeOP, and SmeYZ, and this resistance is rising worldwide with the spread of trimethoprim insensitive dihydrofolate reductase dfrA genes encoded on integrons2,52. In a subset of our S. maltophilia collection, in the four strains with genome sequencing data, a gene encoding dihydrofolate reductase type III (dhfrIII, dfrA3) was found directly downstream of thyA encoding thymidylate synthase. These dfrA3 genes contribute to the high intrinsic resistance to trimethoprim observed in our lab, with all strains tested having an MIC to trimethoprim of 256 µg/mL or greater (Fig. 7, Supplementary Fig. S3). Because of the intrinsic resistance to trimethoprim observed in clinical S. maltophilia samples, the current recommended treatment is a combination of trimethoprim and sulfamethoxazole. Although functionally similar, the AXL1 DHFR protein shares only a maximum of 24.5% identity with the S. maltophilia DfrA3 proteins in our strains, and BLASTn analysis reveals no significant hits to genes outside of the Pamexvirus dhfr gene, suggesting an unknown origin. To determine the contribution of the AXL1-encoded dihydrofolate reductase to its host’s ability to survive in the presence of trimethoprim, we conducted checkerboard assays containing increasing concentrations of trimethoprim and sulfamethoxazole on five S. maltophilia strains carrying the AXL1 dhfr gene on a plasmid and compared the MICs to their corresponding empty vector controls. Under the conditions tested, only two strains, D1585 and D1571, had detectable MICs to trimethoprim alone; in D1585, the expression of AXL1 dhfr increased the MIC of trimethoprim from 256 to 512 µg/mL and in D1571, the MIC increased from 512 to > 512 µg/mL (Fig. 7, Supplementary Fig. S3). In all strains examined except D1585, the addition of sulfamethoxazole effectively reduced the concentration of trimethoprim required to inhibit bacterial growth beyond what was required alone, as expected for these synergistic antibiotics53 (Fig. 7, Supplementary Fig. S3). Examination of the trimethoprim concentration required to reduce bacterial growth to below 10% of the solvent control in the presence of 32 µg/mL sulfamethoxazole shows a substantial increase in MIC for most strains expressing AXL1 dhfr, with fold changes ranging from zero for D1585 to a 64-fold increase for D1571 (Table 4). These results support the conclusion that AXL1 encodes a functional dihydrofolate reductase enzyme that is capable of increasing resistance to a clinically relevant antibiotic combination in its native host.

Expression of AXL1 dhfr in S. maltophilia D1571 increases resistance to trimethoprim–sulfamethoxazole. Colour intensity represents percent bacterial growth relevant to solvent treated controls, with growth below 10% shown as white. Bacterial growth was compared between an empty vector control (pBBR1MCS) and D1571 carrying AXL1 dhfr on a plasmid (pAXL1dhfr). Checkerboard assays were conducted in biological triplicate, with the average of the replicates shown. See also Supplemental Fig. S3.
Discussion
The genomic and functional characterization of S. maltophilia phage AXL1 identifies the eighth member of the genus Pamexvirus based on the high degree of sequence identity with the previously characterized S. maltophilia phage DLP4 and phages that infect the nosocomial pathogen P. aeruginosa and species of agricultural crop pathogens in the genus Xanthomonas. Host range analysis indicates that AXL1 infects a moderate range of S. maltophilia strains, however high EOP associated with productive phage infection was observed for only half of the isolates (Table 1). For strains with apparent non-productive phage infection, 102, 103, 282, 287, D1576, ATCC13637, and SMDP92, these may be observations of abortive infection and/or lysis from without10,54,55 where AXL1 is not actively replicating within the host but is still capable of binding to the bacterial cell surface and affecting bacterial survival at a high multiplicity of infection (MOI). Bacterial internal phage defense mechanisms that block phage infection, such as restriction/modification systems, may also vary between strains and upregulation of such phage defense mechanisms could account for the reduced plating efficiency of AXL1 observed at 37 °C. However, because previously characterized pili-binding phages do not exhibit this temperature sensitivity when infecting strain D1585, this suggests there is something lacking the in AXL1 genome for intracellular replication at 37 °C.
