
Abstract
Raoultella planticola is an emerging pathogen causing several infections in humans, and its roles in the propagation of antibiotic resistance genes (ARGs) remain uncharacterized. In this study, a carbapenem and tigecycline-resistant R. planticola isolate was recovered from hospital sewage. It carried nine plasmids, bearing 30 ARGs, including one blaKPC-2 and two blaNDM-1. It also contained a plasmid-borne efflux pump gene cluster, tmexCD1-toprJ, conferring resistance to tigecycline. Analysis of plasmid sequences revealed that both blaNDM-1-carrying plasmids were highly similar to those recovered from humans, reinforcing the close relatedness of environmental and clinical isolates. We also identified that plasmid bearing blaNDM-1 or tmexCD1-toprJ1 was transferable, and can be stabilized in the host bacteria, indicating that the R. planticola isolate has a considerable potential in the dissemination of ARGs. Besides, we found that this isolate could produce biofilm and was virulent in a Galleria mellonella infection model. In conclusion, our study shows the convergence of virulence and multidrug resistance in a R. planticola isolate. This potentially virulent superbug may disseminate into its receiving rivers, and finally to humans through cross-contamination during recreation activities or daily use of water, which poses a risk to public health.
Similar content being viewed by others
Introduction
The development of antibiotics provided an effective treatment for bacterial infections, and markedly reduced the health and economic burden associated with infectious diseases1. However, the rapid emergence and prevalence of antibiotic resistance has posed a great threat to the human and animal health at the global level2. Especially, the widespread of carbapenem- and colistin-resistant Enterobacterales represents a major challenge due to limited treatment options3,4. In this scenario, tigecycline, classified as a critically important antimicrobial by the WHO (World Health Organization), was considered as one of the last therapeutic options against infections caused by these bacteria5. Unfortunately, the clinical potential of tigecycline has been significantly compromised by the recent emergence of novel mobile tet(X) orthologs that confer high-level tigecycline resistance5,6. In addition, plasmid-borne tmexCD-toprJ homologs, encoding Resistance-Nodulation-Division (RND) family multidrug efflux pumps that confer tigecycline resistance, were also identified in Enterobacterales7,8. The emerging transferable tigecycline resistance determinants represent a new threat to global public health.
Antibiotic resistance is more than a clinical problem. The complex and close interactions between humans, animals, and environments have contributed to the propagation and spread of antibiotic-resistant bacteria (ARB) across all community sectors9. Addressing these issues, a holistic approach known as the One Health concept was proposed at the animal-human-environment interface to tackle the expanding antibiotic resistance and reduce the risk of infectious diseases globally10. The aquatic environment, especially hospital sewage, which is a mixing pool of antibiotic residues and ARB from nosocomial settings and excrement of patients, serves as a vast reservoir of ARGs11. With high concentrations of antibiotics and resistant organisms, hospital sewage also serves as a hotspot for horizontal gene transfer, enabling the exchange of ARGs between bacterial communities, ultimately generating multidrug-resistant (MDR) organisms12. Feng et al. reported the isolation of an Acinetobacter johnsonii strain harboring nine plasmids and encoding NDM-1 and OXA-58 carbapenemases from hospital sewage in 2010 in China13. Besides, hospital sewage also provides an ideal platform to generate new mobile elements, such as hybrid plasmids and novel transposons, via active genetic events, which facilitates a more flexible dissemination of antimicrobial resistance among bacteria14.
Raoultella planticola, belonging to the Enterobacteriaceae family, is a Gram-negative, non-motile, rod-shaped anaerobic bacterium15. It was first found by Freney et al. in 1984 in a patient with sepsis, and was originally classified as the Klebsiella spp., while it was reclassified as a member of the Raoultella genus based on 16S rRNA and rpoB gene analysis in 200116,17,18. This bacterium was primarily considered as harmless, environmental organism that mostly grows in soil and water16. However, in recent years it was identified as an emerging virulent pathogen due to its close association with many reports of severe human infections, including liver abscesses19, cholangitis20, pancreatitis21, conjunctivitis22, acutecholecystitis23 and urinary tract infections24. Though, factors contributing to the pathogenesis of Raoultella spp. remains largely uncharacterized. Besides, a number of MDR R. planticola isolates harboring carbapenemase genes, such as blaKPC-225, blaOXA-4826, blaNDM-127 and blaIMP-828, have been reported in humans, implying a possibly underestimated role of this bacterial species in the spread of antimicrobial resistance. The combination of MDR and hypervirulence would significantly limit options for treating severe infections, causing a particular threat for human health.
In this study, we report a carbapenem- and tigecycline-resistant R. planticola isolate harboring nine plasmids from hospital sewage. We aim to elucidate the transmission mechanisms of carbapenem and tigecycline resistance determinants by analyzing the plasmids carrying them. Also, we investigated the pathogenicity of the MDR R. planticola isolate, to our knowledge, have not been described previously.
Materials and methods
Bacterial isolation
R. planticola SCLZS62 was isolated during a study for the presence of carbapenem-resistant Enterobacteriaceae strains in hospital sewage. 5 ml of water sample was collected from the influx mainstream of hospital sewage at the affiliated hospital of Southwest Medical University, Luzhou in western China, in November 2019. As described in our previous study29, bacterial cells were concentrated by centrifugation at 5000g for 5 min. The sediment was resuspended in sterile 0.9% NaCl solution and plated onto MacConkey agar containing meropenem (2 μg/ml) and incubated for 24 h at 37 ℃. Pink colonies with various morphologies were picked and repeatedly streaked on new MacConkey agar plates to obtain pure isolates. Initial species identification was performed by PCR amplifying of 16S rRNA gene and Sanger sequencing30. The presence of the acquired carbapenemase genes, blaKPC, blaNDM, blaOXA-48, blaOXA-58, blaVIM, and blaIMP was screened via PCR assays using primers as previously described29.
Antimicrobial susceptibility testing
The minimum inhibitory concentrations (MICs) of amikacin, gentamicin, colistin, meropenem, imipenem, cefoxitin, chloramphenicol, ciprofloxacin, cefotaxime and tigecycline against the target strains were determined using the broth microdilution method according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (CLSI, 2017). Breakpoints for colistin and tigecycline were defined by European Committee on Antimicrobial Susceptibility testing (EUCAST) guidelines (http://www.eucast.org/clinical_breakpoints/). Escherichia coli ATCC 25922 was used as a quality control for MIC determination. All antibiotics used in this study were obtained from yuanye Bio-Technology Co. (Shanghai, China).
