
Abstract
Interactions between the gut microbiota and the immune system may be involved in vaccine and infection responses. In the present study, we studied the interactions between caecal microbiota composition and parameters describing the immune response in six experimental inbred chicken lines harboring different MHC haplotypes. Animals were challenge-infected with the infectious bronchitis virus (IBV), and half of them were previously vaccinated against this pathogen. We explored to what extent the gut microbiota composition and the genetic line could be related to the immune response, evaluated through flow cytometry. To do so, we characterized the caecal bacterial communities with a 16S rRNA gene amplicon sequencing approach performed one week after the IBV infectious challenge. We observed significant effects of both the vaccination and the genetic line on the microbiota after the challenge infection with IBV, with a lower bacterial richness in vaccinated chickens. We also observed dissimilar caecal community profiles among the different lines, and between the vaccinated and non-vaccinated animals. The effect of vaccination was similar in all the lines, with a reduced abundance of OTU from the Ruminococcacea UCG-014 and Faecalibacterium genera, and an increased abundance of OTU from the Eisenbergiella genus. The main association between the caecal microbiota and the immune phenotypes involved TCRϒδ expression on TCRϒδ+ T cells. This phenotype was negatively associated with OTU from the Escherichia-Shigella genus that were also less abundant in the lines with the highest responses to the vaccine. We proved that the caecal microbiota composition is associated with the IBV vaccine response level in inbred chicken lines, and that the TCRϒδ+ T cells (judged by TCRϒδ expression) may be an important component involved in this interaction, especially with bacteria from the Escherichia-Shigella genus. We hypothesized that bacteria from the Escherichia-Shigella genus increased the systemic level of bacterial lipid antigens, which subsequently mitigated poultry γδ T cells.
Similar content being viewed by others


Introduction
Microbes are becoming increasingly resistant to the antibiotics used to treat human and animal infections. This is why the use of antibiotics as growth promoters (AGP) in animal production is forbidden in a growing number of countries. In chicken flocks, the ban of antibiotics has been followed by the observation of more frequent gastro-intestinal disorders such as necrotic enteritis1. Moreover, genetic selection on production traits such as growth rate, feed efficiency or meat and carcass yield may have indirectly impacted animal robustness, since immune phenotypes or disease resistance phenotypes were generally not included in selection programs by breeders2,3. For instance in broiler breeding, selection for an increased body weight led to a decrease in the relative weight of primary and secondary immune organs4. Intense selection may also have compromised the host immune response to Escherichia coli vaccination5 or increased the susceptibility to Marek’s disease6,7. Potential trade-offs between productivity and immunity are often hypothesized to explain such results8.
In order to avoid economic losses due to infections by pathogens and to ensure animal health in the absence of antibiotics, reinforced vaccination and the inclusion of probiotics in animal feeding were among the first approaches considered9. In this context, increasing the efficiency of vaccines may also be considered. Vaccines are still suboptimal for many endemic chicken diseases due to the variety of geographical areas, types of production, and poultry species and breeds, which all influence the variability of response to the vaccine and the outcome of the infection10,11,12. Most of efficiency evaluations of experimental vaccines comprise measurements of the immunological response and disease outcome after submitting vaccinated animal to a suitable challenge mimicking natural infection13.
Several approaches have been considered to enhance the animal immune response to vaccination, and some may depend on the intestinal microbiota14,15. Certain strategies focus on the early manipulation of intestinal innate lymphoid cells (ILCs)16. Besides, many new vaccine designs specifically target the antigen-presenting cells (APC) and use bacterial components as adjuvants through ligand-based APC targeting. For instance, TLR ligands have been used as adjuvant in chickens immunized with inactivated avian influenza IAV H5N2 co-administrated with Salmonella enterica flagellin11,17, and an enterobactin conjugate vaccine demonstrated potent ability to elicit high antibody response against Campylobacter jejuni in chicken18.
In parallel, the early modulation of the intestinal microbiota is already promoted to improve intestinal health of chickens19,20. Studies report the effects of microbiota modulatory compounds such as probiotics: Anaerosporobacter mobilis or Lactobacillus reuteri impact the outcome of vaccination against Campylobacter jejuni21,22. Moreover, variations in the commensal gut microbiota composition can affect adaptive immune responses in vaccinated chickens, with higher hemagglutination titers in chicken submitted to a probiotic treatment based on several Lactobacillus species in comparison with antibiotic treated chicken23. A few recent studies report also the effect of vaccination on the gut microbiota, for instance against Salmonella24. In addition, vaccines with attenuated Eimeria have been shown to diminish the microbial diversity and Firmicutes relative abundance in broilers25,26. The interactions between vaccination and the gut microbiota remain to be studied for a larger set of pathogens, and for different types of vaccines.
In addition to the intestinal microbiota, animal genetics is a parameter scarcely considered when developing vaccination strategies. Adjusting vaccines to the host genetics, or genetically selecting animals displaying a better immune response to vaccines, could be a relevant way of improving the vaccine response in farm conditions. For example, in chickens, strong associations occur between many infectious diseases and the genotype at the MHC (Major Histocompatibility Complex), which is a polymorphic region comprising genes coding for cell surface molecules involved in antigen presentation27. Genetic variations at the MHC might also influence the vaccine responses, as observed for instance for the Newcastle disease vaccine24 and IBV28,29.
The Infectious Bronchitis Virus (IBV) is a single-stranded RNA and enveloped Coronavirus causing a respiratory disease in infected chickens. The high rate of mutations of this virus is leading to the existence of many strains with different virulence levels. The morbidity through respiratory symptoms in infected husbandry is high. Despite vaccination schedules, chickens still display suboptimal levels of protection worldwide. In layer hens, IBV leads to economic losses with impaired egg production and quality. Typically, the egg laying declines observed range between 3 and 10%, but reductions of up to 50% have also been observed30.
Based on flow cytometry assessments of peripheral blood cells among experimental inbred MHC-B lines infected with IBV, Larsen et al. reported that the animals with the highest monocyte MHC-II expression had the weakest vaccine-induced challenge protection29. We used the animals from this study to investigate if their caecal microbiota was associated with their immune phenotype profiles. Vaccinated challenged animals displayed a mild infection with a reduced tracheal IBV carriage, in comparison to non-vaccinated challenged animals that displayed an acute infection with higher tracheal IBV carriage. Furthermore, in vaccinated animals, different susceptibility levels to the challenge infection were observed between MHC lines, with chickens of the MHC haplotype B21 displaying low IBV loads after challenge, and thus the best responses to the IBV vaccination.
In the present study, we wondered if the vaccination and the IBV challenge of these six chicken MHC lines were also associated with differences in caecal microbiota composition. To investigate it, we compared vaccinated and non-vaccinated animals to identify associations present during either a sub-clinical infection, or an acute infection. We explored if the caecal microbiota of the MHC lines differed one week after IBV challenge and assessed whether the variations of various immune parameters could be associated with the caecal microbiota composition.