Our results show that this phage binds to the major pilin subunit of host type IV pili and requires cellular-mediated pilus retraction to reach the cell surface for successful infection. S. maltophilia, along with many Xanthomonas and Xylella species within the Xanthomonadaceae family16, encode two neighbouring major pilin genes, whereas type IV pili in other well-studied pathogens encode a single pilA gene. The role of pilA2 in these bacteria is unknown, as deletion of pilA1 is sufficient to abolish twitching motility14, as well as phage infection in S. maltophilia, as shown above (Fig. 4). The frequent use of the type IV pilus as a receptor for infection of S. maltophilia by phages isolated from soil suggests a key role for the pilus for survival in the environment and pathogenesis in humans2. Although AXL1 was isolated on S. maltophilia, comparable phage production was observed on X. axonopodis pv. vasculorum strain FB570. Given the similarities in type IV pili architecture and environmental niches of S. maltophilia and Xanthomonas species16, this ability to infect across host species would be evolutionarily advantageous for phage proliferation in the environment. Few studies have examined Xanthomonas as a potential host for S. maltophilia phages; despite genomic similarity of S. maltophilia phage Smp131 to prophages of Xanthomonas strains, Smp131 was unable to infect any of the 59 Xanthomonas strains of seven pathovars tested56. Previous research by Lee et al. however identified strong lytic activity of Xanthomonas oryzae phage φXo411 encoded lysozyme against S. maltophilia57. Although AXL1 was unable to infect any of the 21 P. aeruginosa strains tested, the ability to cause cell lysis in PA01 expressing the D1585 pilA1 gene (Fig. 4) suggests that there are no intracellular blocks to infection and with a compatible surface receptor, cross taxonomic order infection may also occur. Comparative genome analysis of AXL1 to phages of the Pamexvirus genus shows a high degree of amino acid sequence identity with type IV pili-binding S. maltophilia phage DLP4 and the Xanthomonas phages Bosa and Xp12; these four phages share nearly identical gene order, including the presence of four homologous tail proteins encoded between a structural protein and the lysis operon that are not present in the other Pamexvirus phages (Fig. 6). Three of these proteins were identified in AXL1 virions by mass spectrometry (Table 3). The high relatedness of these tail morphogenesis proteins between known type IV pili phages DLP4 and AXL1 with Bosa and Xp12 (Fig. 6), specifically with AXL1 gp44 that contains an Mtd_N domain involved in receptor specificity, suggests that these phages may also use the type IV pilus virulence factor as their receptor for host infection.
The AXL1 genome is 63,962 bp in length and encodes 83 ORFs (Fig. 5). The resistance of the DNA to digestion by restriction enzymes with G/C bases in their recognition sequences suggest that the AXL1 genome is modified or contains atypical bases that protect it from degradation by host restriction-modification systems. Based on the genomic relatedness to Xanthomonas phage Xp12 and similar enzyme susceptibilities, AXL1 may also contain 5-methylcytosine bases as described for Xp1221, however the percentage of cytosine replacement and genetic mechanism for this modification is unknown. Within the DNA replication and repair module few genes stand out as candidates involved in DNA modification beyond AXL1_62 and AXL1_63 that encode enzymes involved in the thymidylate synthase pathway; however, AXL1_79 encodes a PD-(D/E)XK Cas4-like nuclease. Putative Cas4 nucleases have been identified in other S. maltophilia phages, DLP47 and AXL312, and were functionally characterized in Campylobacter jejuni phages to be capable of incorporating host-derived spacers into host CRISPR arrays to ultimately evade host immune defenses58. Given the apparent lack of CRISPR-Cas defense systems in S. maltophilia however and the restriction enzyme-resistant nature of phage genomes encoding cas4 genes, we have hypothesized a potential role of phage-encoded Cas4 enzymes in defense against host restriction-modification systems12,59.
Functional analysis of the 83 proteins encoded by AXL1 revealed an additional gene of interest encoding a dihydrofolate reductase enzyme. During the phage infection cycle, AXL1-encoded dihydrofolate reductase (gp61, DHFR) likely functions in nucleotide biosynthesis, as described above, based on its genomic location and the functions of neighbouring genes. Antibiotic susceptibility assays show that AXL1 encoded DHFR increases bacterial resistance to the antibiotic combination trimethoprim–sulfamethoxazole, the drug of choice for treatment of S. maltophilia infections (Table 4, Fig. 7). This effect varies between strains. This may be due to slight sequence variation between endogenous DfrA3 host proteins that provide different levels of protection against trimethoprim, or differential expression of efflux pumps that were not examined between strains.
The presence of a gene encoding antimicrobial resistance to trimethoprim in phages that infect nosocomial pathogens such as S. maltophilia and P. aeruginosa that are often found in co-microbial infections in patients with cystic fibrosis2,60 is cause for concern, specifically because the recommended treatment option for S. maltophilia infections is trimethoprim–sulfamethoxazole52. We have shown that DHFR encoded by S. maltophilia phages AXL1 and DLP47 promotes resistance to trimethoprim in their hosts, however the sequence variability between these proteins and other Pamexvirus phage-encoded DHFR proteins suggests that additional testing is needed to determine if they are also insensitive to trimethoprim or capable of conferring resistance in their hosts by a gene dosage effect. Although it is commonly understood in the field that phages seldom encode antibiotic resistance genes and play a lesser role in the spread of antimicrobial resistance through rare generalized transduction events than other mobile genetic elements61,62, our data indicates this is not the case for phages of the Pamexvirus genus. The lack of homology between these DHFR phage proteins and known trimethoprim insensitive DfrA proteins found in bacteria falls below the conservative criteria for identification of antimicrobial resistance when searching against a database61. This limited homology to bacterial dihydrofolate reductase enzymes also indicates an unknown origin of this gene. These results suggest revisiting the potential that phages have in contributing to the spread of antimicrobial resistance. The recent increase in sequencing of phage genomes submitted to public databases without corresponding thorough functional characterization may overlook the potential for phage-spread of multidrug resistance in the environment. Overall, we determine that AXL1 is not a good candidate for use in phage therapy, however further study may provide insight into novel phage mechanisms of DNA modification and regulation of phage gene expression.
