
Abstract
Protein kinase C iota (PKCɩ) is a novel protein containing 596 amino acids and is also a member of atypical kinase family. The role of PKCɩ has been explored in neurodegenerative diseases, neuroblastoma, ovarian and pancreatic cancers. Single nucleotide polymorphisms (SNPs) have not been studied in PKCɩ till date. The purpose of the current study is to scrutinize the deleterious missense variants in PKCɩ and determine the effect of these variants on stability and dynamics of the protein. The structure of protein PKCɩ was predicted for the first time and post translational modifications were determined. Genetic variants of PKCɩ were retrieved from ENSEMBL and only missense variants were further analyzed because of its linkage with diseases. The pathogenicity of missense variants, effect on structure and function of protein, association with cancer and conservancy of the protein residues were determined through computational approaches. It is observed that C1 and the pseudo substrate region has the highest number of pathogenic SNPs. Variations in the kinase domain of the protein are predicted to alter overall phosphorylation of the protein. Molecular dynamic simulations predicted noteworthy change in structural and functional dynamics of the protein because of these variants. The study revealed that nine deleterious variants can possibly contribute to malfunctioning of the protein and can be associated with diseases. This can be useful in diagnostics and developing therapeutics for diseases related to these polymorphisms.
Similar content being viewed by others

Introduction
PKCɩ is a member of a serine threonine kinase family that plays an important role in the phosphorylation of hydroxyl group in protein residues (serine and threonine). The family has been categorized into three classes: Classical PKCs, Atypical PKCs and Novel PKCs. This grouping is based on the requirement of DAG and Ca+2 ions. Activation of classical PKCs require both DAG and Ca+2 ions while the Novel class needs only DAG for its functional activation. Atypical isoforms (PKCɩ and PKCζ) are different from the other PKC family members because of the distinct structural and functional characters. They do not need Ca2+ and diacylglycerol for the functional activation1. The studies show that the family of PKC is engaged in the causation of many diseases, for instance cancer, metabolic dysfunctions and cardiovascular disorders2.
PKC iota that has a significant role in the progression of cell cycle, its inhibition can lead to the obstruction of cell cycle progression. Lately, PKCι has also been studied in cancer cell line growth, metastasis and specific tumor gene amplifications3. One study demonstrated the role of PKCι in carcinogenesis through in vitro and in vivo studies4. The role of PKC in progression of cancer has been studied in later stages of cancer and metastasis. PKC isoforms in neoplastic diseases may transform into hyper or hypo activation5. Elevated expression of PKCɩ was noted in prostate, lung, ovarian and colon cancer2. PKCɩ is directly linked to oncogenic Ras signaling. It plays a significant role in Ras mediated transformation in intestinal epithelium that leads to malignancy in rats4.
The protein PKCι has been associated with numerous diseases but the link of its genetic variants with the diseases has not been established yet. For this purpose, the objective was to use various existing methods that predict the most deleterious missense variants in PKCι. Then, to use various bioinformatics tools on the predicted 3D structure of the protein to understand the possible impact of these variants on the structure and function of the protein, prediction of post translational modification of PKCι along with its involvement with cancer and survival. This is a first comprehensive in silico analysis of missense variants of this protein. The outcomes might be useful in designing precision medicines for associated diseases.
Results
Predicted structure of PKCι and post translational modifications
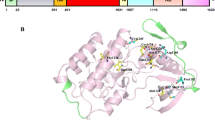
Three dimensional (3D) structures of the protein were predicted via I-TASSER6 which is the most advance and reliable tool. It predicts protein structure based on multiple threading approach. Model 1 with a c-score of − 2.30 was selected7. The protein structure was predicted from I-TASSER because the complete structure with all important domains was not found in the protein structure data bank. The structure was then validated via INTERPRO8 and different domains of the protein were identified (Fig. 1a). Domains of the protein were highlighted through PYMOL. The protein is found to have 596 amino acids with four important domains: PB1 domain, C1 domain (Pseudo substrate domain), protein kinase domain containing the active site of the protein and AGC kinase domain. Alignment of PKCι structure was performed through PYMOL. Protein kinase domain of PKCι was aligned with crystal structure of kinase domain (38AX:ID from protein data base). An RMSD score of 0.944 was obtained. C1 and kinase domain of predicted structure were then aligned against solution structure of PKC-theta (1XJD, C1 and kinase domain). An RMSD score of 0.88 indicates that the structures are well aligned (Supplementary file 4, Fig. 1a,b).

