
Abstract
Access to a comprehensive molecular alteration screening is patchy in Europe and quality of the molecular analysis varies. SPECTAlung was created in 2015 as a pan-European screening platform for patients with thoracic malignancies. Here we report the results of almost 4 years of prospective molecular screening of patients with thoracic malignancies, in terms of quality of the program and molecular alterations identified. Patients with thoracic malignancies at any stage of disease were recruited in SPECTAlung, from June 2015 to May 2019, in 7 different countries. Molecular tumour boards were organised monthly to discuss patients’ molecular and clinical profile and possible biomarker-driven treatments, including clinical trial options. FFPE material was collected and analysed for 576 patients with diagnosis of pleural, lung, or thymic malignancies. Ultimately, 539 patients were eligible (93.6%) and 528 patients were assessable (91.7%). The turn-around time for report generation and molecular tumour board was 214 days (median). Targetable molecular alterations were observed in almost 20% of cases, but treatment adaptation was low (3% of patients). SPECTAlung showed the feasibility of a pan-European screening platform. One fifth of the patients had a targetable molecular alteration. Some operational issues were discovered and adapted to improve efficiency.
Similar content being viewed by others

Introduction
Thoracic malignancies are the most common cause of cancer related death in the world. Lung cancers represent more than 11% of all cancer cases1, with more than 2 million new cases in 2020. Mortality linked to lung cancers reached 18% of all cancer deaths for both genders1, which represents around 388,000 deaths in Europe in 20182. The majority of lung cancers are diagnosed at an advanced stage and the mortality in patients diagnosed with lung cancer is above 80%, including all stages and histologies. Non-small cell lung cancer (NSCLC) is the most common histological type (84%3) comprising mainly of the adenocarcinoma and squamous cell carcinoma subtypes. Rarer thoracic malignancies include thymic carcinoma, mesothelioma, and neuro-endocrine tumours. The discovery of oncogenic driver mutations in NSCLC has improved the treatment landscape, moving it toward biomarker-driven treatments. Driver alterations include predominantly EGFR and KRAS, but also RET, BRAF, MET (exon 14 skipping) mutations, HER2 amplifications and ALK, ROS1, RET, NRG1 and NTRK1/2/3 fusions.
Across Europe, we observe a diversity of guidelines for biomarker testing for advanced NSCLC varying from recommended testing of a few genes and proteins (EGFR, ALK, ROS1 and PD-L1 in England) to testing 11 genes (including NTRK1/2/3 and ROS1) in France and the Netherlands4.
SPECTA, the European Organisation for Research and Treatment of Cancer (EORTC) translational research platform, launched a program in 2015 called SPECTAlung, to perform molecular screening for patients with thoracic malignancies. The goal was not only to better understand the molecular landscape of thoracic malignancies in Europe but also to provide access to biomarker testing for patients with no or limited access to comprehensive molecular screening technology.
Here we are reporting the results of the SPECTAlung program in terms of platform quality metrics, molecular landscape and actionability.
Results
SPECTAlung program quality metrics
The SPECTAlung program ran for almost 4 years and recruited 576 patients. The recruitment took place in 7 countries (Italy, Belgium, France, Slovenia, Spain, Poland and UK, Fig. 1a). At the central biobank, 36 quality failures (6.2%) were recorded for lung cancer (out of 577 samples from 516 screened patients), no FFPE QC failure for thymic carcinoma (38 samples for 36 patients), and only 1 for mesothelioma (3.8%, out of 26 samples received for 24 patients). Therefore 480 patients with NSCLC, 36 patients with thymic malignancy and 23 patients with mesothelioma were eligible in SPECTAlung.

Quality metrics for the SPECTAlung platform. (a) Map of recruiting countries and number of patients enrolled per country (b) Number of registered, eligible, and evaluable patients per disease (c) FFPE QC (left) and sequencing (right) failure per stage in NSCLC, at the sample level. Colour code on the graph and in the legend are in the same order. (d) Number of genes covered by each platform, including overlapping genes. (e) Turnaround time between patient registration and pathology FFPE QC (left) or molecular tumor board (right). The bar below the graph represents the different platform used.