Results
In the present study, we studied the interactions between caecal microbiota composition and parameters describing the immune response in six experimental inbred chicken lines harboring different MHC haplotypes. The chicken lines were challenge-infected with the infectious bronchitis virus (IBV), and half of them were previously vaccinated against this pathogen (Fig. 1). We explored to what extent the gut microbiota composition and the genetic line could be related to the variability of the immune response, evaluated through flow cytometry. To do so, we characterized the caecal bacterial communities with a 16S rRNA gene amplicon sequencing approach performed one week after the IBV infectious challenge. In an associated study on the same animals, Larsen previously categorized the 6 chicken lines according to the levels of their tracheal IBV load 5 days post infection in the vaccinated and non-vaccinated group to assess the capacity of the chicken to dampen the infection29. B21 animals that displayed the lowest viral loads after challenge in the V-Ch group, and had the highest load in the NV-Ch group were thus qualified as susceptible to IBV infection and high responder to vaccination.

Experimental design and IBV load after challenge. A group of 48 vaccinated and challenged (V-Ch, nB12 = 8, nB14 = 8, nB19 = 8, nB15 = 8, nB2 = 8, nB21 = 8) individuals was opposed to a group of 45 non-vaccinated and challenged (NV-Ch, nB12 = 6, nB14 = 7, nB19 = 8, nB15 = 8, nB2 = 8, nB21 = 8) individuals, with 6 lines of White Leghorn laying hens followed in each group. Two pens per vaccine status were used, and the lines were reared together.
Global taxonomic composition of caecal microbiota after IBV challenge in vaccinated and non-vaccinated chickens
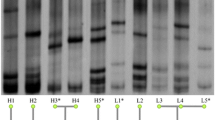
From the total population composed of 93 caecal samples from V-Ch and NV-Ch groups, 16S rRNA gene sequencing provided a total of 12,256,442 reads, reduced to 4,952,178 sequences after quality control with an average of 53,249 sequences per sample (range from 32,560 to 76,819 sequences). We identified 487 OTU from 5 phyla, 12 families and 73 genera (Fig. 2). The massively dominant phylum was Firmicutes (average abundance 96%), with essentially Lachnospiraceae (48.1%), Ruminococcaceae (42.1%), and to a lesser extent Bacilli (2.1%). It was followed by the much less abundant Proteobacteria (average abundance 3.9%) (Supplementary Table S1; Supplementary Fig. S1). Over the 73 genera identified, Ruminococcus torques group (12.4%), Eisenbergiella (9.0%), and Ruminococcaceae UCG-014 (8.0%) were the three dominant ones, and 34 significant correlations were observed between their abundances. Eisenbergiella was negatively associated with Ruminococcaceae UCG-014 (ρ = -0.52), and Escherichia-Shigella, which is often described as pathogenic, was positively associated with Anaerostipes (ρ = 0.54), and with an unknown genus from the Gammaproteobacteria class (ρ = 0.95) (Supplementary Table S2, Supplementary Fig. S2).

Caecal microbial community of 9-week-old layers, one week after IBV infection. Taxonomic composition is given at the genus level among the different lines, and according to their vaccine status: vaccinated and challenged (V-Ch, n = 48) vs non-vaccinated and challenged (NV-Ch, n = 45). Every bar corresponds to one individual. Only taxons with a mean relative abundance above 2% are displayed.
Effect of vaccination on the caecal microbiota after IBV challenge
In vaccinated animals, one week after the IBV challenge, species richness (p = 7.0 × 10–4) and Chao1 estimator (p = 7.2 × 10–4) were reduced in caecal microbiota composition when compared to non-vaccinated animals. But no significant effects, neither on Shannon (p = 0.24) nor on inverse Simpson indexes (p = 0.92), were found (Table 1). As Lachnospiraceae and Ruminococcaceae were the dominant families observed, we wondered whether vaccination had also an effect on species Richness and Chao1 in those two families separately. In the Ruminococcaceae subgroup, vaccination significantly decreased OTU richness (p = 1.1 × 10–7) and Chao1 (p = 9.8 × 10–7), but it had no significant effect in the Lachnospiracea group (Supplementary Fig. S3).
Regarding β-diversity, vaccination had a significant effect on Bray–Curtis distances (p = 1 × 10–4) with V-Ch animals forming a distinct cluster from the NV-Ch animals on NMDS representation (Fig. 3a). Furthermore, it strengthened microbiota homogeneity as it significantly reduced β-diversity (p = 4 × 10–3). Finally, by looking at the centroids displacement on an NMDS from the NV-Ch group to the V-Ch group, we observed that the microbiota was similarly modified with the vaccination in all lines (Fig. 3b).

Effect of IBV vaccination on the caecal microbial community of 9-weeks-old hens infected with IBV. (a) NMDS representation of the caecal microbial communities using Bray–Curtis distances among the vaccinated challenged (V-Ch, n = 48) (orange) and the non-vaccinated challenged (NV-Ch, n = 45) (blue) groups considering all the lines together. (b) NMDS representation of the caecal microbial communities in each MHC line separately. Arrows represent the centroids displacement from the non-vaccinated challenged (NV-Ch) group (blue) to the vaccinated challenged (V-Ch) group (orange).
Differential analysis on OTU abundance between the V-Ch and NV-Ch groups showed that among the differentially abundant OTU, 23 out of 25 from the Ruminococcaceae UGG-014 group genus, and 5 out of 6 OTU from Faecalibacterium genus were less abundant in the vaccinated group. On the contrary, the following components were more abundant in the vaccinated group: most OTU from the Eisenbergiella genus (11 out of 12), the Ruminococcus torques group (5 out of 7), the Ruminoclostridium 9 group ; the Oscillibacter genera (5 out of 6); Escherichia-Shigella, Lachnospiraceae NK4A136 group, Subdoligranulum, and Lactobacillus genus (3 out of 3); and both OTU from the Tyzerella 3 genus (Supplementary Table S3).
Considering the lines separately, the trend for Ruminococcaceae was similar for each line, as most of the OTU significantly different in this family were less abundant in the vaccinated group. Ruminococcaceae UCG-014, which was the most affected genus within this family, was impacted significantly in the B2, B19, and B21 lines. Finally, the statistical effect of vaccination on OTU abundances was the most important in the B2 line (39 OTU differentially abundant), followed by the B19 line (22 OTU), and to a lesser extent by the B15 and B21 lines (9 OTU) (Supplementary Table S4).
Differences in microbiota composition according to the genetic line
Genetic lines were significantly associated with the β-diversity both in the V-Ch and NV-Ch groups (p = 1 × 10–4) (Fig. 4a,b), but not with the α-diversity indices (Supplementary Fig. S4, Supplementary Table S5). The microbial dissimilarity observed between the lines was thus not due to a higher bacterial richness in some of them.

Association between the MHC line and the caecal microbial beta-diversity in laying hens one week after their infection with IBV. NMDS representation of the caecal microbial communities using Bray–Curtis distances among the different MHC lines in the (a) vaccinated and challenged (V-Ch, n = 48) and in the (b) non-vaccinated and challenged (NV-Ch, n = 45) groups. Representation of the community dispersion in each MHC-line through its β-diversity among (c) the total population, (d) the vaccinated challenged (V-Ch) group, and (e) the non-vaccinated challenged (NV-Ch) group. We indicated the p-value of the line effect. Asterisks indicate a significant difference between two lines based on post-hoc comparison.