Materials and methods
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Tables 1 and 5. S. maltophilia strain D1585 was used for phage isolation and as the primary host for propagation. All strains were grown overnight at 30 °C on Lennox (LB; 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) solid medium or in LB broth with shaking at 225 RPM. Media was supplemented with 35 µg/mL chloramphenicol (Cm) antibiotic for plasmid maintenance in E. coli and S. maltophilia D1585, D1571 and ATCC13637, 75 µg/mL Cm for S. maltophilia 280 and SMDP92, or 35 µg/mL gentamicin (Gm) for P. aeruginosa when necessary.
Phage isolation, propagation and host range analysis
Phage AXL1 was isolated from empty planter soil in Edmonton, Alberta, Canada using S. maltophilia strain D1585 and a previously described enrichment protocol19. Briefly, soil was enriched for phage by overnight incubation at 30 °C and shaking at 225 RPM with S. maltophilia D1585 liquid overnight culture and additional LB broth and modified suspension medium (SM) (50 mM Tris–HCl [pH 7.5], 100 mM NaCl, 10 mM MgSO4). Solids were pelleted by centrifugation and the supernatant was filter sterilized using a Millex-HA 0.45 µM syringe-driven filter unit (Millipore, Billerica, MA, USA). After overnight incubation of soft agar overlays with S. maltophilia D1585, a single plaque was picked into 500 µL of SM with 20 µL chloroform to generate an AXL1 stock.
High titre working stocks of AXL1 were propagated using soft agar overlays as previously described12,67 or liquid infections. Briefly, 150 µL of D1585 overnight culture and 150 µL of phage were incubated for 30 min at 30 °C with shaking at 225 RPM before adding 15 mL LB broth and 1.5 mL SM and incubating overnight under the same conditions. 200 µL of chloroform was added the following day and incubated on a platform rocker at room temperature for 30 min. Following centrifugation, the supernatant was collected, filter sterilized as above and stored at 4 °C. Phage titre was determined by soft agar overlays on D1585. Plaques were backlit and viewed under the magnifying glass of a New Brunswick Scientific colony counter (model C110) and plaque size was measured using digital calipers manufactured by Tresna (Guilin, China) and reported as the average from 10 plaques ± standard deviation.
Host range analysis was conducted on a panel of 30 phenotypically distinct clinical S. maltophilia isolates that vary in phage susceptibility profiles7,12,13,19,68,69 and colony morphology, 21 P. aeruginosa isolates and four Xanthomonas strains. Soft agar overlays containing 100 µL of overnight culture mixed with 3 mL of 0.7% 1/2 LB top agar were spotted with 5 µL of a 1011 pfu/mL AXL1 stock at multiple dilutions and scored for clearing and/or plaque formation after incubation at 30 °C for 24 h and 48 h. Efficiency of plating (EOP) was calculated as the ratio of the number of plaques on a given strain to the titre on the isolation host, D1585. Where plaques were not detected, the lowest dilution with evidence of phage activity was considered for EOP. Predicted phage production was scored based on EOPs greater than 0.5 (high) or 0.001 (low)10. The same procedure was conducted for AXL1 efficiency of plating on host mutants for receptor analysis, with antibiotics added to the bottom and top agar as required for plasmid maintenance.
Temperature stability of AXL1 virions was determined after incubation of 100 µL of a 107 PFU/mL lysate at various temperatures (− 20 °C, 22 °C (room temperature), 30 °C, 35 °C, 37 °C, 42 °C, 50 °C, 60 °C, 80 °C, 90 °C) for 1 h. The treated lysate was serially diluted and 5 µL of each dilution was spotted on soft agar overlays containing 100 µL of overnight D1585 culture. Plaques were counted after overnight incubation at 30 °C and reported as the average phage titre from three biological replicates with error bars showing standard deviation.
Transmission electron microscopy
For electron microscopy, phages were purified by cesium chloride density gradient ultracentrifugation and dialysis. CsCl was dissolved in high titre 1011 pfu/mL AXL1 lysate to 1.45 g/mL followed by ultracentrifugation at 35,000 RPM in a 50.2 Ti rotor for 20 h at 4 °C. The phage band was extracted using an 18 G needle into 12 kDa molecular weight cutoff dialysis tubing and dialyzed at 4 °C in 1.5 L SM for 4 days, with the SM buffer changed every 24 h. To visualize phages, 10 µL purified phage lysate was loaded onto a carbon-coated copper grid for 2 min and stained with 4% uranyl acetate for 20 s. Transmission electron micrographs were captured using a Philips/FEI Morgagni transmission electron microscope with charge-coupled device camera at 80 kV (University of Alberta Department of Biological Sciences Advanced Microscopy Facility). The average capsid and tail dimensions ± standard deviation was calculated using Microsoft Excel based on measurements from 10 individual virions taken using ImageJ software (NIH, Bethesda, MD, USA)70.