For lung cancer patients, sequencing failures were also observed: 63 samples failed sequencing in total. However, as sites could re-send new material in case of sequencing failure, only 11 overall failures were observed at the patient level. Similarly, sequencing failure was observed for 1 thymic sample (but another sample was successfully analysed for this patient) and none for mesothelioma samples. Altogether, 469 patients were evaluable in the lung cohort, 36 patients with thymic malignancies and 23 patients with pleural mesothelioma (Fig. 1b). Overall, we observed only 8.6% of failure (at the patient level). Most failures were observed for samples from patients with stage IV NSCLC (15 out of 36 of sample QC failures, 42%, Fig. 1c, left panel and 24% of all screening failures, Fig. 1c, right panel).
Clinical data at baseline (demographics, medical history, current disease status) was fully completed for 96% of patients. For 21 patients (4%), missing data on patient medical history or primary disease and biomarkers was reported. Follow-up data was completed for all patients, with a median OS follow-up time of 21.4 (19.6, 22.6) months (Sup. Figure 1). At time of database lock, 269 patients were alive, 186 dead and 84 were lost to follow-up.
Samples were successfully analysed for 31 patients using the 14MG platform, 342 patients using the Oncomine focus panel and 145 patients with the Illumina TST-170 NGS assay. Samples from six patients (5 lung, 1 mesothelioma) were successfully analysed by both 14MG and Illumina and 4 lung cancer samples with Oncomine and Illumina.
Data on 45 genes were available across all three diagnostic panels (Fig. 1d and Sup. Table 1 for gene list). Therefore, for the combined molecular analysis, we focussed on those 45 genes, with the addition of TP53, which was not sequenced with Oncomine focus panel.
The median turn-around time (TAT) between patient registration in the program and sample passing QC at the central biobank was 18 days (Fig. 1e, left panel), but with a wide range (from 1 to 1405 days). This variability was mostly linked with delay in site shipping the FFPE material to the central biobank and the change in biobank that happened in August 2017. Similarly, the TAT from patient registration to molecular tumour board (MTB) was 214 days, ranging from 56 to 1545 days (Fig. 1e, right panel). This very long TAT can also be explained by the switch in central lab (14MG, Gustave Roussy, Almac Diagnostic) and was clearly an issue to inform treatment of advanced stage patients.
Patients characteristics
The SPECTAlung program enrolled samples form 539 individual patients, diagnosed with NSCLC (n = 480, 89%), thymic malignancies (n = 36, 6.7%) and pleural mesothelioma (n = 23, 4.3%). Patient characteristics can be found in Table 1. Overall survival (from date of cancer diagnosis) is displayed in Supplementary Fig. 1.
For NSCLC, median age at diagnosis was 65, the gender ratio of males to females was 1.5, and more than 75% were either current or former smokers, as expected for this patient population. 62.5% of NSCLCs were adenocarcinomas and 21.7% were of the squamous cell subtype. All stages were analysed within this program, and equally distributed (stage at registration: 28.5% stage I, 24.4% stage II, 23.8% stage III, 21% stage IV).
For thymic malignancies, median age at diagnosis was 49, with a 1.4 male to female ratio and around 50% of the patients were current or former smokers. Despite the small number of cases recruited in the program, we analysed samples representing most subtypes and stages.
Finally, our limited pleural mesothelioma population had a median age at diagnosis of 63, and a similar male to female ratio (1.55). Around 50% of the patients were former smokers, and 50% of the population had a known exposure to asbestos, the most important risk factor for this disease. Different subtypes of mesothelioma were analysed, from all stages.
Overall survival (OS, from date of cancer diagnosis) is displayed in Fig. 2. OS for NSCLC is as expected, however, due to few events in thymoma and mesothelioma populations, the curves are less informative.

OS Kaplan Meier curve by AJCC v7 Stage. together with a summary of associated statistics (median OS, 3-year OS rate estimates including the corresponding two-sided 95% confidence interval intervals (calculated by Greenwood formula’s estimation of the standard deviation for rates and by Brookmeyer and Crowley technique for the median). OS censoring markers were displayed. Cox’s proportional hazards model was fit by AJCC v7 stage. Hazard ratios (HR) with 95% confidence intervals were computed with AJCC v.7 stage I as reference stratum (HR = 1.00). Log-rank test was computed at 5% significance. NE: Not Estimated. (a) NSCLC. There was a significant difference between stage (p < 0.0001). (b) Mesothelioma. There was no significant difference between stage (p = 0.70). (c) Thymic malignancies. There was a significant difference between stage (p = 0.0498).