(a) Predicted Structure of Protein PKCι; It contains 4 domains. Red color represents PB1 domain (2–108 AA), C1 domain (123–192 AA) is highlighted in blue color, pink color shows Protein kinase domain (254–522) while dark grey color represents AGC kinase domain (523–596). (b) PKCɩ structure with predicted post translational modification sites distributed through its domains. Yellow pentagon shape is the representation of phosphorylation sites, red triangle is depicting ubiquitination sites, while blue circle is for methylation and acetylation is illustrated by brown oval shape.
Protein kinase domain is predicted to have maximum number of post translational modifications 17 phosphorylation sites (yellow pentagon), 6 ubiquitination sites (red triangles) and 1 acetylation (brown oval). AGC kinase and PB1 domain hosted only phosphorylation sites that were five and eleven in number respectively, while in C1 domain a total of three phosphorylation, four ubiquitination and two acetylation sites are observed. One methylation, one phosphorylation and a few phosphorylation sites were noticed in region 193–254 that comes in the regulatory domain (Fig. 1b) (Supplementary file 2; Data Tables 3, 4 and 5).
Identification of variants in PKCɩ and calculation of %SNP effect
A total of 1317 SNPs of PKCɩ were collected from ENSEMBL data base (Fig. 2a). ENSEMBL data consists of variant information, protein functional annotations, disease association, and sequence data. The coding SNPs are found across 596 amino acid residues in PKCɩ. Only missense SNPs (301) were selected for the further analysis, because mostly missense variants are found to be associated with diseases. A frequency of non-sense variants is very less as compared to missense variants and are concentrated in the Protein kinase and AGC kinase domain (Fig. 2b).

(a) Count of synonymous, stop gained, missense, frame shift, 5 and 3 prime UTR variants included in the study, (b) Mutational allocation of missense (green) and nonsense (blue) variation across protein co-ordinates of PKCɩ, (c) Distribution of missense (blue) and nonsense (orange) variations across 18 exons of PKCɩ, (d) Heat map drawn for missense SNPs (i), non-sense variations are illustrated in row (ii), while (iii) is the exons over which SNPs distribution are highlighted. Row (iv) is the null set of SNPs where probability of occurrence of SNPs is not high.
Exon wise relative abundance analysis of coding SNPs illustrated that exon one has the highest number of mutations (thirty-two in number), all of which are missense SNPs. Exon one encodes the PB1 domain of the protein. The lowest number of variations are displayed by fifteen exons containing a total of eight SNPs out of which seven are missense and one is non-sense. Exons that encode PB1 and C1 domain contained the highest number of variations. AGC domain has the lowest number of variations (Fig. 2c,d).
Deleterious SNPs in PKCι
The missense SNPs were analyzed on seven tools SIFT (≤ 0.05), POLYPHEN (> 0.9), REVEL (> 0.5), Mutation Accessor (≥ 0.8 = Medium, > 1.9 = high), CADD (≥ 30), MetaLR (> 0.5), and PROVEAN (≤ − 2.5), the cut off criterion for deleteriousness is shown in parenthesis (Supplementary file 1). The percentage SNP effect of missense variations were determined in each residue of the protein (Fig. 3a). The total number of identified SNPs varied in all the tools. SNP was considered as deleterious if pathogenicity was confirmed by > 75% tools (Table 1). After final scrutiny of deleterious and non-deleterious nine missense SNPs were identified to be highly deleterious with two SNPs residing in the PB1 domain, five in the C1 domain, one in the AGC domain while one in the protein kinase domain (Table 2) (Fig. 3b). The highest number of deleterious SNPs are found in C1 and pseudo kinase domain. This region is encoded by four, five, six, seven and eight exons. This region of the protein can be regarded as mutationally sensitive region of the protein. In atypical type of PKCs C1 domain is not responsive to DAG, it is catalytically activated by phospholipids that releases it from its membrane bound state to perform activities like wound healing, chemotaxis, and migration. The C1 domain can be targeted for drug development along with its hinge region9.