S. maltophilia D1585 ΔpilT bacterial cells were prepared for electron microscopy as follows. Cells grown on ½ LB agarose plates overnight at 30 °C were collected and washed in 1× phosphate-buffered saline (PBS), pH 7.2, and fixed at room temperature in EM fixative (2.5% glutaraldehyde, 2% paraformaldehyde, 0.1 M phosphate buffer, pH 7.2) for 15 min. The fixed cells were pelleted and resuspended in 1× PBS. 5 µL of this sample was incubated on a copper grid for 30 s and stained with 2% phosphotungstic acid (PTA) for 10 s and imaged as above.
One-step growth curve
To determine burst size and latent period of AXL1, one-step phage growth analysis of AXL1 on S. maltophilia D1585 was conducted as previously described12,71, with modifications. Overnight liquid cultures of D1585 were subcultured in LB broth and grown to an OD600 of 0.2 at 30 °C. AXL1 lysate was added at an MOI of ~ 1 and allowed to adsorb for 5 min at room temperature followed by incubation at 30 °C with aeration at 225 RPM for 6 h. Samples were taken in triplicate at 30 min intervals and serially diluted in SM for spotting on soft agar overlays containing D1585. Plaques were counted after overnight incubation at 30 °C. Resulting data from four biological replicates was analyzed using GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, US).
Growth reduction assay and phage lifestyle analysis
To analyze the killing effect of AXL1 phage in liquid culture, growth reduction assays were conducted. Three D1585 overnight liquid cultures were subcultured in LB broth and grown at 30 °C to an OD600 of 0.2, corresponding to 4.0 × 108 CFU/mL. 100 µL of each culture was added to wells of a 96 well plate containing 100 µL of AXL1 phage lysate at multiple concentrations to give MOIs of approximately 30, 6, 0.6, and 0.06, or LB broth as a control, resulting in biological triplicate with three replicates each. The plate was incubated at 30 °C with continuous orbital shaking at 237 cpm in an Epoch 2 microplate spectrophotometer (Bio Tek Instruments, Inc., VT, USA), with the OD600 measured every 30 min for 48 h. Data from three biological replicates was analyzed using GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, US).
To investigate AXL1 lifestyle, the isolation of lysogens was attempted from both confluent lysis plate infections and liquid infections as described above at 30 °C and 37 °C. Surviving bacterial cells were washed three times to remove contaminating phage and plated for single colonies. Individual colonies were tested for superinfection resistance by spotting with phage lysate and resistant colonies were analyzed by colony PCR with AllTaq DNA polymerase (Qiagen, Inc., Germantown, MD, USA) following manufacturer protocols using primers specific to AXL1 gDNA (F 5′-GACTACGACGCCTTCTCCGC-3′; R 5′-TTTGCCTGCCTCGACGCCAG-3′).
Twitching motility
As an indirect measurement of type IV pili function, twitching motility assays were conducted as previously described14. Single colonies were suspended in 100 µL LB and stab inoculated through a 3 mm thick LB 1% agar layer containing 0.3% porcine mucin to the bottom of the petri plate and incubated with humidity at 30 °C or 37 °C for 48 h. Twitching zones stained with crystal violet were imaged and measurements using ImageJ software (NIH, Bethesda, MD, USA)70 are reported as average twitching area ± standard deviation from nine twitching zones representing results in biological triplicate for each strain.
Phage DNA isolation, RFLP analysis and genome sequencing
AXL1 genomic DNA (gDNA) was isolated by phenol/chloroform extraction and ethanol precipitation as previously described12. Following incubation with proteinase K, gDNA from a nuclease-treated high titre phage lysate was isolated with three phenol:chloroform extractions and a single chloroform wash. Phage DNA was ethanol precipitated and dissolved in sterile milli-Q water. A NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA) was used to determine the purity and concentration of phage gDNA.
Restriction fragment length polymorphism (RFLP) analysis was conducted using 31 FastDigest (Thermo Scientific) restriction enzymes: AccI, AseI, MspI, HpaII, HhaI, Bsh1236I, MauBI, PdmI, HaeIII, NheI, AciI, Eam1105I, SmaI, XbaI, BamHI, KpnI, ApaI, SacI, HindIII, SalI, PstI, ClaI, XhoI, NotI, StuI, BglII, AvrII, MscI, StyI, TasI, and Tru1I. Digests were set up in 20 µL volumes using 1 µL of enzyme, 2 µL of restriction buffer and 1 µg of AXL1 gDNA. Reactions were incubated at 37 °C for 1 h, separated on a 0.8% (wt/vol) agarose gel in 1× TAE (pH 8.0) and DNA visualized with ethidium bromide post-staining.
Sequencing of AXL1 was performed at The Applied Genomics Core at the University of Alberta. A DNA genomic library was constructed using a Nextera XT library prep kit followed by paired-end sequencing on a MiSeq (Illumina, San Diego, CA) platform using a MiSeq v3 reagent kit.