Molecular characterisation of the NSCLC, mesothelioma and thymic malignancies
Molecular analysis was successful for 480 patients, with clinically relevant molecular alterations (SNVs + INDELs + CNVs) detected in 286 samples. The top 20 altered genes (19 included in all 3 panels and TP53) are presented on the oncoplot (Fig. 3a). The most frequently altered genes were KRAS (mutated in 29% of adenocarcinoma and 1.9% of squamous cell carcinoma patients), EGFR (mutation in 6.7% of adenocarcinoma and 1.9% of squamous cell carcinoma but amplification in 2.3% of adenocarcinoma and 8.6% of squamous cell carcinoma patients) and TP53 (9.3% of adenocarcinoma and 21.1% of squamous cell carcinoma). PIK3CA is amplified in 13.4% of squamous cell carcinoma only. On the contrary, CDK4 amplification is only found in 5.3% of adenocarcinoma. The mutational pattern observed in our population is independent of stage for all histologies. Comparing with two other cohorts (TCGA5, composed mainly of early stage disease and MSKCC6, composed mainly of advanced stage disease), the top alterations from SPECTAlung are found within similar range, with the exception of TP53, that was altered more often in the TCGA and MSKCC cohort. Numbers of targetable driver alterations are comparable within the 3 cohorts, except for the low number of fusions identified within the SPECTAlung program (Fig. 3b). No co-mutations were observed between KRAS and EGFR in this cohort.

Molecular landscape of patients. Only clinically relevant molecular alterations (SNVs, indels, CNVs) are represented. (a) Top 20 altered genes (y axis) in NSCLC patients (sorted by histology on the x axis). The platform used to sequence each sample is also represented on the x axis. The bar plots above the graph represent the mutation rate for each sample. Alterations are color coded by type (SNV in green, deletion in dark blue, insertion in purple, amplification in red, loss in clear blue and multi-hit in brown. Note: (*) TP53 gene is not covered by Oncomine platform. (b) Alteration frequency for clinically actionable alterations for NSCLC patients from SPECTAlung (grey bar), MSK (yellow bar) or TCGA (blue bar) cohorts. Frequency calculations for SPECTAlung cohort are adjusted for TP53, ATM and KEAP1 genes to include only samples covered by platforms that screened those genes (Illumina &14MG for TP53 and ATM, 14MG only for KEAP1) (c) Comparison of the mutational landscape of mesothelioma samples in SPECTAlung with top altered genes in TCGA cohort (87 patients). Note: (*) TP53 gene is not covered by Oncomine platform, NF2 and BAP1 covered only by 14MG. Frequency calculations are adjusted for those genes. (d) Top altered genes in the SPECTAlung thymoma population and TCGA thymoma cohort (123 patients). Note: GTF2I oncogene is not covered by any panel used in this study.
A limited number of patients entering the SPECTAlung program had a diagnosis of mesothelioma (n = 23, 25 samples analysed) or thymic malignancies (n = 36, 37 samples analysed).
For mesothelioma, alterations were found in 12 out of 25 samples (48%). The most common alterations were BAP1 mutations (n = 5; of note, BAP1 was assessed only by the 14MG panel and only in the tumour specimen, no germline analysis), CDKN2A loss (n = 2) and NF2 mutation (n = 2; to note, NF2 was assessed only by the 14MG panel) (Fig. 3c). Despite the low number of mesothelioma samples in this cohort, the results are similar to the one from the TCGA cohort7 (Fig. 3c), where high prevalence for alterations in those 4 genes was also found.
For thymic malignancies, clinically relevant alterations were found in only 10 samples (out of 37 samples, 27%) (Fig. 3d): 3 MYC amplification, 2 amplifications in the cell cycle pathway (1 for CDK4 and 1 for CDK6), and 1 pathogenic alteration in KIT (p.Asp579del). However, when comparing with the top genes altered in the TCGA cohort7, no overlap was seen. Our cohort is extremely limited and importantly, GTF2I, the most altered gene in the TCGA cohort, was not assessed in any of the panels used in this program.