Results from pairwise comparisons of BC distances between lines showed that the B14 line displayed both the greatest dissimilarity with the other ones (all pairwise p < 0.0002), and the greatest homogeneity within its bacterial community (Table 2; Fig. 4c–e). The B14 line was followed by the B21 line in terms of dissimilarity with the other lines, and then by B2 and 12 to a lesser extent. Furthermore, we observed a closer proximity between B12 and B14, and between B19 and B21. The number of OTU with significant differential abundance between each couple of lines ranged from none between lines that did not display significant dissimilarity (B12–B15 and B2–B21), to 126 between the B14 and B21 lines that displayed the greatest dissimilarity (Table 2; Supplementary Table S6). Globally, the B14 line had more abundant OTU from the Ruminococcus torques group, and lower abundances of OTU from Ruminococcaceae UCG-014 and Ruminiclostridium 5 genus (Supplementary Table S7).
The B21 line is mostly dissimilar from B12 and B14, and it globally harbored a larger abundance of OTU from Ruminococcaceae UCG-014 and GCA-900066575 genera and a lower abundance of OTU from Flavonifractor, Escherichia-Shigella, and Oscillibacter genera. Finally, the B12 line harbored a higher abundance of OTU from Escherichia-Shigella genus in comparison with the B2, B21 and B19 lines (Supplementary Table S6).
The same pattern was observed considering the V-Ch and NV-Ch groups separately. There was no line effect on α-diversity in any group, and once again, the B14 and B21 lines displayed the most dissimilar microbiota in both groups (Fig. 4a,b; Supplementary Table S6). However, vaccination brought these lines closer: they were less dissimilar in the V-Ch group than in the NV-Ch group, especially for B21 line. All p-values were lower than 0.006 in pairwise comparison on BC distance involving B21 line in the NV-Ch group, while only comparisons between B21 line with B12 and B14 lines displayed significant p-value in the V-Ch group.
As microbiota dissimilarity between the lines could be due to weight differences, we considered lines weight. There were only slight variations in weight between genetic lines, with the exception of line B12, which was on average heavier than other lines, in both the vaccinated group and the non-vaccinated group (Supplementary Fig. S5). It is to notice that the microbiota from the B12 line was not the most dissimilar. Furthermore, there was no significant effect of weight on gut microbiota composition, neither with alpha, nor with beta-diversity. Thus, we conclude that the dissimilarity in microbiota composition we observed between the lines was not due to weight differences.
Associations between immune phenotypes and microbiota according to the genetic line
Globally, in the total population, using Adonis analysis we identified four immune parameters that shared a significant association with the caecal microbiota from the whole cohort. TCRϒδ expression on TCRγδ T cells was the immune parameter with the most significant association (p = 5 × 10–4). Three other associated immune parameters displayed weaker associations: Bu1 expression on B cells, IgM expression on B cells, and CD45 expression on monocytes (Table 3). The analysis of each line separately showed that for TCRϒδ expression on TCRγδ T cells, the B21 line displayed the strongest association (p = 0.008), followed by the lines B14 and B15 (p values of 0.02 and 0.04 respectively), while no association was found within B2, B12, and B19 lines. In order to increase the statistical power of our analysis, we performed again this model on a subset of the three lines (B21, B14 and B15) considered together and on a subset of the three other lines (B19, B12, and B2), and represented the results on the total population on an NMDS plot (Supplementary Fig. S6). As expected, TCRϒδ expression on TCRγδ T cells was significantly associated to the Bray–Curtis distances of the caecal microbiota only in the total population (p = 0.0002) and the B21–B14–B15 sub-group (p = 0.0025). Considering the other immune parameters, the B21 and B19 lines were the only ones that displayed a significant association with Bu1 expression on B cells (respective p-values of 0.03 and 0.01), while the line B19 had an association with serum titer of IBV-specific antibodies (p = 0.02), and the line B21 had an association with CD45 expression on monocytes (p = 0.02).
Associations between immune phenotypes and microbiota composition of chicken with a varying infection severity after an IBV challenge
Finally, we searched for the bacteria that could be involved in these associations through Spearman correlation analysis that we calculated between the level of each immune parameters and the relative abundance of each OTU. We first calculated them on the entire population of vaccinated and non-vaccinated birds to search for associations present whatever the level of IBV infection (Fig. 5a; Supplementary Table S10). Then, we calculated it separately on the animal from the V-Ch group (Fig. 5b; Supplementary Table S11) and from the NV-Ch group (Fig. 5c; Supplementary Table S12) to identify associations specific of an acute infection with an inflammatory state group (NV-Ch) or of a rather sub-clinical infection (V-Ch).

Network representing the associations observed between the immune parameters and the OTU abundances in laying hens, one week after their infection with IBV. Associations were calculated with Spearman correlation in (a) the total population, (b) the vaccinated and challenged (V-Ch) group, and (c) the non-vaccinated and challenged (NV-Ch) group, with positive associations represented with green segments, and negatives ones with red segments. Only significant (p < 0.05), intermediate (total population: 0.4 < r < 0.6; V-Ch and NV-Ch groups: 0.5 < r < 0.7) and high (r > 0.6, thick segments) correlations are shown.
We only got few common associations between these three analyses. We observed negative associations between TCRϒδ expression on TCRγδ T cells and OTU from Escherichia-Shigella genus in the three datasets. Especially, Cluster 544 from this genus was observed in the three datasets, whatever the level of infection.
All the other associations we observed in the V-Ch group and the NV-Ch group implied quite different immune parameters and different OTU. The highest correlations were in the V-Ch group with, for instance, two strong positive correlations implying MHC-II expression on monocytes: one with Cluster 93 from Ruminoccocaceae UGC-014 genus (ρC = 0.63, adjusted-p = 0.001), and one with Cluster 279 from Ruminococcus 1 genus (ρ = 0.62, adjusted-p = 0.001).
Discussion
In the present study, we wondered if the vaccination and the IBV challenge of six chicken MHC lines were associated with differences in caecal microbiota composition. We report the observation of statistical associations between both the vaccination to IBV and the genetic line, and the caecal microbiota in a context of IBV infection. Animals were studied one week after an intranasal viral infection. This experimental IBV challenge enabled us to fully follow the individual capacity of birds to decrease the viral load and thus their level of protection and recovery, as serum IBV-specific immunoglobulin level per se is not a good predictor of vaccine-induced protective immunity31,32. Based on the tracheal IBV load after experimental infection, the B21 line appeared to show the most efficient response to IBV vaccination. We wondered whether the caecal microbiota could also display associations with the cellular immune profiles in peripheral blood. We first identified significant associations between the vaccine status (vaccinated challenged vs non vaccinated challenged) and the caecal microbiota composition on the one hand, and between the genetic line and the caecal microbiota on the other hand. We then investigated the associations between the caecal microbiota composition and immune phenotypes of the challenged animals in the total population and in the V-Ch and NV-Ch groups separately. A recent study reported the effect of nephropathogenic IBV (NIBV) infection on the ileal microbiota of Hy-Line brown chickens33. It was observed that NIBV disturbed the intestinal epithelium, with an hyperplasia of goblet cells, and a lowered ratio of the villus height to crypt depth. NIBV also disrupted the ileal microbiota with a reduction in the OTU richness, Firmicutes and Cyanobacteria phyla and of E.coli, and with an enrichment of phylum Proteobacteria and Bacteroidetes. Based on those results, it was hypothesized that the intestinal inflammation should have increased the availability of oxygen, which must have favored Proteobacteria bloom and intestinal dysbiosis. Although a direct comparison of our results with this study is not very relevant since the experimental design differs from our design in many ways, such results confirm our hypothesis that the gut microbiota composition may actually interact with the host response to IBV.