Bioinformatic analysis
Quality control analysis was completed using FastQC v0.11.972 and the 1,266,570 paired-end reads were processed using Trimmomatic v0.3873, with 80.07% of both read pairs surviving. SPAdes v3.11.174 was used to assemble a 64,089 bp contig with 2,023,812 reads mapping to the contig to give a mean coverage of 5850 reads. Regions of low coverage, random sites, and ends of the contig were confirmed with PCR using seven primer pairs followed by Sanger sequencing of the PCR products to confirm the complete genome of 63,962 bp in length due to a duplication of 127 bp between AXL1_67 and AXL1_68 at the assembly ends. Exploration of this region as direct terminal repeats did not produce confirmatory results. In the absence of data supporting physical genomic termini, the genome start site was determined by convention and placed upstream of the small terminase, similar to related phage genomes PaMx28 (accession: NC_028931) and PaMx74 (accession: NC_028809)24.
Predicted protein coding genes were identified using the GLIMMER plugin75 for Geneious using the Bacteria and Archaea setting, as well as GeneMarkS for phage76 and Prodigal77. Annotations to the contig and visualization of the genome was done using Geneious Prime v2022.0.125. BLASTn was used to identify relatives based on genomic data and putative protein functions were assigned using BLASTp limited to Viruses (taxid:10239) on the NCBI non-redundant protein sequence and nucleotide collection databases (update date: 2021/11/04)78. Conserved domain searches were performed using CD-Search against the CDD v3.19-58235 PSSMs database and default options32 to support functional annotation. TMHMM79 and LipoP 1.080 were used to identify transmembrane regions and predict lipoproteins, respectively, in putative lysis proteins. tRNAscan-SE software with the general tRNA model81 and Aragorn v1.2.3682 were used to identify potential tRNA genes. Rho-independent transcription terminator sequences were identified using ARNold83 and 13 putative intergenic terminators with ΔG values less than − 10 kcal/mol were included. Phage promoters were identified using the PhagePromoter tool in the CPT Galaxy webserver84. Protein alignments were accomplished using MUSCLE85. Protein structural and functional predictions were conducted using I-TASSER35, COACH34 and COFACTOR33.
Genomic comparison of phages in the Pamexvirus genus was conducted using Clinker v0.0.2326 on phage genomes that were first oriented to have the same start site as AXL1 upstream of the small terminase protein. Only links with 30–100% identity are shown.
Proteomic analysis of virion-associated proteins
Phage particles were purified for proteomic analysis by CsCl density gradient ultracentrifugation as described above. The equivalent of ~ 2 × 109 PFU was boiled for 5 min in Laemmli sample buffer and loaded into a single lane of an SDS-PAGE gel with 5% stacking and 10% resolving sections and run at 80 V for 2 h followed by Coomassie R-250 staining. Whole lane in gel trypsin digestion and protein identification was conducted by the Alberta Proteomics and Mass Spectrometry (APM) facility at the University of Alberta as previously described13, with modifications. The lane was cut into four equal gel sections and following processing, proteins were trypsin digested (6 ng/µL) at 37 °C overnight. Tryptic peptides were extracted from the gel and fractions containing tryptic peptides were resolved and ionized using nanoflow high-performance liquid chromatography (HPLC) (Easy-nLC 1000; Thermo Scientific) coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Scientific). A PepMap RSLC C18 EASY-Spray column (Thermo Scientific) with a 75-μm inner diameter (100 Å, 3 μm pore size) was used for nanoflow chromatography and electrospray ionization. Peptide mixtures were injected onto the column at a flow rate of 3000 nL/min and resolved at 350 nL/min using 60-min step gradients of 4% to 37% (vol/vol) aqueous ACN with 0.2% (vol/vol) formic acid. The mass spectrometer was operated in data-dependent acquisition mode, recording high-accuracy and high-resolution Orbitrap survey spectra using external mass calibration, with a resolution of 35,000 and m/z range of 400 to 2,000. The 12 most intensely multiply charged ions were sequentially fragmented by HCD fragmentation and their spectra were collected at a resolution of 17,500. After two fragmentations, all precursors selected for dissociation were dynamically excluded for 30 s. Data were processed using Proteome Discoverer 1.4 (Thermo Scientific) and all AXL1 and S. maltophilia D1585 proteins were searched using SEQUEST (Thermo Scientific).
Antibacterial susceptibility checkerboard assays
The AXL1 dhfr gene was amplified from phage genomic DNA using Phusion high-fidelity DNA polymerase (New England Biolabs) with GC buffer and 3% DMSO according to manufacturer protocols and using primer pair AXL1gp63F (TTCTAAGCTTTACCCATCACCTACATTGCG) and AXL1gp63R (TTATTCTAGAGAGCTCACCAGGTTCTCGAC). Restriction enzyme recognition sites are underlined. The resulting product was purified by gel extraction using the QIAquick gel extraction kit (Qiagen, Inc., Germantown, MD, USA), digested with HindIII and XbaI Fast Digest restriction endonucleases (Thermo Scientific) and ligated into the similarly digested vector pBBR1MCS using T4 DNA ligase (New England Biolabs), producing the construct pAXL1dhfr. This plasmid was transformed into electrocompetent E. coli DH5α and verified by Sanger sequencing before transforming S. maltophilia strains by electroporation as previously described14.