Molecular tumour board and treatment orientation
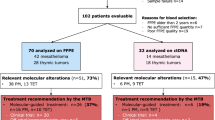
Molecular tumour board meetings were organised to discuss treatment options for every patient and were composed of expert clinicians, molecular biologists and pathologists. Treatment recommendations were possible only for 107 patients (19.9%) (Fig. 4a), mainly for lung malignancies (103 patients) but also for 1 mesothelioma patient and 3 thymic malignancies.

Actionability and limits of the platform. (a) Attrition rate between patient enrolled to treatment adaptation. Thymoma in red, NSCLC in clear blue and mesothelioma in dark blue. (b) Treatment recommendations for NSCLC patients. The different targetable alterations are color-coded and enlarged in the pie chart below. (c) Main reason for treatment adaptation (right) or absence of adaptation (left) per stage, for NSCLC patients and globally for other diseases.
These recommendations were based on a defined spectrum of molecular alterations that have well-known treatment options for NSCLC such as EGFR mutations, HER2 amplifications, MET exon 14 skipping or RET/ALK/ROS1 fusions (Fig. 4b). The IO recommendations (PD-(L)1 inhibitors) were based on a TMB above 16 (calculated only for Illumina analysis), following findings from the POPLAR and OAK trials.
From the 103 recommendations, adaptation of treatment was reported only for 16 patients (15.5% of cases). Reasons for not adapting treatment are displayed in Fig. 4c. The main reason for early stages (I to IIb) was that few recurrences were observed: 2 out of 15 patients with stage Ia (13.3%); 2 out of 10 patients with stage Ib (20%); none for patients with stage IIa; and 1 out of 10 patients for stage IIb (10%). While in later stages (IIIb and IV), the main reason was due to patient death: 12 out of 28 patients with stage IV (42.8%) and 1 out of 5 patients with stage IIIb (20%). For stage IIIa, both reasons applied: 4 out of 20 patients died (20%) and 5 out of 20 patients had no progression (25%).
Molecular alterations and smoking status in the population of patients with NSCLC
In our population, 81.6% of the patients were either former or current smokers at time of diagnosis. Our cohort confirmed the fact that most patients harbouring an EGFR mutation in exons 18, 19 or 21, are either never smokers (Fig. 5a, dark blue) or former smokers that stopped more than 15 years prior to diagnosis (Fig. 5a, light blue). Furthermore, statistical analysis confirmed association between non-smoking status and EGFR p.E746_A750del (p = 0.000028) and p.L858R (p = 0.0019) mutation. Only 2 active smokers with NSCLC (squamous cell histology) showed aberrations in EGFR, one with an EGFR amplification, the other a with a mutation in exon 6 (Fig. 5a, red). For EGFR exon 20, mutations were seen in both former smoker and never smoker population (Fig. 3a, orange and blue): 2 p.T790M mutation, 1 p.S768I mutation and 2 p.Ala767_Ser768insSerValAsp mutations. EGFR mutations were observed across all stages and no co-mutations in the EGFR gene were observed in this cohort.

Smoking status and molecular alterations. (a) EGFR mutations in exon 6, 18, 19 and 21 found in the current smokers (red), former smokers (yellow) and never smokers (blue). (b) KRAS mutations in exon 12, 13 and 61 found in the current smokers (red), former smokers (yellow) and never smokers (blue). (c) BRAF, TP53, MET mutations, JAK2 mutations and loss and fusions in ALK, RET and ROS1 found in the current smokers (red), former smokers (yellow) and never smokers (blue). NOTE: For alterations with *, BH adjusted p value from Fisher’s exact test was < 0.05.