Conjugation assay
Conjugation experiments were performed using broth-based methods. The azide-resistant E. coli strain J53 was used as the recipient, and transconjugants were selected on Luria–Bertani (LB) agar plates containing 4 μg/ml meropenem or 4 μg/ml tigecycline plus 150 μg/ml sodium azide. Sixty-four and twenty-three transconjugants were randomly selected from meropenem- and tigecycline- containing plates for PCR assay, respectively. The presence of plasmids p1_SCLZS62, p2_SCLZS62, p3_SCLZS62, p4_SCLZS62, p5_SCLZS62, p6_SCLZS62, p7_SCLZS62, p8_SCLZS62 and p9_SCLZS62 in transconjugants was confirmed by PCR using the primers p1-F/R, p2-F/R, p3-F/R, p4-F/R, p5-F/R, p6-F/R, p7-F/R, p8-F/R and p9-F/R in Table S1.
Genome sequencing and analysis
Genomic DNA of R. planticola SCLZS62 was extracted and purified using the QIAamp DNA Mini Kit (Qiagen). The generated DNA was sheared with an average size of 10 kb and submitted for whole genome sequencing using a PacBio RSII sequencer (Pacific Biosciences, Menlo Park, USA). Meanwhile, the genomic DNA was sequenced on a HiSeq 2000 sequencer (Illumina, San Diego, CA, USA), using a paired-end library with an insert size of 150 bp. The de novo assembly of the PacBio reads was carried out with the Link v5.0.1. BWA-MEM was employed for mapping Illumina reads over the PacBio-generated contigs to correct the assembled contigs31. Library construction and sequencing was carried out at Beijing Novogene Bioinformatics Technology Co. Ltd. Annotation was carried out using Prokka32 and BLASTP searches against the UniProtKB/Swiss-Prot database. The species identification was carried out by average nucleotide identity (ANI) analysis with the online software JSpeciesWS (http://jspecies.ribohost.com/jspeciesws/#analyse). Digital DNA-DNA hybridization (dDDH) values were calculated using GGDC 3.0 server (http://ggdc.dsmz.de/distcalc2.php) by means of genome-to-genome sequence comparison33. Plasmid incompatibility types were analysed using the PlasmidFinder tool (95%, minimum threshold for identity; 60%, minimum coverage)34, and antimicrobial resistance genes were predicted using ResFinder (90%, minimum threshold for identity; 60%, minimum coverage)35. Insertion elements (ISs) and integrons were predicted using ISfinder36 and INTEGRALL37. The presence of virulence genes was investigated by searching the virulence factor database (VFDB) with an E value cutoff of 0.000138. The retrieved virulence genes were further screened with a cutoff of > 50% query coverage and > 75% identity. Multiple and pairwise sequence comparisons were carried out using the BRIG tool39. Gene organization diagrams were visualized with Inkscape 0.92.4 (https://inkscape.org/en/).
Phylogenetic analysis
Genomes were annotated using Prokka and the generated GFF3 files were used to create a core genome alignment with Roary. Single nucleotide polymorphisms (SNPs) were obtained with snp-sites v2.3.240. Based on the SNPs, A maximum-likelihood phylogenetic tree was constructed with FastTree version 2.1.1041. The presence of β-lactamase genes was detected by Abricate (https://github.com/tseemann/abricate). Carriage of β-lactamase genes and detail information of isolates were annotated on the tree using iTOL42.
Gene cloning
DNA fragment containing baeSR-tmexAB-toprM, tmexAB-toprM and baeSR-tmexAB were amplified from R. planticola SCLZS62 using the primers F1/R1, F2/R1 and F1/R2 in Table S1. The resulting PCR product was then ligated into a cloning vector pMD19-T using a ClonExpress® II One Step Cloning Kit (Vazyme, China) to give pMD19-baeSR-tmexAB-toprM, pMD19-tmexAB-toprM, pMD19-baeSR-tmexAB. The recombinant vectors were then electroporated into E. coli DH5α and verified by PCR assays. E. coli DH5α containing empty pUC19 served as a negative control for antimicrobial susceptibility assay.
Plasmid stability
The stability of p2_SCLZS62, p5_SCLZS62, p7_SCLZS62 and p8_SCLZS62 in R. planticola SCLZS62 was studied as previously described with little modification7. SCLZS62 was grown overnight in 3 ml LB (antibiotic-free) broth at 37 °C, and 3 µl overnight culture was then incubated into 3 ml fresh LB broth each day, yielding ~ 10 generations of growth per passage. The serial passage was lasted for 21 days. Every three days, cultures were serially diluted and plated on LB agar plates without antibiotics. To determine the percentage of plasmid-containing cells, ~ 100 colonies were screened on LB agar plates containing 4 μg/ml tigecycline or meropenem. The presence of p2_SCLZS62, p5_SCLZS62, p7_SCLZS62 and p8_SCLZS62 was confirmed by PCR assays using primers p2-F/R, p5-F/R, p7-F/R and p8-F/R, respectively, in Table S1. This experiment was performed in triplicate.
In vitro growth assays
Three independent cultures of J53 and transconjugants were grown overnight and diluted to 1:100 in LB broth. Bacteria cultures were incubated while shaking at 37℃. In the total period of 12 h, the value of optical density (OD) at 600 nm (OD600) was consistently recorded at an interval of 1 h with the iMark microplate Reader (Bio-Rad).
Biofilm formation assays
Biofilm formation assays were performed as described previously with minor changes43. Overnight cultures of R. planticola SCLZS62 was diluted 1/100 into fresh LB broth, and 200 μl of bacterial suspensions was inoculated into sterile 96-well microplates plates in triplicate. After 24 h at 37 ℃, culture supernatant was removed and the wells were washed with phosphate-buffered saline (PBS) and stained with 200 μl 0.1% (w/v) crystal violet solution at room temperature for 15 min. After that, the wells were washed with PBS for three times to remove excess stain and dry for 1 h. The bound dye was released by adding 100 ul of acetic acid (33%, v/v) and the optical density was measured at 595 nm using a microplate reader. NTUH-K2044, a hypermucoviscous and hypervirulent Klebsiella pneumoniae isolate belonging to the sequence type 23 and capsular type K1 from a patient with primary liver abscess44, and MG1655, a non-pathogenic E. coli K-12 strain45, were included as the positive and negative control strains, respectively. LB broth without any inoculation serves as a blank control. This experiment was performed in triplicate.