Overnight cultures of S. maltophilia strains carrying pBBR1MCS or pAXL1dhfr were grown in LB with chloramphenicol for 18 h at 30 °C before subculturing 1:100 in Mueller Hinton (MH) broth and growing to an OD600 corresponding to 108 CFU/mL. 190 µL of each subculture was added to 96-well plates, followed by 5 µL each of serially diluted sulfamethoxazole (Sigma-Aldrich) and trimethoprim (MP Biomedicals), with DMSO added in place of either antibiotic as a solvent growth control. One lane contained MH and DMSO to serve as a negative control. Plates were incubated at 30 °C with shaking at 225 RPM for 20–24 h and OD600 was measured using a Wallac 1420 VICTOR2 multilabel counter (PerkinElmer, Waltham, MA). Data from three biological replicates was analysed using GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, US), with blank values subtracted from the absorbance and data normalized to the no antibiotic, solvent control well.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files. The genome of vB_SmaS-AXL_1 is available on Genbank under the accession number OL674541.
References
Berg, G. & Martinez, J. L. Friends or foes: Can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex?. Front. Microbiol. 6, 241 (2015).
McCutcheon, J. G. & Dennis, J. J. The potential of phage therapy against the emerging opportunistic pathogen Stenotrophomonas maltophilia. Viruses 13, 1057 (2021).
Ryan, R. P. et al. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525 (2009).
Brooke, J. S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25, 2–41 (2012).
Giles, A., Foushee, J., Lantz, E. & Gumina, G. Sulfonamide allergies. Pharmacy 7, 132 (2019).
Gil-Gil, T., Martínez, J. L. & Blanco, P. Mechanisms of antimicrobial resistance in Stenotrophomonas maltophilia: A review of current knowledge. Expert Rev. Anti. Infect. Ther. 18, 335–347 (2020).
Peters, D. L., McCutcheon, J. G., Stothard, P. & Dennis, J. J. Novel Stenotrophomonas maltophilia temperate phage DLP4 is capable of lysogenic conversion. BMC Genomics 20, 300 (2019).
Fan, H. et al. Complete genome sequence of IME13, a Stenotrophomonas maltophilia bacteriophage with large burst size and unique plaque polymorphism. J. Virol. 86, 11392–11393 (2012).
Ackermann, H. W. & Eisenstark, A. The present state of phage taxonomy. Intervirology 3, 201–219 (1974).
Mirzaei, M. K. & Nilsson, A. S. Isolation of phages for phage therapy: A comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 10, e0118557 (2015).
Pirnay, J. P. et al. Introducing yesterday’s phage therapy in today’s medicine. Future Virol. 7, 379–390 (2012).
McCutcheon, J. G., Lin, A. & Dennis, J. J. Isolation and characterization of the novel bacteriophage AXL3 against Stenotrophomonas maltophilia. Int. J. Mol. Sci. 21, 1–20 (2020).
Peters, D. L., McCutcheon, J. G. & Dennis, J. J. Characterization of novel broad-host-range bacteriophage DLP3 specific to Stenotrophomonas maltophilia as a potential therapeutic agent. Front. Microbiol. 11, 1358 (2020).
McCutcheon, J. G., Peters, D. L. & Dennis, J. J. Identification and characterization of type IV pili as the cellular receptor of broad host range Stenotrophomonas maltophilia bacteriophages DLP1 and DLP2. Viruses 10, 338 (2018).
Burrows, L. L. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu. Rev. Microbiol. 66, 493–520 (2012).
Dunger, G., Llontop, E., Guzzo, C. R. & Farah, C. S. The Xanthomonas type IV pilus. Curr. Opin. Microbiol. 30, 88–97 (2016).
Craig, L., Pique, M. E. & Tainer, J. A. Type IV pilus structure and bacterial pathogenicity. Nat. Rev. Microbiol. 2, 363–378 (2004).
Harvey, H. et al. Pseudomonas aeruginosa defends against phages through type IV pilus glycosylation. Nat. Microbiol. 3, 47–52 (2018).
Peters, D. L., Lynch, K. H., Stothard, P. & Dennis, J. J. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genomics 16, 664 (2015).
Clavijo-Coppens, F. et al. Novel virulent bacteriophages infecting mediterranean isolates of the plant pest Xylella fastidiosa and Xanthomonas albilineans. Viruses 13, 725 (2021).
Flodman, K., Corrêa, I. R., Dai, N., Weigele, P. & Xu, S. Y. In vitro Type II restriction of bacteriophage DNA With modified pyrimidines. Front. Microbiol. 11, 604618 (2020).
Dong, Z. et al. Isolation and characterization of a novel phage Xoo-sp2 that infects Xanthomonas oryzae pv. oryzae. J. Gen. Virol. 99, 1453–1462 (2018).