The association between smoking status and KRAS mutations is different than for EGFR. 94 mutations were identified in KRAS, most of them on exon 2, either at the codon 12 position (85 out of 94), codon 13 (n = 5) or codon 61 (n = 4). The most prevalent mutations were p.G12C (n = 34), p.G12V (n = 25) and p.G12D (n = 18). The most prevalent mutations were observed in both smokers and non-smokers, with a higher prevalence in the first subgroup (Fig. 5b). As example, the p.G12C mutation, 17.6% of carriers were never smokers and 82.4% were current or former smokers, in line with the smoking profile of the global lung population of this program. The p.G12V mutation has a similar profile as the p.G12C mutation but with no specific trend toward smoking status (24% of carriers are never smokers).
For the p.G12D mutation, almost half of carriers are never smokers (44%). Looking in the TCGA cohort, this mutation was observed in 50% of non-smokers (10 out of 20) whereas the other codon 12 mutations were carried in 73% (p.G12C) and 74% (p.G12V) of smokers, in line with the global smoking prevalence of the TCGA population (28.2% of non-smoker and reformed smokers for longer than 15 years) (Supplementary Fig. 2a). Finally, a similar trend is observed in the MSKCC cohort with the p.G12D mutations found in 30% of non-smokers, whereas the p.G12C mutation was present only in 12% of non-smokers and p.G12V in 17% (Supplementary Fig. 2b).
Unlike with EGFR alterations, the association between other driver alterations and smoking status remains unclear (Fig. 5c). ALK and RET fusions seem to be predominantly associated with non-smokers, but only a limited number of fusions were detected in SPECTAlung (3 ALK, 8 RET and 1 ROS1). The BRAF mutations (1 p.V600E, 1 p.K601N, 1 p.G466A, 1 p.D594N, 1 p.G469R, 1 p.P299ThrfsTer) and JAK2 alterations were all identified in smokers, whereas TP53, MET and JAK3 mutations were identified independently of smoking habits.
Discussion
This pan-European platform, focusing on thoracic malignancies, ran for almost 4 years (June 2015–May 2019) and recruited 576 patients from 10 clinical sites, in 7 European countries.
The attrition rate due to sample QC failure or sequencing failure was limited, with more than 80% of the registered patients evaluable. This low failure rate might be explained by the relatively high volume of surgical specimens submitted for analysis (26.4% of samples) and repartition of disease stage with less than 25% of NSCLC patients with advanced disease. The completion of the database for patient baseline characteristics and follow-up is of good quality, with a median overall survival follow-up of almost 2 years and only 15.5% of patients lost to follow-up.
The molecular landscape of NSCLC was comparable to other studies (TCGA or MSKCC). EGFR mutations were found in 6.5% of NSCLC patients, which is somewhat lower than the general prevalence of EGFR alterations in NSCLC (around 10–15% in European cohorts8,9). In the MSKCC study, they found 26.9% of patients with EGFR alterations, probably due to a higher rate of never smokers in this study or preselection of subjects. One explanation for the lower prevalence in our cohort was that patients with a high chance of an EGFR mutation (e.g. adenocarcinoma and never smokers) might have had a prior local testing10, and only those with negative local testing or in the unusual event of no testing for EGFR being available were included in SPECTA4,11.
Very few genomic alterations were identified in patients with pleural mesothelioma and thymic malignancies due to the limited number of patients included and the small size of the NGS panel run in SPECTAlung. For mesothelioma, we found the same top altered genes as in the TCGA cohort, such as loss of CDKN2A or TP53, NF2 or BAP1 mutations, none of which have currently been linked to a possible treatment. CDKN2A loss has been correlated with worst outcome12 or proposed as a diagnostic tool, as observed in more than 75% of mesothelioma13. BAP1 is also frequently altered in mesothelioma, however a recent study highlights no difference in response to treatment (chemotherapy, immune checkpoint inhibitors or PARP inhibitors) between BAP1 wild-type or mutant mesothelioma14. For thymic malignancies, we were unable to confirm findings from TCGA as GTF2I was not part of any of the panels used in SPECTAlung and the sample size was very small. Moreover, we were also unable to repeats the molecular profile of the Arcagen studies (n = 23) for which several mutations were found in the RAS/RAF pathway15, similarly to what was found in the TCGA cohort. This clearly highlight the need for further fundamental and clinical research for rare cancers and possibly NGS panels adaptations.