Galleria mellonella infection assays
The virulence potential of the R. planticola SCLZS62 was assessed using wax moth (Galleria mellonella) larvae weighing 250 to 350 mg (Tianjin Huiyude Biotech Company, Tianjin, China) as described previously with minor changes46. Overnight culture of R. planticola SCLZS62 from LB agar plates was harvested and adjusted using PBS to final concentrations of 1 × 106 CFU/ml, 1 × 107 CFU/ml, 1 × 108 CFU/ml and 1 × 109 CFU/ml. Ten larvae were chosen randomly as a group, and 10 μl aliquots of bacterial suspension was injected into the last left proleg of each larvae using a 50 μl Hamilton syringe. Larvae injected with the K. pneumoniae NTUH-K2044 were used as hypervirulence-positive controls, the E. coli MG1655 as low-virulence controls, and PBS as negative controls. The larvae were then incubated at 37 °C, and the number of live larvae was counted at 12 h intervals for 3 days. In all cases, no dead larvae were observed in the PBS groups. This experiment was performed in triplicate, and a representative result of three independent experiments was used to generate survival curves.
Statistical analysis
Data of biofilm formation assays were analyzed with a student t test performed in the GraphPad Prism version 8.3.0 (GraphPad Software, CA, USA). A two-way analysis of variance (ANOVA) was used to evaluate statistical significance in the bacterial growth assays. The survival rate of the G. mellonella was analyzed using the log-rank test. Differences were considered statistically significant at P < 0.05.
Results and discussion
Genome characterization of R. planticola SCLZS62
SCLZS62 was a MDR strain as it exhibited resistance to all tested antibiotics (≥ 3 classes of antimicrobial agents), except for colistin47 (Table 1). Whole genome sequencing showed that SCLZS62 had a chromosome of 5,490,846 bp, with a GC content of 55.88% and a total of 5185 open reading frames (ORFs). It also carried nine plasmids designated as p1_SCLZS62 to p9_SCLZS62, ranging in size from 10.5 to 334.2 kb and encoding 14 to 382 ORFs (Table 2). SCLZS62 belongs to R. planticola as it had 99.02% identity (89.81% query coverage) to the R. planticola reference strain FDAARGOS_64 by ANI analysis, and the dDDH value between them was 93.10%, both above the suggested cut-off for defining a bacterial species. In consistence with its multidrug resistance profile, SCLZS62 had 30 antibiotic resistance genes, mediating resistance to aminoglycosides (aac(6′)-Ib3, aadA5, ant(2″)-Ia, armA and rmtC), β-lactams (blaKPC-2, blaNDM-1, blaCTX-M-14, blaPLA2a and blaPER-1), macrolide (mph(A), mph(E) and msr(E)), fosfomycin (fosA and fosA3), quinolones (aac(6′)-Ib-cr, qnrA1 and qnrS1), rifampicin (ARR-3), sulfonamides (sul1), trimethoprim (dfrA1), and tetracycline (tet(A)). The detection of carbapenemase genes blaKPC-2 and blaNDM-1 explained the resistance to all the cephalosporin and carbapenem drugs tested. It was notable that R. planticola SCLZS62 contained three genes conferring resistance to carbapenems, including two blaNDM-1 and one blaKPC-2, which were carried by three different plasmids. In addition, it was observed that R. planticola SCLZS62 was resistant to tigecycline, and the recently characterized plasmid-mediated tigecycline resistance genes tet(X) variants were absent. Further analysis of the genome data showed that the newly identified efflux pump gene cluster, tmexCD1-toprJ1, involved in tigecycline resistance was identified on a plasmid in R. planticola SCLZS62, which probably contributed to the resistance of this isolate to tigecycline.
Phylogenetic analysis of R. planticola SCLZS62
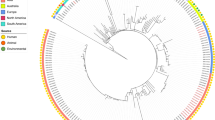
To determine a possible clinical relevance of SCLZS62, whole-genome sequences of 57 publicly R. planticola strains retrieved from GenBank (on 2021/09/02) were aligned with that of SCLZS62. A core genome-based phylogeny showed a diverse set of genomes, with a total of 655 core genes and 39,331 SNPs. These R. planticola isolates were mainly recovered from humans and the environment, and also found in animals, plant and insects across various countries. Phylogenetic tree showed that SCLZS62 was present in a cluster with two isolates AS012264 and AS012263 (Accession no. GCA_010598615.1 and GCA_010598665.1) from patients with lung disease in USA in 2016, with 1430 and 1428 SNPs, respectively (Fig. 1, Table S2). This result revealed a potential close clinical relevance of R. planticola SCLZS62. Resistance gene profiles of all R. planticola isolates showed a sporadical acquisitions of carbapenemase genes, with blaKPC-2 being the most frequent one, followed by blaNDM-1. SCLZS62 and another clinical isolate (GCA_013462275.1), also from China in 2018, represent the only two strains coharboring blaKPC-2 and blaNDM-1, with 1508 SNPs differences.

Phylogenetic tree of SCLZS62 with other 57 R. planticola genomes available from GenBank. The tree is based on 655 core genes with 39,331 SNPs, and the tree scale indicates substitutions per site. SCLZS62 is indicated in red. Resistance gene profiles are visualized in compliance to the tree. The annotation denotes (from left to right) isolation sources, locations, and years of strains. NA, not available. Detailed information of these R. planticola strains is presented in Table S2.