Effect of vaccination on caecal microbiota composition after IBV challenge
In the present study, the bacterial richness and the Chao1 estimator were lower in V-Ch animals, as compared to NV-Ch animals, especially in the Ruminococcaceae family. As we did not observe a significant effect on the Shannon index that is more sensitive to the more abundant OTU, we can hypothesize that the greater richness from the NV-Ch group could be due to its higher carriage of low abundant OTU. Our results are in accordance with other reports on viral infections caused by Influenza or the Respiratory Syncytial Virus (RSV)34,35. We also noticed that NV-Ch and V-Ch groups had a dissimilar microbiota, with differences observed on the OTU relative abundances between the two groups that were similar in all the lines. Globally, the vaccinated and challenged animals displayed a significant reduction of the abundance of OTU from the Ruminococcacea UCG-014 and Faecalibacterium genera, and a significant increase of the relative abundance of OTU from the Eisenbergiella genus, and to a lesser extent of Ruminococcus torques group, Tyzzerella 3, and Ruminoclostridium 9 or 5 genera. Due to the IBV viral challenge, we can assume that the vaccination effect we observed was mostly the consequence of a different level of infection between the unprotected control and vaccinated groups. Thus, we cannot exclude that the differences we observed on the caecal microbiota between the control and the vaccinated group may have resulted from the differences of those clinical states. There are only a few reports on the effect of vaccination on the intestinal microbiota, with observations made on uninfected individuals, and the results are contradictory34,36,37. Investigating the effect of vaccination on gut microbiota in a context of infection is thus of interest to improve vaccine design. Beside vaccination and animal clinical state, another possible source of microbiota variation that cannot be excluded is a possible difference in the environmental microbiota to which animals were exposed, since vaccinated and non-vaccinated animals had to be reared into two different units.
An association between genetic lines and caecal microbiota composition
We observed that the microbial communities of the six genetic lines were dissimilar in their global composition, as assessed by the comparisons of their Bray–Curtis distances and the differential analysis performed on their OTU abundances. In our experimental design, animals from the different lines were reared in the same pens and were thus pecking a litter containing faeces from all the lines38,39. The differences observed between lines therefore do not come from a different exposure to environmental microbes, thus strengthening the relevance of the association we observed between the host genetic background and the microbiota. Intestinal microbial dissimilarities have been reported among different poultry breeds and different broiler lines, for instance between lines genetically divergent for their digestive efficiency, or differing for their susceptibility to Salmonella Enteritidis and Campylobacter jejuni40,41. These microbial dissimilarities between lines of animals reared in the same environment could have resulted from physiological particularities related to their immune system42, or from differences in their digestive anatomy43,44,45,46. In the present study, the differences observed between lines could be due to genetic variations in the MHC locus but not only: the lines we studied are not isogenic. They may differ genetically for genomic regions other than the MHC locus, which could also have an effect on the caecal microbiota composition. Progenitors of the studied animals were reared in the same environment, so that we can discard the hypothesis made in other studies of a selecting pressure exerted by the environment through yolk antibodies38,47. This reinforces our hypothesis that genetic differences shaped the differences of caecal microbiota between lines through an action on immunity. At least one genetics study showed that several quantitative trait loci (QTL) controlling higher abundance of specific bacterial species might be involved in immunity48. The present lines differ for their MHC, which has a pivotal role in the adaptive immune response. In fact, MHC may shape the intestinal microbiota through its influence on the antibody response, on some CD4+ Th cell subsets, or on innate lymphoid cells that recognize commensal antigens48. Its involvement in the gut enterocytes, that secrete antimicrobial molecules able to influence microbiota composition, is also suspected48,49,50. Furthermore, differences in both affinity and number of amino acids recognized in binding pockets between alleles of MHC class I molecules in birds is a new concept that could explain the association between MHC haplotypes and some disease resistances51. According to this concept, MHC motifs can be considered as either “promiscuous” or “fastidious”. Alleles leading to a lower expression of MHC class I molecules having a lower affinity but a greater ability to bind a large variety of peptides are identified as promiscuous (generalist) against a wide variety of infectious pathogens. At the opposite, fastidious alleles display a higher expression of MHC class I molecules that can bind to a reduced variety of peptides, which can support an advantage against some specific pathogens. In a study on wild seabirds, but with limited sampling, an effect of the MHC on the gut microbiota was observed52. In stickleback fish, 6 out of 14 MHC motifs from class II molecules were significantly associated with the abundance of 7 bacterial families, and fishes with more diverse MHC motifs had less diverse gut microbiota49. Even if it is difficult to compare effects of the MHC on the gut microbiota between poultry and fish, which harbor quite dissimilar bacterial communities and environments, we can notice that the Enterobacteriaceae family was prone to sex-specific MHC effects in stickleback fish. Interestingly, in a similar way, we also observed in the present study that OTU from this family displayed higher relative abundance in the B14 and B12 lines. Furthermore, Enterobacteriaceae is a group of interest as many bacteria from this family are pathogenic and lead to intestinal inflammation53. In mice, 189 OTU displayed differential abundances between animals differing genetically at the MHC locus50. In poultry, and more specifically in layers, only one similar study was performed on the caecal microbiota from two congenic inbred lines that differ by their MHC genotypes54. Alpha-diversity was the only metric reported to describe the MHC effect, and as we observed in the present study, they did not notice any significant difference in Shannon’s diversity in the microbiota composition of the two lines. To our knowledge, there is no other publication about the association between MHC haplotypes and gut microbiota in poultry and the present study thus brings some promising elements to deeply investigate the effect and role of MHC in outbred populations, segregating for different MHC haplotypes.