The SPECTAlung platform faced important operational challenges, the main one being the long turn-around time from patient registration to delivery of molecular results (above 200 days on average). Workflow adaptations were made to try improving the overall quality and TAT. Three successive central laboratories performed the molecular analysis throughout the project and some improvement was noticed regarding workflow uniformity and robustness with a decrease of TAT variability, once the latest sample workflow was established (SD of 104 for Oncomine vs 62.3 for Illumina). However, despite those adaptations, the slow TAT impaired the actionability of treatment recommendations and in fine, the treatment adaptations based on molecular tumor board recommendations. Even though treatment recommendations were suggested for 103 patients (19.5%), mostly for NSCLC, adaptation was limited to 15.5% of the recommendations. The very high attrition rate between number of patients eligible in the platform (n = 480) to treatment adaptation (n = 16, 3.3%) might be linked to the fact that half of SPECTAlung patients had early-stage disease and did not progress within the timeframe of the project, or died prior to receiving the results (for patients with advanced disease) due to the very long TAT. Another important aspect is limitation to treatment access when outside standard of care and clinical trials. Finally, it is important to note that the use of three different molecular tests hinder the comparability of the SPECTAlung patients and therefore the final analysis.
SPECTAlung was one of the first pan-European academic screening platform and here we highlighted the challenges and successes encountered. Developing a pan-European screening platform, albeit limited to thoracic malignancies, paved the way for a more global pan-tumor screening program, which could help with personalised treatment and new platform trials, by reducing costs, ensuring quality and reproducibility of analysis, and minimizing turn-around time.
The platform was set-up and functional in more than 7 countries and was useful, especially in alterations present in multiple tumour types with a low prevalence. However, the operational set-up and the TAT were long and hindered the relevance of testing for patient treatment. A centralised testing system with timely MTB needs to be more efficient than local labs, should deliver a broader spectrum of genes being tested, have more flexibility in choice of panel and panel content, including the use of liquid biopsy to decrease TAT and help with FFPE sample failure, and include option for the patients to be recruited into clinical trials. During the course of the project, we adapted the screening panel, to increase the number of sequenced genes. However, in SPECTAlung, we never used liquid biopsy, even if its role is now well-established for lung cancers. Moreover, novel biomarkers are emerging, especially linked with Antibody–drug Conjugates and new targeted therapy. Panel adaptation, but also use of new technologies and combination of other screening methods (such as IHC for protein expression) might be needed in the future.
Based on this experience and from other European platforms (MAPPYACTS (Gustave Roussy, France), Master and INFORM protocol (DKFZ, Germany)), the SPECTA platform has been adapted toward a translational research platform, to increase the scope and improve its global efficiency. Beyond providing molecular results to cancer patients, SPECTAlung is also a source of clinically annotated molecular data, close to real-world data, that could prove extremely valuable as control-arm for future clinical research on biomarker of rare incidence.
The area of personalized medicine and complex trials is developing actively. Even in the light of immunotherapy, biomarkers identifying best responders and rationale for potential combination therapies are being developed. Drugs are now being approved based on tumor agnostic molecular abnormalities, so that screening has to cover all indications, not only the drivers related to a specific tumor type. Therefore, the need for and harmonized screening access in Europe, especially for smaller hospitals or countries where the screening is not systemic is crucial to allow access to such therapies to all patients. This is especially true for non-small cell lung cancer, as a disease with a high number of potential actionable molecular alterations.
Methods
Patients older than 18, with a diagnosis of any thoracic malignancy (including NSCLC, pleural mesothelioma and thymic malignancies) at any stage were enrolled in the program (NCT 02214134). All patients had provided written informed consent at the time of sample collection for genomic analysis. The SPECTAlung study was approved by several ethical committees (Ethische Commissie Onderzoek UZ/KU Leuven, in Belgium (S57513), Comité de protection des personnes "Ile-de-France VII", in France (15-030 (PP 15-001))).
Sample workflow
At registration, a FFPE sample was sent to the central biobank (Gustave Roussy, France or IBBL, Luxembourg). A pathology review was carried out to assess sufficient quality for molecular analysis, based on tumour content (more than 100 viable tumour cells and more than 10% tumour content). Samples used in this project were processed and stored under the management of the Integrated Biobank of Luxembourg (IBBL) following ISO17025:2005 standards and the ISBER Best Practices.