Andrade-Domínguez, A. & Kolter, R. Complete genome sequence of Pseudomonas aeruginosa phage AAT-1. Genome Announc. 4, 165–181 (2016).
Sepúlveda-Robles, O., Kameyama, L. & Guarneros, G. High diversity and novel species of Pseudomonas aeruginosa bacteriophages. Appl. Environ. Microbiol. 78, 4510–4515 (2012).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Gilchrist, C. L. M. & Chooi, Y. H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 37, 2473–2475 (2021).
Ravichandran, K., Olshansky, L., Nocera, D. G. & Stubbe, J. A. Subunit interaction dynamics of class Ia ribonucleotide reductases: in search of a robust assay. Biochemistry 59, 1442–1453 (2020).
Maley, F. & Maley, G. F. A tale of two enzymes, deoxycytidylate deaminase and thymidylate synthase. Prog. Nucleic Acid Res. Mol. Biol. 39, 49–80 (1990).
Hardy, L. W. et al. Atomic structure of thymidylate synthase: Target for rational drug design. Science 235, 448–455 (1987).
Raimondi, M. V. et al. DHFR inhibitors: Reading the past for discovering novel anticancer agents. Molecules 24, 1140 (2019).
Iyer, L. M., Babu, M. M. & Aravind, L. The HIRAN domain and recruitment of chromatin remodeling and repair activities to damaged DNA. Cell Cycle 5, 775–782 (2006).
Lu, S. et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268 (2020).
Roy, A., Yang, J. & Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 40, W471–W477 (2012).
Yang, J., Roy, A. & Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 41, D1096–D1103 (2013).
Yang, J. & Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Hatfull, G. F. Bacteriophage genomics. Curr. Opin. Microbiol. 11, 447–453 (2008).
Simon, N. C., Aktories, K. & Barbieri, J. T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 12, 599–611 (2014).
Jacobs-Sera, D. et al. Genomic diversity of bacteriophages infecting Microbacterium spp. PLoS ONE 15, e0234636 (2020).
Jamet, A. et al. A widespread family of polymorphic toxins encoded by temperate phages. BMC Biol. 15, 75 (2017).
Wilkens, K., Tiemann, B., Bazan, F. & Rüger, W. ADP-ribosylation and early transcription regulation by bacteriophage T4. Adv. Exp. Med. Biol. 419, 71–82 (1997).
Chen, L. et al. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328 (2005).
Ahern, S. J., Das, M., Bhowmick, T. S., Young, R. & Gonzalez, C. F. Characterization of novel virulent broad-host-range phages of Xylella fastidiosa and Xanthomonas. J. Bacteriol. 196, 459–471 (2014).
Liu, M. et al. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science 295, 2091–2094 (2002).
Inoue, T. et al. Genome-wide screening of genes required for swarming motility in Escherichia coli K-12. J. Bacteriol. 189, 950–957 (2007).
de Souza, R. F. & Aravind, L. Identification of novel components of NAD-utilizing metabolic pathways and prediction of their biochemical functions. Mol. Biosyst. 8, 1661–1677 (2012).
Frelin, O. et al. A directed-overflow and damage-control N-glycosidase in riboflavin biosynthesis. Biochem. J 466, 137–145 (2015).
Young, R., Wang, I. N. & Roof, W. D. Phages will out: Strategies of host cell lysis. Trends Microbiol. 8, 120–128 (2000).
Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 52, 243–258 (2014).
Shan, J. et al. Temperature dependent bacteriophages of a tropical bacterial pathogen. Front. Microbiol. 5, 599 (2014).
Egilmez, H. I. et al. Temperature-dependent virus lifecycle choices may reveal and predict facets of the biology of opportunistic pathogenic bacteria. Sci. Rep. 8, 1–13 (2018).
Gleckman, R., Blagg, N. & Joubert, D. W. Trimethoprim: Mechanisms of action, antimicrobial activity, bacterial resistance, pharmacokinetics, adverse reactions, and therapeutic indications. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1, 14–19 (1981).
Hu, L. F. et al. Stenotrophomonas maltophilia resistance to trimethoprim/sulfamethoxazole mediated by acquisition of sul and dfrA genes in a plasmid-mediated class 1 integron. Int. J. Antimicrob. Agents 37, 230–234 (2011).
Bushby, S. R. M. Synergy of trimethoprim sulfamethoxazole. Can. Med. Assoc. J. 112, 63–66 (1975).
Kutter, E. Phage host range and efficiency of plating. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Charcterization, and Interactions Vol. 501 (eds Clokie, M. R. J. & Kropinski, A. M.) 141–149 (Humana Press, 2009).
Abedon, S. T. Lysis from without. Bacteriophage 1, 46–49 (2011).
Lee, C.-N., Tseng, T.-T., Chang, H.-C., Lin, J.-W. & Weng, S.-F. Genomic sequence of temperate phage Smp131 of Stenotrophomonas maltophilia that has similar prophages in xanthomonads. BMC Microbiol. 14, 17 (2014).