A set of correlations between immune phenotypes and caecal microbiota composition
Larsen et al. reported a large variation in blast transformation between MHC-B lines for γδ T cells considering the same bird population29. In the present study, TCRϒδ expression on TCRγδ T cells was also the immune parameter with the greatest consistency among the different association analyses we performed between the microbiota and the immune phenotypes. It was particularly associated with the caecal microbiota of the B21, B14 and B15 lines that are of interest to further study potential interactions. TCRϒδ expression on TCRγδ T cells also shared negative associations with many OTU from the Escherichia-Shigella genus, whatever the level of infection in both the control and the vaccinated group, and especially with Cluster 544 from this same genus. Such consistency strengthens the likelihood of a biological link between a specific bacteria from the Escherichia-Shigella genus and TCRϒδ expression on TCRγδ T cells. Gamma delta T cells are a subpopulation of T lymphocytes with TCR composed by ϒ and δ chains. In adult chickens, ϒδ T cells are the dominant population among intestinal epithelial lymphocytes (20 to 60% of the lymphocytes)55, circulating T cells (20 to 50%), and splenocytes (30%)56. They are also contributing to the first line of defense against pathogens55. This population grows during infectious challenges with Salmonella enterica57, and demonstrates in vitro a capacity for cytokine and interferon production (IFNϒ, TNFα, IL17). Their dependency on APC and MHC restriction for antigen recognition remains uncertain, and some ϒδ T cells recognizing directly bacterial and viral epitopes were identified55. In mice, intestinal lamina propria, γδ T cells are the major source of IL-17, which is gut protective as it promotes the repair of damaged intestinal epithelium through the regulation of occludin subcellular localization to cell junctions58,59. Furthermore, some reports in mouse show that lipid antigens from E. coli are recognized by the hepatocytes CD1d, which alleviates hepatic γδ T-17 cell clones60. In the present study, we observed that several OTU from Eschericha-Shigella genus were negatively correlated with TCRϒδ expression on TCRγδ T cells, and were also less abundant in the B2 and B21 lines that were characterized as high responders to the IBV vaccination. Despite the absence of known orthologous gene of CD1d in the chicken genome, we can wonder if the CD1 paralogous genes could be involved in a similar mechanism61. Thus, in the present study, we can hypothesize that bacteria from the Escherichia-Shigella genus increased the systemic level of bacterial lipid antigens, which subsequently mitigated poultry γδ T cells.
Conclusion
In the present study, performed on inbred chicken lines with contrasted abilities to respond to IBV vaccination, we observed after an IBV infection that the different genetic lines harbored dissimilar microbiota. In addition to this role of the host genetics on the microbiota composition, we observed that the microbiota from the vaccinated chickens was highly dissimilar from the microbiota of the non-vaccinated ones. We hypothesized that a bi-directional crosstalk may have occurred between immunity and the gut microbiota. Vaccinated animals, that had developed good protective immunity, displayed a different immune response upon challenge, which may have dampened the shift of their gut microbiota during the infection. On the contrary, the non-vaccinated animals, which encountered IBV for the first time, encountered a higher immune perturbation, which in conjunction with dyspnea, polydipsia, and a reduced appetite may have fostered the shift of their gut microbiota. Regardless of the vaccine status and the ability to respond to IBV vaccination, we observed that some bacteria from the Escherichia-Shigella genus were always negatively correlated with TCRϒδ expression on circulating TCRγδ T cells and were less abundant in the B2 and B21 lines, that displayed the best vaccine efficiency. Those bacteria may affect the γδ T cell differentiation or proliferation, and thus their subsequent function e.g. cytokine production.
Methods
Animals
Offspring from 6 different inbred lines kept at Aarhus University and homozygous for the MHC haplotypes B2, B12, B14, B15, B19, and B21 were used for this experiment, as described previously26. Briefly, a group of 48 vaccinated chickens, with 8 chickens per line, was reared in two pens (24 chickens per pen, 4 chickens of each line per pen) in a unit of a bio-secure IBV-free facility. Vaccination was performed nasally at 2 week of age with a live attenuated IBV vaccine (Nobilis IB Ma5, MSD Animal Health) with a booster vaccination at 5 weeks of age. Another group of chickens with the same composition (n = 48; 8 chickens per line) was reared in another unit from the same facility, with separate entrances and strict biosafety precautions, but not vaccinated. All the vaccinated and non-vaccinated chickens were nasally challenged at 8 weeks of age with the IBV strain M41, as described previously29, generating two groups: vaccinated and challenged (V-Ch), and non-vaccinated and challenged (NV-Ch) (Fig. 1). Within the present population of 96 animals, all but 6 were females. The 6 males were distributed in different groups: 3 males from the B21 line (2 NV-Ch and 1 V-Ch) and 3 males from the B2 line (1 NV-Ch and 2 NV-Ch). Progenitors of the studied animals were reared in the same environment. Chickens were fed ad libitum with a wheat, soy, oat, corn, and barley diet (Paco Start 19, Forrådshuset, dlg, Denmark) adapted to their nutritional requirements as defined by the National Research Council requirements (NRC, 1994) (18.5% of crude proteins; 3.3% of crude lipids, 4.7% of crude fiber).
The experiment was conducted under protocols approved by the Danish Animal Experiments Inspectorate and complied with the Danish Ministry of Justice Law number 382 (10th June 1987) and Acts 739 (6thDecember 1988) and 333 (19th May 1990) concerning animal experimentation and care of experimental animals, following the described ethical guidelines. Ethics committee of Aarhus University approved the study, and license to conduct the animal experiment was obtained by DVM Ricarda Engberg, Aarhus University (license number 2017-15-0201-01211). The study was carried out in compliance with ARRIVE guidelines.
Samples collection
Tracheal swabs were collected at 0, 3, 4, and 5 days post IBV infection to assess IBV viral load. At 9 weeks of age, and 7 days post infection, the 96 animals were slaughtered by cervical dislocation in conformity with the Danish regulation for experimental animals. Animals were weighted before slaughter. After slaughter, caecal contents were gently removed from the caecal bags to avoid collection of mucosa and immediately frozen in liquid nitrogen before being stored at − 80 °C. EDTA-preserved blood samples were collected at the same time from the jugular vein for the immuno-phenotyping of all the chickens.
Immunoprofiling, serum titers and viral load
As reported previously29, serum IBV antibody titers were assessed by ELISA and peripheral blood leukocyte counts by flow cytometry one week post IBV infection. The following cell subsets were included in the analyses: heterophils (CD45+ SSChigh), monocytes (CD45+ Kul01+), thrombocytes (CD45+ CD41/61+), B cells (CD45 + Bu1 + MHCII + IgM +) and various T cell subsets (TCRγδ-CD4 + CD25 ± , TCRγδ-CD8β + CD25 ± , TCRγδ + CD8β + /− CD25 ±).
The tracheal IBV viral load was measured from tracheal swabs collected at 0, 3, 4, and 5 days post IBV infection, by RT-qPCR.