Workflows and screening platforms
Three screening platforms were used for SPECTAlung: 14 MG (spin off from the Sanger Institute, June to December 2015), Oncomine focus panel (ThermoFisher, December 2015 to August 2017) and Illumina TST-170 (September 2017 to November 2018). Molecular alterations were called using the 14MG proprietary pipeline for samples analysed by 14MG1. For samples analysed with Oncomine focus panel bioinformatics analysis were performed with Torrent Suite™ Server and Ion Reporter™ Server, which includes the Oncomine™ Variant Annotation Tool, according to Thermofisher standard recommendation. For samples analysed by Illumina, CNVs were called by the Illumina official pipeline but the EORTC pipeline was used for somatic variants and fusion calling. All methods were carried out in accordance with relevant guidelines and regulations. All genomic data generated for this study is available on EGA under study ID: EGAS00001004485 (https://ega-archive.org/studies/EGAS00001004485).
Genomic and clinical data can be shared upon request submitted to EORTC (Data sharing—EORTC : EORTC).
Clinical database
All clinical information was collected centrally, in EORTC clinical database. For patient registration, complete information on patient demographics, medical history, and current disease situation was required. Follow-up visits were done according to local standards, and follow-up data were submitted to EORTC.
Reporting and molecular tumour board
For all patients with molecular analysis results, a molecular report was shared with the treating-clinician and the case was discussed within an international virtual molecular tumour board (monthly), with local and expert clinicians, bioinformaticians and molecular biologists.
See supplementary material and methods for more information.
Data availability
All genomic data generated for this study is available on EGA under study ID: EGAS00001004485 (https://ega-archive.org/studies/EGAS00001004485). Genomic and clinical data can be shared upon request submitted to EORTC (Data sharing—EORTC : EORTC).
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. https://doi.org/10.3322/caac.21660 (2021).
Ferlay, J. et al. Cancer statistics for the year 2020: An overview. Int. J. Cancer https://doi.org/10.1002/ijc.33588 (2021).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. https://doi.org/10.3322/caac.21654 (2021).
Kerr, K. M. et al. The evolving landscape of biomarker testing for non-small cell lung cancer in Europe. Lung Cancer https://doi.org/10.1016/j.lungcan.2021.02.026 (2021).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. https://doi.org/10.1038/ng.3564 (2016).
Jordan, E. J. et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-16-1337 (2017).
Hoadley, K. A. et al. Cell-of-Origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell https://doi.org/10.1016/j.cell.2018.03.022 (2018).
Esteban, E. et al. Prevalence of EGFR mutations in newly diagnosed locally advanced or metastatic non-small cell lung cancer Spanish patients and its association with histological subtypes and clinical features: The Spanish REASON study. Cancer Epidemiol. https://doi.org/10.1016/j.canep.2015.02.003 (2015).
Zhang, Y. L. et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget https://doi.org/10.18632/oncotarget.12587 (2016).
Lindeman, N. I. et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Patho. J. Thorac. Oncol. https://doi.org/10.1097/JTO.0b013e318290868f (2013).
Janssens, A. et al. Routine implementation of EGFR mutation testing in clinical practice in Flanders: ‘HERMES’ project. Acta Clin. Belg. https://doi.org/10.1179/0001551214Z.00000000029 (2014).
Dacic, S. et al. Prognostic significance of p16/cdkn2a loss in pleural malignant mesotheliomas. Virchows Arch. https://doi.org/10.1007/s00428-008-0689-3 (2008).
Illei, P. B., Ladanyi, M., Rusch, V. W. & Zakowski, M. F. The use of CDKN2A deletion as a diagnostic marker for malignant mesothelioma in body cavity effusions. Cancer https://doi.org/10.1002/cncr.10923 (2003).
Dudnik, E. et al. BAP1-altered malignant pleural mesothelioma: outcomes with chemotherapy, immune check-point inhibitors and poly(ADP-Ribose) polymerase inhibitors. Front. Oncol. https://doi.org/10.3389/fonc.2021.603223 (2021).