Genetic features of the bla NDM-1-harbouring plasmid p2_SCLZS62
p2_SCLZS62 is a 110,781-bp circular plasmid belonging to the IncFII(Yp) group, with a total of 125 annotated ORFs and an average GC content of 54.85%. BLAST search of the plasmid sequences against the GenBank database showed p2_SCLZS62 is highly similar (100% query coverage and > 99.9% nucleotide identity) to several blaNDM-harboring plasmids recovered from humans (Figure S1), such as pRJF866 (GenBank accession no. KF732966) from K. pneumoniae (China, 2014), pNDM-SCNJ07 (Accession no. MK933278) from Enterobacter hormaechei (China, 2019), pNDM-Ec1GN574 (Accession no. KJ812998) from Enterobacter cloacae (Canada, 2014), pEC4-NDM-6 (Accession no. KC887916) from E. coli (New Zealand, 2013), and pNDM_4TM (Accession no. MF042352) from Serratia marcescens (Romania, 2015). In all of these plasmids, blaNDM was located in a ~ 6.0 kb region bracketed by two copies of Tn3-derived inverted-repeat transposable elements (TIMEs, bases 6434 to 6689 and 23,920 to 24,175 of p2_SCLZS62), which contribute to the mobilization of blaNDM. In addition to blaNDM-1, p2_SCLZS62 also harbored resistance genes rmtC and sul1, which were located in a 11.5-kb region upstream of blaNDM-1 that was also flanked by two copies of TIMEs.
Genetic features of the bla NDM-1-harbouring plasmid p5_SCLZS62
Sequence analysis of p5_SCLZS62 indicated that it was a circular IncC-type plasmid of 173,509 bp with 213 predicted ORFs. Based on the presence or absence of orf1832/orf1847, rhs1/rhs2, i1, and i2, which are key features distinguishing between type 1 and type 2 IncC plasmids, p5_SCLZS62 was recognized as the type 1 IncC plasmid48. A BLAST search against the GenBank database showed that p5_SCLZS62 was almost identical (100% coverage and ≥ 99.98% identity) to several plasmids recovered from humans, including pGD31-NDM (Accession no. CP031297) in E. coli from Viet Nam and pSAL-19–0623 (Accession no. MN604267) in Salmonella enterica from Singapore (Fig. 2A).

Genetic features of p5_SCLZS62. (A) Circular comparison of p5_SCLZS62 with type 1 IncC plasmids. The complete sequence of p5_SCLZS62 was used as the reference. Arrows on the outer ring indicate deduced ORFs and their orientations. The replication gene repA is highlighted in red, and genes for conjugal transfer are indicated in blue. Two accessory resistance regions (ARI and Efflux pump region) are indicated by green curves. (B) Organization of the ARI of p5_SCLZS62, and comparisons to related regions. Genes are denoted by arrows, and are colored based on their functional classification. Regions of > 85% homology are indicated by grey shadings. The accession numbers of Tn1696 and Tn125 for reference are U12338 and JN872328, respectively. Δ represents truncated genes. CS, conserved segment; VR, variable region. (C) Comparison of the efflux pump region of p5_SCLZS62 with those of closely related sequences. The baeSR-tmexAB-toprM gene cluster is highlighted in red, and remaining genes in this region are indicated in green. The gene uvrD, being indicated in yellow, is interrupted by the insertion of the efflux pump region, leaving 6-bp direct target repeats (CTCTAC). Other plasmid backbone genes and hypothetical genes are colored gray. Regions of > 80% homology are indicated by grey shadings.
Compared with the type 1 prototype plasmid pR148, p5_SCLZS62 shared a 128-kb backbone, but the content of antibiotic resistance island, designated ARI, was varied (Fig. 2A). It is notable that all resistance genes of p5_SCLZS62, including ant(2″)-Ia, qnrA1, sul1 and blaNDM-1 are located in the ARI, with the structure of a novel unit transposons, designated Tn7353, being inserted at a site within a gene named orf3 encoding a putative permease in the plasmid pKPHS3 (Accession no. NC_016839) from clinical K. pneumoniae isolate (Fig. 2B). The Tn7353 was a derivative of Tn1696, with identical tnpA-tnpR and 89.57% nucleotide identity of the mer gene cluster, but differed from it mainly by insertion of a complex class 1 integron instead of In4 in Tn1696. The complex class 1 integron consists of one 5′-CS, three 3′-CSs, two ISCR1s, and three VRs bearing resistance genes. blaNDM-1 was located in the ISCR1-1-linked VR-2, with the structure of ΔTn125 consisting of ΔgroL, groS, ΔcutA, dsbD, trpF, ble, blaNDM-1 and ΔISAba125 in order, within In7p5_SCLZS62. According to a previous report, ISCR27 may be involved in the initial acquisition and mobilization of blaNDM-1, and then Tn125 was commonly associated with the wide dissemination of blaNDM-1 in bacteria49. The presence of ISCR1 downstream of the truncated Tn125 in this complex class 1 integron suggests an important role of ISCR1 in the evolution and further movement of the blaNDM-1 gene, by mobilizing the ΔTn125 segment in this situation. The construction of the complex class 1 integron harboring multiple resistance genes by the introduction of two ISCR1 elements revealed significant roles of mobile elements like ISCR1 in the creation and evolution of multidrug- and pandrug-resistant regions47, which poses a big challenge in fighting against antibiotic resistance. By sequence comparison, the ARI of p5_SCLZS62 showed high similarity (99% query coverage and 99.98% identity) to the corresponding region of pGD31-NDM, which was also identified as a derivative of Tn1696 termed Tn7352, with some deletions: (1) A 3577-bp deletion of the cassette array, blaOXA-4-aadA2-strA-strB, within the class 1 integron, which probably resulted from homologous recombination via the 5′-CS of the class 1 integron; (2) two deletions at the ISCR1-1-associated VR-2, including a 7856-bp deletion downstream of the ISCR1-1 and a 1780-bp deletion downstream of blaNDM-1. We speculated that ARI of p5_SCLZS62 might have progressively evolved from the genetic structure like Tn7352 by experiencing multiple genetic events.
In addition to the ARI, p5_SCLZS62 also contained an 15-kb region harboring a novel efflux pump gene cluster, designated tmexA-tmexB-toprM (“t” for transferrable7), when it was compared with pR148. The efflux pump region had an ents-toprM-tmexB-tmexA-baeR-baeS-dgcT-rhaS-ywfM-orf-tyrS structure, which was inserted into the uvrD gene, splitting it into two separate parts and meanwhile leaving 6-bp direct repeats (DRs, CTCTAC) at both ends (Fig. 2C). A BLASTn search in the GenBank database showed that the efflux pump region was also found in plasmids pIncHIB (Accession no. CP036336) in K. pneumoniae from India, and pM216_AC2 (Accession no. AP018145) in E. coli from Japan. Specially, the plasmid-borne efflux pump gene cluster baeS-baeR-tmexAB-toprM was found to be an 88.87% match to a chromosomal DNA fragment of Shewanella baltica strain CW2 (Accession no. CP028355), raising the possibility that this efflux pump gene cluster originates from certain species of Shewanella, such as S. baltica. Despite the presence of perfect DRs, there was no mobile element found in the efflux pump region. The transposition mechanism of this region requires further elucidation.