DNA extraction, amplification and sequencing
DNA was extracted from individual caecal contents, using a standardized protocol adapted from Godon et al62. DNA quantities were measured using a Qubit analyser, and DNA qualities using a Nanodrop analyser by measuring ratios of absorbances at 230, 260 and 280 nm. DNA samples were diluted at 15 ng/µL before PCR amplification. A 464 bp fragment targeting the hypervariable V3–V4 region from bacterial 16S rRNA gene was first amplified with the following primers: 460-F-iII-16S (adapter-CTTTCCCTACACGACGCTCTTCCGATCT-343-ACGGRAGGCAGCAG-357), and 460-R-iII-16S (adapter-GGAGTTCAGACGTGTGCTCTTCCGATCT-781-TACCAGGGTATCTAATCCT-806), in accordance with primer adapter previously reported by Lluch et al63. The first PCR reaction was conducted with 1 µL of genomic DNA and 0.5 µL of each primer (10 µM), 0.5 µL of dNTP mix (10 mM), 2.5 µL of 10X MTP Taq buffer mix (10 mM), 0.25 µL of MTP Taq DNA Polymerase (SIGMA-ALDRICH) and water qsp 25 µL. PCR conditions were: initial denaturation at 94 °C for 1 min, followed by 30 cycles of 94 °C for 1 min, annealing at 65 °C for 1 min, extension at 72 °C for 1 min, then a final elongation step at 72 °C for 10 min. We checked by agarose gel electrophoreses that each PCR generated a unique product size at the expected length. Purifications, second PCR with the Illumina adapters, and sequencing using a 2 × 300 bp MiSeq Illumina sequencer were performed by the GeT-PlaGe platform (INRAE, Toulouse). DNA from three samples could not be extracted, leading to a set of 93 animals with exploitable microbiota DNA. Raw sequences are available through the accession PRJNA658073 on the NCBI SRA (Sequence Read Archive).
Bioinformatic analyses
We used the FROGS pipeline (v3.0)64 to assemble read pairs, trim the 460-F-iII-16S and 460-R-iII-16S primers, and filter sequences to keep only amplicons with a length ranging from 300 to 490 bp. Unique sequences with their associated abundances in each sample were clustered with FROGS using Swarm (v 1.4.1)65, first with an aggregation distance of 1 and secondly of 3. FROGS removes chimera thanks to Vsearch (v2.6.0)66. Rare OTU were filtered out (abundance < 0.005% of total abundance)67 and the remaining sequences were annotated by NCBI Blast + (2.6.0)68 with an alignment on SILVA database restricted to 16S references (version 132 from 2017 update)69. We obtained an abundance table and then produced a phylogenetic tree using FROGS Tree tool with Mafft (v7.310)70 and Fasttree (2.1.10)71. We rarefied counts for α- and β-diversity analyses at 10,000 counts per sample. The unrarefied OTU table, the corresponding taxonomic classifications, and the metadata are available in the Supplementary Tables S13, S14, and S15 respectively.
Bio-statistical analyses
Microbiota taxonomic composition and diversity
Bio-statistical analyses were performed using R 3.5.2 and the Phyloseq 1.26.1 package. From the distribution of OTU counts, we described taxonomic composition at phylum, family and genus levels. To identify excluding or co-abundant genera, we calculated Spearman correlations between the abundances of each possible pair of genera on the overall dataset with the Hmisc 4.2.0 package. We represented on a network the moderate and high associations present in at least 20% of the sampled chicken, using the cutoff of 0.5 for the Spearman coefficient, and 0.05 for the adjusted p-value.
Effect of vaccination and chicken line on microbiota composition
We used the Vegan 2.5.5 package to evaluate the α-diversity by calculating the species richness, which is the number of observed OTU, and the Chao1, Shannon, and Inverse Simpson indexes that we represented for each vaccine group and each line. β-diversity indices were calculated using the Bray–Curtis (BC) dissimilarities. We described β-diversity in each line and represented it using a NMDS analysis (Non metrical Multidimensional Scaling). We first depicted the effect of vaccination by gathering the 6 chicken lines, and then in each line separately. We represented the vaccination impact in the different lines, adding on the NMDS ordination the centroids displacements from the control group to the vaccinated one in each line. To assess the effects of line and vaccination on α-diversity, we performed an ANOVA on α-indices. We used the vaccine status as a co-variable to assess the line effect, and the line as a co-variable to assess the vaccine-status effect. We did not correct the data for a “sex” effect, because : only 6 out of 93 animals were males, they were distributed in different groups, and we assessed with an NMDS analysis that the microbial communities of males and females were not dissimilar (Adonis R2 = 0.008, p = 0.54). Similarly, we did not correct the data for a putative pen effect, because in this experiment it was confounded with the vaccine status group. Significance threshold was set at 0.05 for the p-values. To assess the line and the vaccination effects on β-diversity, we used an Adonis model in Vegan. Similarly, we used the MetagenomeSeq 1.24.1 package to compare OTU abundances between each couple of lines, and between each couple of vaccine status groups. This package enables the normalization of raw sequences counts through a cumulative-sum scaling (CSS) method to reduce biases due to uneven sequencing depth72. We then used a zero-inflated Gaussian mixture model with vaccine status or line as main effect to identify, respectively, OTU with an abundance differing between each pair of lines, or rather between each vaccine status, with a threshold of 0.05 on the adjusted p-value (FDR). Analyses were performed independently at the species and genus levels.
Association between microbiota and immune phenotypes
We searched for associations between individual caecal microbiotas (OTU) and the immune phenotypes by calculating Spearman correlations between each immune parameter and the abundance of each OTU. To perform it, we used as starting point the normalized abundances from previous MetagenomeSeq normalization. As both the vaccination and the line had a significant effect on the microbiota, we collected residuals from linear models implemented on those normalized OTU counts with lines and vaccine status as main effects. Similarly, we collected residuals from linear models implemented on each immune parameter with the vaccine status and line as main effects. Then, these two sets of residuals were used to calculate Spearman correlation coefficients, and we set a cutoff of 0.05 on their adjusted p-values.
We first performed this analysis on the whole dataset, and then on the vaccinated challenged (V-Ch) and on the non vaccinated challenged groups (NV-Ch) separately. To analyse the V-Ch and NV-Ch groups separately, we collected residuals from linear models implemented on each normalized OTU count and each immune parameter with only the line as main effect. We thus assessed the vaccine effect from the comparison of the separate results. We represented the intermediate and high associations on a network, adjusting the cutoff on the Spearman correlation in order to keep at least 5 significant associations. Thus, we set a cutoff of 0.4 on the entire dataset, and a cutoff raised to 0.5 on the analysis performed in the V-Ch and the NV-Ch groups separately.
We also searched for associations between the microbiota and the immune phenotypes regarding the effect of microbiota β-diversity calculated with Bray–Curtis distances on each immune parameter. We used an Adonis model on the entire dataset with line and vaccine status as main effects. In order to identify the lines that contributed the most to the observed effects, we performed these analyses on each line separately by implementing the Adonis model only with the vaccine status as main effect. For the immune parameters that displayed the highest association with the microbiota, we represented their level according to the microbiota ordination on a NMDS plot.
Data availability
Raw sequences are available through the accession PRJNA658073 on the NCBI SRA (Sequence Read Archive), and the sequences of negative control were included as Additional File 1. The unrarefied OTU table, the corresponding taxonomic classifications and the metadata table have been included as Additional Files 2, 3, and 4 respectively.