Morfouace, M. et al. First results of the EORTC-SPECTA/Arcagen study exploring the genomics of rare cancers in collaboration with the European reference network EURACAN. ESMO Open https://doi.org/10.1136/esmoopen-2020-001075 (2020).
Acknowledgements
This publication was supported by Fonds Baillet Latour from Belgium and Walgreens Boots Alliance.
We would like to thank the patients, physicians, nurses, and trial coordinators who participated in the SPECTAlung project. From the EORTC headquarters, we would like to acknowledge Maarten Spruyt (DM), Alexandra Jouret (COM), and Maira Seini (RA).
Author information
Authors and Affiliations
Contributions
M.M., S.N. and A.S. lead the data analysis and the writing of the publication. C.D., U.J., T.B., R.D., T.G., E.F., L.P.A., J.M., M.O’B., P.B., J.V., L.L., A.C.D. participated to the conduct of the project and the writing and reviewing of the manuscript. V.G. and B.B. developed the project and found the funding. They also participated in the writing and reviewing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Dr. Morfouace, A. Stevovic, Dr. Gorlia, Dr. Golfinopoulos, Dr. Dooms, Dr. Janžič, Dr. Berghmans, Dr. O’Brien, Dr. Bironzo, Dr. Vansteenkiste and Dr. Lacroix declare no competing interests. Dr. MAZIERES reports: Personal fees from Roche, Astra Zeneca, Pierre Fabre, Takeda, BMS, MSD, Jiangsu Hengrui, Blueprint, Daiichi, Novartis, Amgen. Grants from Roche, Astra Zeneca, Pierre Fabre, BMS. Dr. DINGEMANS reports: Attending advisory boards and/or provided lectures for: Roche, Eli Lilly, Boehringer Ingelheim, Astra Zeneca, Pfizer, BMS, Amgen, Novartis, MSD, Takeda, Pharmamar. Receiving research support from BMS, AbbVie, Amgen (all paid to her institute). Dr. NOVELLO reports: Being an advisor/Speaker bureau: AZ, AMG, BI, Beigene, MSD, Eli Lilly, Roche, Takeda, Pfizer, Roche, Sanofi, Novartis. Dr. FELIP reports: Advisory Role or Speaker’s Bureau: Amgen, Astra Zeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, F. Hoffmann-La Roche, Glaxo Smith Kline, Janssen, Medscape, Merck KGaA, Merck Sharp & Dohme, Novartis, Peptomyc, PeerVoice, Pfizer, Regeneron, Sanofi Genzyme, Syneos Health, Seattle Genetics, Takeda, Touch Medical. Board member: Grifols, Independent member. Research Funding: Fundación Merck Salud, Grant for Oncology Innovation, Merck Healthcare KGaA. Dr. PAZ-ARES reports: Grants or contracts from any entity: MSD, Astrazeneca, Pfizer, BMS. Consulting fees: Lilly, MSD, Roche, Pharmamar, Merck, Astrazeneca, Novartis, Servier, Amgen, Pfizer, Ipsen, Sanofi, Bayer, Blueprint, BMS, Mirati. Payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events: Astrazeneca, Janssen, Merck, Mirati, Sanofi. Leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Genomica, Altum sequency. Dr. DZIADZUSZKO reports: Advisory role/compensated: AstraZeneca, Roche, Novartis, MSD, Pfizer, Boehringer Ingelheim, FoundationMedicine, Karyopharm, Takeda. Dr. BESSE reports: Sponsored Research at Gustave Roussy Cancer Center: 4D Pharma, Abbvie, Amgen, Aptitude Health, AstraZeneca, BeiGene, Blueprint Medicines, BMS, Boehringer Ingelheim, Celgene, Cergentis, Cristal Therapeutics, Daiichi-Sankyo, Eli Lilly, GSK, Inivata, Janssen, Onxeo, OSE immunotherapeutics, Pfizer, Roche-Genentech, Sanofi, Takeda, Tolero Pharmaceuticals.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morfouace, M., Novello, S., Stevovic, A. et al. Results of screening in early and advanced thoracic malignancies in the EORTC pan-European SPECTAlung platform. Sci Rep 12, 8342 (2022). https://doi.org/10.1038/s41598-022-12056-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12056-0
- Springer Nature Limited