Sequence analysis showed that this novel efflux pump tMexA-tMexB-tOprM, showed 36.02%, 55.6%, 46.04% amino acid identity to the MexA, MexB, OprM encoded on the chromosome of Pseudomonas aeruginosa, respectively. This gene cluster tmexAB-toprM is adjacent to ORFs annotated as a two-component regulatory system encoded by baeS and baeR, indicating an intact gene cluster encoding an efflux pump. To validate the function of tmexAB-toprM, three recombinant plasmids: pMD19-baeSR-tmexAB-toprM, pMD19-tmexAB-toprM (missing baeSR), pMD19-baeSR-tmexAB (missing toprM) were constructed. Relative to those with the empty vector pUC-19, none of the E. coli DH5α strain carrying recombinant plasmid showed increase in the MICs of the tested antimicrobial agents (Table S3), suggesting that this tmexAB-toprM failed to function as an efflux pump system. Additional studies are needed to fully characterize the biological functions of the tmexAB-toprM gene cluster.
Genetic features of plasmid p7_SCLZS62
p7_SCLZS62 has 334,207-bp circularly closed DNA sequences, and carries 541 predicted ORFs in total. p7_SCLZS62 belongs to IncHI5 group because it contains a replication gene repHI5B, and an additional repFIB-like gene. A BLASTn search against the GenBank database showed the highest 87% query coverage, and 99.93% identity to the plasmid pJNQH579-2 (Accession no. CP078148) in Klebsiella variicola from a clinical sputum specimen in China. Genomic comparison of p7_SCLZS62 with two IncHI5 reference plasmid pKOX_R1 (Accession no. CP003684) and p11219-IMP (Accession no.MF344561) showed that it possess conserved backbones, including repHI5B (together with its iterons) and repFIB-like gene for replication, parAB for partition, and two tra regions (tra1 and tra2) for conjugal transfer50(Fig. 3A). This plasmid harbors numerous antibiotic resistance genes (Table 2), as well as a newly identified RND efflux pump gene cluster, tmexCD1-toprJ1, which confers transferable resistance to tigecycline7.

Genetic features of p7_SCLZS62. (A) Circular comparison of p7_SCLZS62 with IncHI5 plasmids. The complete sequence of p7_SCLZS62 served as the reference. Arrows on the outer ring indicate deduced ORFs and their orientations. The replication gene repA is highlighted in red, conjugal transfer genes in blue, and parAB for partition in yellow. Three accessory regions MDR-1, MDR-2, and Efflux pump region are indicated by green curves. (B) Organization of the MDR-1 of p7_SCLZS62, and comparison to related regions. The accession numbers of In469 and Tn6381 for reference are KP076293 and MF344566, respectively. (C) Organization of the MDR-2 of p7_SCLZS62, and comparison to related regions. The accession numbers of Tn6279 and Tn6400 for reference are KT317075 and KU318421, respectively. (D) Organization of the efflux pump region of p7_SCLZS62, and comparison to related regions. The accession numbers of Tn5393, Tn1721, Tn1696, Tn6347, Tn6361 and Tn6344 for reference are M96392, X61367, U12338, MF344562, KM660724 and MF344567, respectively. Genes are denoted by arrows, and are colored based on their functional classification. Regions of > 90% homology are indicated by grey shadings. Δ represents truncated genes.
p7_SCLZS62 carries three accessory modules, namely the MDR-1 region, MDR-2 region, and efflux pump region. The MDR-1 region was identified as a Tn6381 derivative, which was inserted at a site within an intl1 gene, breaking it into two separate parts Δintl1-5′ and Δintl1-3′ (Fig. 3B). The Tn6381 derivative was generated from insertion of IS26-ΔIn469-IS26 unit into a site downstream of tnpA, leading to a 651-bp deletion in the Tn3-family core transposition module tnpA-res-tnpR, and a truncation of both tnpA and tnpR. The MDR-2 region consists largely of a ΔTn6279-like transposon, and the rest was a tnpA-tnpR module with its terminal 38-bp IRL interrupted by IS5075 and a res remnant res-5′ (Fig. 3C). As a derivative of Tn6279, the ΔTn6279-like transposon had major modifications including an insertion of ISKpn21 and ISAs27, a deletion of ISAba24 and the terminal IS26-aph(3′)-Ia-IS26 unit, and a replacement of In438 by In797. The efflux pump region was identified as a complex chimera structure composed of a Tn6347-like remnant, two IS26-composite transposon-like units (IS26-mph(A)-IS26 and IS26-fosA-IS26), ΔTn1721, ΔTn6361, In268, ΔTn1696, Tn6344 and the efflux pump module (ΔtnpA-int-int-infB-psrP-tnfxB1-tmexC1- tmexD1-toprJ1-ΔtnpA- tnpR) that was most likely to be derived from Tn5393 (Fig. 3D). The efflux pump module in p7_SCLZS62 shared high similarity (95% coverage, 99.97% identity) to that in pHNAH8I-1, the first reported tmexCD1-toprJ1-harboring plasmid7. Unlike the strA-strB of Tn5393-3′ retained in the pHNAH8I-1, forming tnpR-strA-strB, a Tn6361 remnant was located downstream of tnpR in p7_SCLZS62, and formed the tnpR-qnrS1-IS26 structure. The acquisition of ΔTn6361 in p7_SCLZS62 was most likely to result from massive recombination events between two copies of tnpR.