Abbreviations
- AGP:
-
Antibiotics as growth promoters
- APC:
-
Antigen-presenting cells
- BC:
-
Bray–Curtis
- CAP:
-
Cationic antimicrobial peptide
- FDR:
-
False discovery rate
- IAV:
-
Influenza A virus
- IBV:
-
Infectious bronchitis virus
- ILCs:
-
Intestinal innate lymphoid cells
- IFN:
-
Interferon
- LPS:
-
Lipopolysaccharide
- MHC:
-
Major histocompatibility complex
- NMDS:
-
Non metrical multidimensional scaling
- NV-Ch:
-
Non vaccinated challenged
- PRRs:
-
Pattern recognition receptors
- QTL:
-
Quantitative trait loci
- RSV:
-
Respiratory syncytial virus
- TLR:
-
Toll like receptors
- V-Ch:
-
Vaccinated challenged
References
Hughes, L., Hermans, P. & Morgan, K. Risk factors for the use of prescription antibiotics on UK broiler farms. J. Antimicrob. Chemother. 61, 947–952 (2008).
Hocking, P. M. Unexpected consequences of genetic selection in broilers and turkeys: Problems and solutions. Br. Poult. Sci. 55, 1–12 (2014).
Saxena, V. K. Selection methods in poultry breeding: from genetics to genomics. In Application of Genetics and Genomics in Poultry Science (ed. Liu, G. K.) Ch. 2 (IntechOpen, 2018). https://doi.org/10.5772/intechopen.77966.
Cheema, M. A., Qureshi, M. A. & Havenstein, G. B. A comparison of the immune response of a 2001 commercial broiler with a 1957 randombred broiler strain when fed representative 1957 and 2001 broiler diets. Poult. Sci. 82, 1519–1529 (2003).
Yunis, R., Ben-David, A., Heller, E. D. & Cahaner, A. Immunocompetence and viability under commercial conditions of broiler groups differing in growth rate and in antibody response to Escherichia coli vaccine. Poult. Sci. 79, 810–816 (2000).
Han, F.-S. & Smyth, J. R. The influence of growth rate on the development of Marek’s disease in chickens. Poult. Sci. 51, 975–985 (1972).
van der Most, P. J., de Jong, B., Parmentier, H. K. & Verhulst, S. Trade-off between growth and immune function: A meta-analysis of selection experiments. Funct. Ecol. 25, 74–80 (2011).
Zerjal, T. et al. Assessment of trade-offs between feed efficiency, growth-related traits, and immune activity in experimental lines of layer chickens. Genetics Sel. Evol. 53, 1 (2021).
Mahfuz, S. U. et al. Inclusion of probiotic on chicken performance and immunity: A review. Int. J. Poult. Sci. 16, 328–335 (2017).
Marangon, S. & Busani, L. The use of vaccination in poultry production. Rev. Sci. Tech. Off. Int. Epiz 26, 265 (2006).
Shrestha, A., Sadeyen, J. R. & Iqbal, M. Enhancing protective efficacy of poultry vaccines through targeted delivery of antigens to antigen-presenting cells. Vaccines 6, 75 (2018).
Tarigan, S. et al. Field effectiveness of highly pathogenic avian influenza H5N1 vaccination in commercial layers in Indonesia. PLoS ONE 13, e0190947 (2018).
Francis, M. J. A veterinary vaccine development process map to assist in the development of new vaccines. Vaccine 38, 4512–4515 (2020).
Ciabattini, A., Olivieri, R., Lazzeri, E. & Medaglini, D. Role of the microbiota in the modulation of vaccine immune responses. Front. Microbiol. 10, 1305 (2019).
Lynn, D. J., Benson, S. C., Lynn, M. A. & Pulendran, B. Modulation of immune responses to vaccination by the microbiota: Implications and potential mechanisms. Nat. Rev. Immunol. 22, 33–46 (2022).
Cording, S., Medvedovic, J., Lecuyer, E., Aychek, T. & Eberl, G. Control of pathogens and microbiota by innate lymphoid cells. Microbes Infect. 20, 317–322 (2018).
Chaung, H.-C. et al. Salmonella flagellin enhances mucosal immunity of avian influenza vaccine in chickens. Vet. Microbiol. 157, 69–77 (2012).
Cui, Y. et al. Immunization of chickens with the enterobactin conjugate vaccine reduced campylobacter jejuni colonization in the intestine. Vaccines 8, 1–13 (2020).
Carrasco, J. M. D., Casanova, N. A. & Miyakawa, M. E. F. Microbiota, gut health and chicken productivity: What is the connection?. Microorganisms 7, 374 (2019).
Rubio, L. A. Possibilities of early life programming in broiler chickens via intestinal microbiota modulation. Poult. Sci. 98, 695–706 (2019).
Nothaft, H. et al. Coadministration of the Campylobacter jejuni N-Glycan-based vaccine with probiotics improves vaccine performance in broiler chickens. Appl. Environ. Microbiol. https://doi.org/10.1128/AEM (2017).
Tarabees, R., El-Sayed, M. S., Shehata, A. A. & Diab, M. S. Effects of the probiotic candidate E. faecalis-1, the Poulvac E. coli vaccine, and their combination on growth performance, caecal microbial composition, immune response, and protection against E. coli O78 challenge in broiler chickens. Probiotics Antimicrobial Proteins 12, 860–872 (2020).
Yitbarek, A. et al. Commensal gut microbiota can modulate adaptive immune responses in chickens vaccinated with whole inactivated avian influenza virus subtype H9N2. Vaccine 37, 6640–6647 (2019).
Loywyck, V., der Laan, M. H. P. & van, Goldringer, I. & Verrier, E.,. On the need for combining complementary analyses to assess the effect of a candidate gene and the evolution of its polymorphism: The example of the Major Histocompatibility Complex in chicken. Genet. Res. 87, 125–131 (2006).
Oviedo-Rondón, E. O., Hume, M. E., Hernández, C. & Clemente-Hernández, S. Intestinal microbial ecology of broilers vaccinated and challenged with mixed eimeria species, and supplemented with essential oil blends. Poult. Sci. 85, 854–860 (2006).
Wang, X. et al. Effects of Bacillus subtilis and coccidial vaccination on cecal microbial diversity and composition of Eimeria-challenged male broilers. Poult. Sci. 98, 3839–3849 (2019).
Miller, M. M. & Taylor, R. L. Brief review of the chicken Major Histocompatibility Complex: The genes, their distribution on chromosome 16, and their contributions to disease resistance. Poult. Sci. 95, 375–392 (2016).
Banat, G. R. et al. Association of the chicken MHC B haplotypes with resistance to avian coronavirus. Dev. Comp. Immunol. 39, 430–437 (2013).
Larsen, F. T. et al. Immunoprofiling of peripheral blood from infectious bronchitis virus vaccinated MHC-B chicken lines—Monocyte MHC-II expression as a potential correlate of protection. Dev. Comp. Immunol. 96, 93–102 (2019).
Ignjatovic, J. & Sapats, S. Avian infectious bronchitis virus. Rev. sci. tech. Off. int. Epiz 19, 493 (2000).
da Silva, A. P., Hauck, R., Zhou, H. & Gallardo, R. A. Understanding immune resistance to infectious bronchitis using major histocompatibility complex chicken lines. Avian Dis. 61, 358–365 (2017).
Gelb, J., Nix, W. A. & Gellman, S. D. Infectious bronchitis virus antibodies in tears and their relationship to immunity. Avian Dis. 42, 364–374 (1998).
Xu, P. et al. 16S rRNA gene sequencing reveals an altered composition of the gut microbiota in chickens infected with a nephropathogenic infectious bronchitis virus. Sci. Rep. 10, 1 (2020).