Lee, C. N., Lin, J. W., Chow, T. Y., Tseng, Y. H. & Weng, S. F. A novel lysozyme from Xanthomonas oryzae phage φXo411 active against Xanthomonas and Stenotrophomonas. Protein Expr. Purif. 50, 229–237 (2006).
Hooton, S. P. T. & Connerton, I. F. Campylobacter jejuni acquire new host-derived CRISPR spacers when in association with bacteriophages harboring a CRISPR-like Cas4 protein. Front. Microbiol. 5, 744 (2015).
Tsai, R., Corrêa, I. R., Xu, M. Y. & Xu, S. Y. Restriction and modification of deoxyarchaeosine (dG+)-containing phage 9 g DNA. Sci. Rep. 7, 8348 (2017).
Yin, C. et al. Co-infection of Pseudomonas aeruginosa and Stenotrophomonas maltophilia in hospitalised pneumonia patients has a synergic and significant impact on clinical outcomes. Eur. J. Clin. Microbiol. Infect. Dis. 36, 2231–2235 (2017).
Enault, F. et al. Phages rarely encode antibiotic resistance genes: A cautionary tale for virome analyses. ISME J. 11(1), 237–247 (2017).
Torres-Barceló, C. The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerg. Microbes Infect. 7, 168 (2018).
Jacobs, M. A. et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. 100, 14339–14344 (2003).
Hanahan, D., Jessee, J. & Bloom, F. R. Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol. 204, 63–113 (1991).
Kovach, M. E., Phillips, R. W., Elzer, P. H., Roop, R. M. 2nd. & Peterson, K. M. pBBR1MCS: A broad-host-range cloning vector. Biotechniques 16, 800–802 (1994).
West, S. E. H., Schweizer, H. P., Dall, C., Sample, A. K. & Runyen-Janecky, L. J. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 128, 81–86 (1994).
Kropinski, A. M., Mazzocco, A., Waddell, T. E., Lingohr, E. & Johnson, R. P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Charcterization, and Interactions (eds Clokie, M. R. J. & Kropinski, A. M.) 69–76 (Humana Press, 2009). https://doi.org/10.1007/978-1-60327-164-6.
Peters, D. L., Stothard, P. & Dennis, J. J. The isolation and characterization of Stenotrophomonas maltophilia T4-like bacteriophage DLP6. PLoS ONE 12, e0173341 (2017).
Peters, D. L. & Dennis, J. J. Complete genome sequence of temperate Stenotrophomonas maltophilia bacteriophage DLP5. Genome Announc. 6, e00073-e118 (2018).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Carlson, K. Working with bacteriophages: common techniques and methodological approaches. In Bacteriophages: Biology and Applications (eds Kutter, E. M. & Sulakvelidze, A.) 437–494 (CRC Press, 2005).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007).
Besemer, J., Lomsadze, A. & Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618 (2001).
Hyatt, D. et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Juncker, A. S. et al. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662 (2003).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: searching for tRNA genes in genomic sequences. In Gene Prediction: Methods and Protocols, Methods in Molecular Biology Vol. 1962 (ed. Kollmar, M.) 1–14 (Humana, 2019).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004).
Naville, M., Ghuillot-Gaudeffroy, A., Marchais, A. & Gautheret, D. ARNold: A web tool for the prediction of rho-independent transcription terminators. RNA Biol. 8, 11–13 (2011).
Ramsey, J. et al. Galaxy and Apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PLOS Comput. Biol. 16, e1008214 (2020).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Acknowledgements
The authors thank Arlene Oatway from the University of Alberta Department of Biological Sciences Advanced Microscopy Facility for her assistance with TEM and Brent Weber for advice and thoughtful discussion regarding antibiotic susceptibility assays. We also thank James Zlosnik and the Canadian Burkholderia cepacia complex Research and Referral Repository (CBCCRRR) and LeeAnn Turnbull of the Provincial Laboratory for Public Health -North, Alberta Health Services for providing S. maltophilia clinical isolates, as well as Jorge Girón for gifting strains SMDP92 and ATCC13637. We thank Guus Bakkeren of Agriculture and Agri-Foods Canada for contributing Xanthomonas strains from an AAFC strain collection. JGM was kindly supported by a CGS-D award from NSERC and an AIGSS award from Alberta Innovates. This research was supported by operating grants to JJD from the Natural Sciences and Engineering Research Council of Canada (NSERC-238414-2018), and the Alberta Major Innovation Fund (RCP-19-003-MIF).
Author information
Authors and Affiliations
Contributions
A.L. isolated and performed initial characterization of AXL1. J.G.M. fully characterized AXL1, including genomic, bioinformatic, and functional analysis, and drafted the manuscript. J.J.D. directed and supervised the research, provided financial support, and edited the manuscript. All authors reviewed the manuscript and approve the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McCutcheon, J.G., Lin, A. & Dennis, J.J. Characterization of Stenotrophomonas maltophilia phage AXL1 as a member of the genus Pamexvirus encoding resistance to trimethoprim–sulfamethoxazole. Sci Rep 12, 10299 (2022). https://doi.org/10.1038/s41598-022-14025-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14025-z
- Springer Nature Limited