Genetic features of the bla KPC-2-harbouring plasmid p8_SCLZS62
Plasmid p8_SCLZS62 is a circular molecule of 42,847 bp with an average GC content of 49.72%, and harbors 62 predicted ORF. It could not be assigned to any known incompatibility group, and the blaKPC-2 gene was the sole resistance determinant of this plasmid. Full plasmid sequence queries of p8_SCLZS62 against the NCBI GenBank showed that it has high similarity (> 99% query coverage and > 99% nucleotide identity) to pHS062105-3 (Accession no. 023331), pCP40 (Accession no. MH328006), pP10159-3 (Accession no. MF072963) and pCKPC18-1(Accession no. CP022276), which were recovered from humans and the environment in different regions of China (Figure S2). This result reinforced the important role of the pCKPC18-1-like untypeable plasmids in the dissemination of blaKPC-2 in Enterobacteriaceae in China51,52,53. The backbone of these plasmids include an untypeable replication initiation gene (repA), parA and topB for stability, relEB genes for maintenance, and tviB for conjugation. In the accessory region, blaKPC-2 was carried by a ΔTn6296 genetic platform, in which ΔTn1722-3′ was lost from the prototype Tn6296 (Fig. 4).

Organization of genetic context of blaKPC-2 and comparison to related regions. Genes are denoted by arrows, and are colored based on their functional classification. Regions of > 90% homology are indicated by grey shadings. The accession numbers of Tn1721 and Tn6296 for reference are X61367 and FJ628167, respectively. Δ indicates truncated genes.
Plasmid transferability, stability and host fitness
Conjugal transfer experiments showed that carbapenem resistance determinants could be transferred into E. coli J53 at the frequency of ~ 10–6 (transconjugant/recipient). Among the transconjugants, cells containing p5_SCLZS62 accounted for 73.44%, and p8_SCLZS62 only for 4.69%. Specially, all the transconjugants harbored p2_SCLZS62. This result indicates that these three plasmids carrying carbapenemase genes are mobilizable, with the highest transferability of p2_SCLZS62, and the least efficiency of p8_SCLZS62. MICs of carbapenem (including meropenem and imipenem) and cephalosporin (including cefoxitin and cefotaxime), of these transconjugants, were increased at least 128-fold (Table 1). In addition, p7_SCLZS62 was able to conjugate into E. coli J53 at a frequency of ~ 10–8, which is consistent with its intact set of conjugative transfer genes. Antimicrobial susceptibility analysis revealed that the acquisition of p7_SCLZS62 enabled E. coli J53 to become resistant to tigecycline by at least 8-fold (Table 1). To estimate the fitness cost of these resistant plasmids, we compared growth characteristics of the transconjugants harboring resistant plasmids, and the plasmid-free recipient strain J53. Results showed that transconjugants JM-5 and JT-4, which bear plasmids p2_SCLZS62, p5_SCLZS62 and p7_SCLZS62, exhibited a significant growth retardation compared to J53, indicating that the coexistence of these three plasmids confers a fitness cost on the host (Fig. 5A).

Characterization of the host fitness and stability of resistant plasmids. (A) Growth curves for the transconjugants and the recipient strain J53. (B) Stability of plasmids p2_SCLZS62, p5_SCLZS62, p7_SCLZS62 and p8_SCLZS62. Data were expressed as means ± standard deviations. Error bars show SDs, and asterisks indicate statistical significance using two-way ANOVA (***P < 0.001, ****P < 0.0001).
Stability assays showed that both carbapenem and tigecycline resistance determinants were stable for ≥ 21 d (~ 210 generations) in the R. planticola SCLZS62 in antibiotic-free medium. Among the carbapenem-resistant colonies, plasmids p2_SCLZS62 and p5_SCLZS62 were stably maintained with 100% retention, while p8_SCLZS62 was gradually lost during serial passage with 35.76 ± 5.35% retention after 21 days (Fig. 5B). The transferability and highly stability of plasmids containing blaNDM-1 or efflux pump gene cluster tmexCD1-toprJ1 reinforced the idea that MDR strains like R. planticola SCLZS62 in the aquatic environment serve as a reservoir of ARGs, which has serious public health implications.
Virulence characteristics of R. planticola SCLZS62
Biofilm formation assays showed that the biofilm-forming ability of SCLZS62 was significantly higher than that of E. coli MG1655, and comparable to that of K. pneumoniae NTUH-K2044 (Fig. 6A). The pathogenic potential of SCLZS62 was determined by Galleria mellonella infection testing. Results showed that the survival rates of G. mellonella significantly decreased when infected with strain SCLZS62 or NTUH-K2044 in relative to those with MG1655 under various infection concentrations (Fig. 6B–E). These findings suggested that SCLZS62 is a biofilm producer, and potentially virulent. In silico analysis detected 38 virulence genes in R. planticola SCLZS62 (Table S4), including gene clusters encoding type 3 fimbriae (mrkABC) and type 1 fimbriae (fimACDEGH), which contribute to biofilm formation, and adherence to host cells54. Also, genes encoding aerobactin (iutA) and ent siderophore (entABCES and fepABCG) for iron acquisition, and gnd for serum resistance were identified, which could enhance the ability of the bacteria to survive and colonize within the host55. Additionally, several sets of genes encoding type VI secretion system were included, such as dotU/tssL, hcp/tssD, icmF/tssM and impA/tssA, vipB/tssC, vasE/tssK, which play an important role in the invasion and pathogenicity during the infection process of pathogens56.

Virulence characteristics of R. planticola SCLZS62. (A) Biofilm formation assays of R. planticola SCLZS62, K. pneumoniae NTUH-K2044 (positive control) and E. coli MG1655 (negative control). Biofilms in the 96-well plates, which were stained using crystal violet and released by acetic acid, were shown above the barchart. Quantitative analysis of biofilms was performed by measuring the optical density at 595 nm using a microplate reader. Data presented here are means of triplicate experiments, and error bars indicate the SDs. *, statistical significance (P < 0.05). (B–E) Virulence potential of R. planticola SCLZS62 in a G. mellonella infection model. Survival curves for G. mellonella larvae inoculated with 1 × 104 CFU (E), 1 × 105 CFU (D), 1 × 106 CFU (C), and 1 × 107 CFU (B) of each strain are shown. K. pneumoniae NTUH-K2044 serves as the hypervirulence-positive control, and E. coli MG1655 as the negative control.
Conclusions
In this study, we report a carbapenem- and tigecycline-resistant R. planticola isolate carrying nine plasmids from hospital sewage. The coexistence of nine plasmids provides great genetic plasticity for further spread of resistance genes, and the fitness of host bacteria to its environment. This isolate contains two types of carbapenemase genes, including one blaKPC-2 and two blaNDM-1 genes, which confirms the dissemination of clinically important resistance genes into the environment by sewage discharged from hospitals. The coproduction of multiple determinants for the same resistance is mystifying, while it hints at the important host role of R. planticola in the propagation of ARGs. Finally, we identified that this MDR isolate is potentially virulent, which poses a potential public health risk. The occurrence of virulent superbug in the environment should be closely monitored.