Groves, H. T. et al. Respiratory disease following viral lung infection alters the murine gut microbiota. Front. Immunol. 9, 182 (2018).
Nelson, A. M. et al. Disruption of the human gut microbiota following Norovirus infection. PLoS ONE 7, e48224 (2012).
Abdo, Z. et al. Longitudinal impact of oral probiotic Lactobacillus acidophilus vaccine strains on the immune response and gut microbiome of mice. bioRxiv https://doi.org/10.1101/691709 (2019).
Kajikawa, A. et al. Mucosal immunogenicity of genetically modified Lactobacillus acidophilus expressing an HIV-1 epitope within the surface layer protein. PLoS ONE 10, e0141713 (2015).
Kers, J. G. et al. Host and environmental factors affecting the intestinal microbiota in chickens. Front. Microbiol. 9, 1 (2018).
Wang, L., Lilburn, M. & Yu, Z. Intestinal microbiota of broiler chickens as affected by litter management regimens. Front. Microbiol. 7, 593 (2016).
Mignon-Grasteau, S. et al. Impact of selection for digestive efficiency on microbiota composition in the chicken. PLoS ONE 10, e0135488 (2015).
Schokker, D. et al. Early life microbial colonization of the gut and intestinal development differ between genetically divergent broiler lines. BMC Genomics 16, 1 (2015).
Nakajima, A. et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J. Exp. Med. 215, 2019–2034 (2018).
Rougière, N., Gomez, J., Mignon-Grasteau, S. & Carré, B. Effects of diet particle size on digestive parameters in D+ and D− genetic chicken lines selected for divergent digestion efficiency. Poult. Sci. 88, 1206–1215 (2009).
Schroeder, B. O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 7, 3–12 (2019).
Adolph, T. E., Mayr, L., Grabherr, F., Schwärzler, J. & Tilg, H. Pancreas–microbiota cross talk in health and disease. Annu. Rev. Nutr. 39, 249–266 (2019).
Ridlon, J. M., Kang, D. J., Hylemon, P. B. & Bajaj, J. S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 30, 332–338 (2014).
Hamal, K. R., Burgess, S. C., Pevzner, I. Y. & Erf, G. F. Maternal antibody transfer from dams to their egg yolks, egg whites, and chicks in meat lines of chickens. Poult. Sci. 85, 1364–1372 (2006).
Kubinak, J. L. et al. MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat. Commun. 6, 1 (2015).
Bolnick, D. I. et al. Major histocompatibility complex class IIb polymorphism influences gut microbiota composition and diversity. Mol. Ecol. 23, 4831–4845 (2014).
Wadud Khan, M. A., Zac Stephens, W., Mohammed, A. D., Round, J. L. & Kubinak, J. L. Does MHC heterozygosity influence microbiota form and function?. PLoS ONE 14, e0215946 (2019).
Kaufman, J. Generalists and specialists: A new view of how MHC class I molecules fight infectious pathogens. Trends Immunol. 39, 367–379 (2018).
Pearce, D. S., Hoover, B. A., Jennings, S., Nevitt, G. A. & Docherty, K. M. Morphological and genetic factors shape the microbiome of a seabird species (Oceanodroma leucorhoa) more than environmental and social factors. Microbiome 5, 1 (2017).
Ducatelle, R. et al. Biomarkers for monitoring intestinal health in poultry: Present status and future perspectives. Vet. Res. 49, 1 (2018).
Mon, K. K. Z. et al. Salmonella enterica serovars enteritidis infection alters the indigenous microbiota diversity in young layer chicks. Front. Vet. Sci. 2, 61 (2015).
Smith, A. L., Powers, C. & Beal, R. K. Chapter 13—The avian enteric immune system in health and disease. In Avian Immunology, 2nd edn (eds. Schat, K. A., Kaspers, B. & Kaiser, P.) 227–250 (Academic Press, 2014). https://doi.org/10.1016/B978-0-12-396965-1.00013-3.
Cooper, M. D., Chen, C.-L. H., Bucy, R. P. & Thompson, C. B. Avian T cell ontogeny. In Advances in Immunology (eds. Dixon, F. J. et al.) vol. 50, 87–117 (Academic Press, 1991).
Berndt, A., Pieper, J. & Methner, U. Circulating γδ T cells in response to Salmonella enterica serovar enteritidis exposure in chickens. Infect. Immunol. 74, 3967–3978 (2006).
Li, M. et al. Upregulation of intestinal barrier function in mice with DSS-induced colitis by a defined bacterial consortium is associated with expansion of IL-17A producing gamma delta T cells. Front. Immunol. 8, 824 (2017).
Lee, J. S. et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43, 727–738 (2015).
Li, F. et al. The microbiota maintain homeostasis of liver-resident γδ T-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 7, 1 (2017).
Salomonsen, J. et al. Two CD1 genes map to the chicken MHC, indicating that CD1 genes are ancient and likely to have been present in the primordial MHC. PNAS 102, 8668–8673 (2005).
Godon, J.-J. et al. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 63, 2802–2813 (1997).
Lluch, J. et al. The characterization of novel tissue microbiota using an optimized 16S metagenomic sequencing pipeline. PLoS ONE 10, e0142334 (2015).
Escudié, F. et al. FROGS: find, rapidly, OTUs with galaxy solution. Bioinformatics 34, 1287–1294 (2018).
Mahé, F., Rognes, T., Quince, C., de Vargas, C. & Dunthorn, M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2, e593 (2014).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59 (2013).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 1 (2009).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590 (2013).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202 (2013).
Acknowledgements
We are grateful to the INRAE MIGALE bioinformatics facility (MIGALE, INRAE, 2020. Migale bioinformatics Facility, https://doi.org/10.15454/1.5572390655343293E12) for providing help in computing, and storage resources. We thank Maria Bernard for support in bioinformatics with FROGS workflow, and GeT-PlaGe platform (INRAE) for 16S rRNA gene sequencing.
Funding
Marion Borey had a postdoctoral fellowship funded by Crédit Agricole d’Ile de France Mécénat. The Danish Council for Independent Research funded the animal experiments (Grant number 6111-00304B), and INRAE funded the 16S rRNA gene sequencing.
Author information
Authors and Affiliations
Contributions
T.D. conceived, designed, and coordinated the vaccination and challenge experiment (by Danish Council for Independent Research, grant number 6111-00304B). N.B. and M.B. produced the data relative to the microbiota, and F.L. to the immune phenotype. M.B. performed the bioinformatics and statistical analyses with J.E. supervision, M.B. realized the Figures and Tables. M.B. and F.C. contributed to the results interpretation, and M.B. wrote the manuscript. M.B., F.C., F.B., J.E., M.H.P., T.D., and B.B. contributed to the manuscript revision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borey, M., Bed’Hom, B., Bruneau, N. et al. Caecal microbiota composition of experimental inbred MHC-B lines infected with IBV differs according to genetics and vaccination. Sci Rep 12, 9995 (2022). https://doi.org/10.1038/s41598-022-13512-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13512-7
- Springer Nature Limited