Data availability
The complete sequences of the chromosome of SCLZS62 and its plasmids have been deposited in GenBank under the Project No. PRJNA757536.
References
MacLean, R. C. & San Millan, A. The evolution of antibiotic resistance. Science 365, 1082–1083. https://doi.org/10.1126/science.aax3879 (2019).
Chokshi, A., Sifri, Z., Cennimo, D. & Horng, H. Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 11, 36–42. https://doi.org/10.4103/jgid.jgid_110_18 (2019).
Aghapour, Z. et al. Molecular mechanisms related to colistin resistance in Enterobacteriaceae. Infect. Drug Resist. 12, 965–975. https://doi.org/10.2147/idr.S199844 (2019).
Iredell, J., Brown, J. & Tagg, K. Antibiotic resistance in Enterobacteriaceae: Mechanisms and clinical implications. BMJ 352, h6420. https://doi.org/10.1136/bmj.h6420 (2016).
He, T. et al. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat. Microbiol. 4, 1450–1456. https://doi.org/10.1038/s41564-019-0445-2 (2019).
Sun, J. et al. Plasmid-encoded tet(X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol. 4, 1457–1464. https://doi.org/10.1038/s41564-019-0496-4 (2019).
Lv, L. et al. Emergence of a plasmid-encoded resistance-nodulation-division efflux pump conferring resistance to multiple drugs, including tigecycline, in Klebsiella pneumoniae. MBio https://doi.org/10.1128/mBio.02930-19 (2020).
Wang, C. Z. et al. A novel transferable resistance-nodulation-division pump gene cluster, tmexCD2-toprJ2, confers tigecycline resistance in Raoultella ornithinolytica. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.02229-20 (2021).
Fletcher, S. Understanding the contribution of environmental factors in the spread of antimicrobial resistance. Environ. Health Prev. Med. 20, 243–252. https://doi.org/10.1007/s12199-015-0468-0 (2015).
Walsh, T. R. A one-health approach to antimicrobial resistance. Nat. Microbiol. 3, 854–855. https://doi.org/10.1038/s41564-018-0208-5 (2018).
Singer, A. C., Shaw, H., Rhodes, V. & Hart, A. Review of antimicrobial resistance in the environment and its relevance to environmental regulators. Front. Microbiol. 7, 1728. https://doi.org/10.3389/fmicb.2016.01728 (2016).
Parvez, S. & Khan, A. U. Hospital sewage water: A reservoir for variants of New Delhi metallo-beta-lactamase (NDM)- and extended-spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae. Int. J. Antimicrob. Agents 51, 82–88. https://doi.org/10.1016/j.ijantimicag.2017.08.032 (2018).
Feng, Y., Yang, P., Wang, X. & Zong, Z. Characterization of Acinetobacter johnsonii isolate XBB1 carrying nine plasmids and encoding NDM-1, OXA-58 and PER-1 by genome sequencing. J. Antimicrob. Chemother. 71, 71–75. https://doi.org/10.1093/jac/dkv324 (2016).
Zhao, F., Feng, Y., Lu, X., McNally, A. & Zong, Z. Remarkable diversity of Escherichia coli carrying mcr-1 from hospital sewage with the identification of two new mcr-1 variants. Front. Microbiol. 8, 2094. https://doi.org/10.3389/fmicb.2017.02094 (2017).
Howell, C. & Fakhoury, J. A case of Raoultella planticola causing a urinary tract infection in a pediatric patient. Transl. Pediatr. 6, 102–103. https://doi.org/10.21037/tp.2017.04.02 (2017).
Tufa, T. B. et al. CTX-M-9 group ESBL-producing Raoultella planticola nosocomial infection: First report from sub-Saharan Africa. Ann. Clin. Microbiol. Antimicrob. 19, 36. https://doi.org/10.1186/s12941-020-00380-0 (2020).
Ershadi, A., Weiss, E., Verduzco, E., Chia, D. & Sadigh, M. Emerging pathogen: A case and review of Raoultella planticola. Infection 42, 1043–1046. https://doi.org/10.1007/s15010-014-0638-9 (2014).
Castanheira, M. et al. First descriptions of blaKPC in Raoultella spp. (R. planticola and R. ornithinolytica): Report from the SENTRY Antimicrobial Surveillance Program. J. Clin. Microbiol. 47, 4129–4130. https://doi.org/10.1128/JCM.01502-09 (2009).
Sitaula, S., Shahrrava, A., Al Zoubi, M. & Malow, J. The first case report of Raoultella planticola liver abscess. IDCases 5, 69–71. https://doi.org/10.1016/j.idcr.2016.07.013 (2016).
Yokota, K., Gomi, H., Miura, Y., Sugano, K. & Morisawa, Y. Cholangitis with septic shock caused by Raoultella planticola. J Med Microbiol 61, 446–449. https://doi.org/10.1099/jmm.0.032946-0 (2012).
Alves, M. S., Riley, L. W. & Moreira, B. M. A case of severe pancreatitis complicated by Raoultella planticola infection. J Med Microbiol 56, 696–698. https://doi.org/10.1099/jmm.0.46889-0 (2007).
Vassallo, J., Vella, M., Cassar, R. & Caruana, P. Four cases of Raoultella planticola conjunctivitis. Eye (Lond) 30, 632–634. https://doi.org/10.1038/eye.2015.260 (2016).
Teo, I., Wild, J., Ray, S. & Chadwick, D. A rare case of cholecystitis caused by Raoultella planticola. Case Rep Med 2012, 601641. https://doi.org/10.1155/2012/601641 (2012).
Skelton, W. P. T., Taylor, Z. & Hsu, J. A rare case of Raoultella planticola urinary tract infection in an immunocompromised patient with multiple myeloma. IDCases 8, 9–11. https://doi.org/10.1016/j.idcr.2017.02.002 (2017).
Zhao, Y. et al. Co-existence of the carbapenem resistance genes blaKPC-2 and blaNDM-1 in a Raoultella planticola isolate in China. J. Glob. Antimicrob. Resist 23, 327–328. https://doi.org/10.1016/j.jgar.2020.09.015 (2020).
Demiray, T., Koroglu, M., Ozbek, A. & Altindis, M. A rare cause of infection, Raoultella planticola: Emerging threat and new reservoir for carbapenem resistance. Infection 44, 713–717. https://doi.org/10.1007/s15010-016-0900-4 (2016).
Pan, Z. et al. Combination of tigecycline and levofloxacin for successful treatment of nosocomial pneumonia caused by New Delhi Metallo-beta-Lactamase-1-producing Raoultella planticola. Microb. Drug Resist. 23, 127–131. https://doi.org/10.1089/mdr.2015.0346 (2017).
Tseng, S. P. et al. First report of bla(IMP-8) in Raoultella planticola. Antimicrob. Agents Chemother. 58, 593–595. https://doi.org/10.1128/AAC.00231-13 (2014).
Zhang, L. et al. The prevalence and characterization of extended-spectrum beta-lactamase- and carbapenemase-producing bacteria from hospital sewage, treated effluents and receiving rivers. Int. J. Environ. Res. Public Health. https://doi.org/10.3390/ijerph17041183 (2020).
Lane, D. J. 16S/23S rRNA sequencing. Nucleic acid techniques in bacterial systematics, 115–175 (1991).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. https://doi.org/10.1093/bioinformatics/btu153 (2014).
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L. & Goker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. https://doi.org/10.1093/nar/gkab902 (2022).
Carattoli, A. & Hasman, H. PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). Methods Mol. Biol. 2075, 285–294. https://doi.org/10.1007/978-1-4939-9877-7_20 (2020).
Bortolaia, V. et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. https://doi.org/10.1093/jac/dkaa345 (2020).
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J. & Chandler, M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32-36. https://doi.org/10.1093/nar/gkj014 (2006).
Moura, A. et al. INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics 25, 1096–1098. https://doi.org/10.1093/bioinformatics/btp105 (2009).
Chen, L. et al. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325-328. https://doi.org/10.1093/nar/gki008 (2005).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genomics 12, 402. https://doi.org/10.1186/1471-2164-12-402 (2011).
Page, A. J. et al. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom 2, e000056. https://doi.org/10.1099/mgen.0.000056 (2016).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47, W256–W259. https://doi.org/10.1093/nar/gkz239 (2019).
Li, Y. et al. A TolC-like protein of Actinobacillus pleuropneumoniae is involved in antibiotic resistance and biofilm formation. Front. Microbiol. 7, 1618. https://doi.org/10.3389/fmicb.2016.01618 (2016).
Chou, H. C. et al. Isolation of a chromosomal region of Klebsiella pneumoniae associated with allantoin metabolism and liver infection. Infect. Immun. 72, 3783–3792. https://doi.org/10.1128/IAI.72.7.3783-3792.2004 (2004).
Blattner, F. R. et al. The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. https://doi.org/10.1126/science.277.5331.1453 (1997).
Yuan, Y. et al. blaNDM-5 carried by a hypervirulent Klebsiella pneumoniae with sequence type 29. Antimicrob Resist Infect Control 8, 140. https://doi.org/10.1186/s13756-019-0596-1 (2019).
Falagas, M. E. & Karageorgopoulos, D. E. Pandrug resistance (PDR), extensive drug resistance (XDR), and multidrug resistance (MDR) among Gram-negative bacilli: Need for international harmonization in terminology. Clin. Infect. Dis. 46, 1121–1122. https://doi.org/10.1086/528867 (2008) (author reply 1122).
Cheng, Q. et al. Type 1, 2, and 1/2-hybrid IncC plasmids from China. Front. Microbiol. 10, 2508. https://doi.org/10.3389/fmicb.2019.02508 (2019).
Wu, W. et al. NDM metallo-β-lactamases and their bacterial producers in health care settings. Clin. Microbiol. Rev. 32, e00115-00118. https://doi.org/10.1128/CMR (2019).
Liang, Q. et al. Sequencing and genomic diversity analysis of IncHI5 plasmids. Front. Microbiol. 9, 3318. https://doi.org/10.3389/fmicb.2018.03318 (2018).
Huang, J. et al. Further spread of a blaKPC-harboring untypeable plasmid in Enterobacteriaceae in China. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.01938 (2018).
Ouyang, J. et al. Comparative genomics of five different resistance plasmids coexisting in a clinical multi-drug resistant Citrobacter freundii isolate. Infect Drug Resist. 11, 1447–1460. https://doi.org/10.2147/IDR.S165818 (2018).
Zheng, B. et al. Complete nucleotide sequences of two KPC-2-encoding plasmids from the same Citrobacter freundii isolate. J. Antimicrob. Chemother. 73, 531–533. https://doi.org/10.1093/jac/dkx381 (2018).
Stahlhut, S. G., Struve, C., Krogfelt, K. A. & Reisner, A. Biofilm formation of Klebsiella pneumoniae on urethral catheters requires either type 1 or type 3 fimbriae. FEMS Immunol. Med. Microbiol. 65, 350–359. https://doi.org/10.1111/j.1574-695X.2012.00965.x (2012).
Miethke, M. & Marahiel, M. A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71, 413–451. https://doi.org/10.1128/MMBR.00012-07 (2007).
Cianfanelli, F. R., Monlezun, L. & Coulthurst, S. J. Aim, load, fire: The type VI secretion system, a bacterial nanoweapon. Trends Microbiol. 24, 51–62. https://doi.org/10.1016/j.tim.2015.10.005 (2016).
Acknowledgements
This work was supported by National Natural Science Foundation of China (31900125), Scientific and technological project in Sichuan Province (22MZGC0069), the Joint Funds of the Luzhou and Southwest Medical University Natural Science Foundation (2019LZXNYDJ47 and 2020LZXNYDJ34). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
Y.L.: conceptualization, formal analysis and writing- original draft. Y.Q.: methodology, data curation and formal analysis. Y.G., W.C. and C.L.: methodology, data curation and resources. X.D.: software. L.Z.: conceptualization, writing- review and editing, and supervision. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Qiu, Y., Gao, Y. et al. Genetic and virulence characteristics of a Raoultella planticola isolate resistant to carbapenem and tigecycline. Sci Rep 12, 3858 (2022). https://doi.org/10.1038/s41598-022-07778-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07778-0
- Springer Nature Limited