(a) Summary of percentage SNP effect of missense variations in each residue of the protein. (b) PKCɩ protein structure with domain wise distributed variants; PB1 domain (G34W, F66Y), C1 domain (R127K, R130C, R130H), Kinase domain of the protein (G398S) and AGC kinase domain (G581V). (c) Stability change DDG values of SNPs depicting destabilizing effect of selected SNPs.
Stability changes in mutant structures of protein
The effect of change in the stability of protein was predicted for nine selected SNPs. The stability prediction is based on DDG (delta delta G) values, it is metric of prediction of how single nucleotide polymorphism can affect the stability of a protein. It is the change in Gibbs free energy. A DDG score less than zero indicates lower stability. G34W, R127k, R130C, Y169H, and G398S are found to decrease the stability and F66Y, R130H, G165E, and G581V were found to destabilize the protein (Fig. 3c) (Supplementary file 3; Data Tables 1 and 2). Destabilization of a protein structure can alter its functional dynamics and can affect the normal pathways of the protein.
Functional and physio-chemical analysis of selected SNPs
Project HOPE predicts the effects of Amino acid substitutions on the structural confirmations and functions of the protein. Project HOPE reveals some structural and functional changes in the protein because of these mutations that cause heritable diseases. Protein sequence and mutation are inserted as input query for HOPE. In case of 6 SNPs, the resultant residues are bigger than the wild one while in three SNPs the size has been reduced. This change can affect the overall structure and function of the protein. Most of the SNPs are in the regulatory region of the protein, making it mutationally sensitive region and affecting the regulatory function of the protein. In case of R130C, R130H and G165E the charge of the residue is also affected, changing from positive to neutral in case of R130C and R130H (Tables 3 and 4).
The most evolutionary conversed domain
According to ConSurf results for the PKCɩ, the protein kinase domain is found to be evolutionary more conserved with a greater number of conserved amino acid residues. Literature also suggests that protein kinase domain is most conserved domain in PKC family members10. Few residues in the hinge region are conserved but its most residues were are variable. PB1 domain and pseudo substrate domain are found to be least conserved with very less evolutionary conserved residues (Fig. 4). Mutations in the conserved region of the protein are expected to be more damaging as compared to those in the less conserved region. Surface accessibility analysis gives an insight into the structure and function of Amino acid. The buried residues usually play a role in maintaining the structural integrity while the exposed residues are important for the protein- protein interactions. The SNPs G34W, R127K, R130C, R130H and G581V were found in exposed confirmations while F66Y, G165E, Y169H and G398S were found buried in the structure.

Protein conservation and surface accessibility analysis performed for 596 residues of protein PKCɩ on ConSurf tool.
Flexibility analysis of wild and mutant protein
The change in flexibility of the protein caused by nine SNPs was analyzed by DynaMut. The stabilizing effect was analyzed through ENcom values. According to the ENCom, values all of them are found to have destabilizing effect on the protein structure and function. Five variations (G34W, R127K, R130C, R130H, and Y169H) had increased molecular flexibility that was caused by increased vibrational entropy. Four variations (F66Y, G165E, G398S, G581V) were within the cut off value > 0.5 with decreased molecular flexibility. None of them has increased molecular rigidity. This change in the overall flexibility of the protein can affect the intramolecular interactions of the protein. A comparison of intramolecular interactions of wild and mutated structures is done in Fig. 5.

A comparison of molecular flexibility and destabilizing effect of mutants along with interatomic interactions in wild type and mutants by DynaMut tool.
Molecular dynamic simulations
Molecular dynamic simulations for nine SNPs were performed for 20 ns to get an insight into the confirmational changes in the protein structure due to missense mutations. The time scale of 20 ns is enough for the rearrangements of side chain in the wild structure and various parameters such as RMSF, RMSD, Radius of gyration, total number of intra-molecular hydrogen bonds, and SASA. Domain wise comparison of changes in mutants with wild structure was performed.
Molecular dynamic simulation analysis in PB1 domain of the protein
Two variants (G34W and F66Y) occupy the PB1 domain. The compactness of protein and mutants was examined by the radius of gyration. Wild protein has radius of gyration around 2.8 nm while highest gyration value of 2.87 nm is shown by F66Y at 3 ns. It is illustrated by the data that these structural destabilizations can lead to the loss of compactness to the protein structure as compared to the wild type PKCι (Fig. 6a). In wildtype protein as well in mutants, the total number of intramolecular hydrogen bonds contributes to the stability of the structure. Lowest number of hydrogen bonds are observed in F66Y around 310, followed by G34W having a mean of 320 H-bonds, with wild structure having around 400 bonds. The data suggest lower flexibility in structure with F66Y and G34W mutations (Fig. 6b).

Simulation analysis of the missense SNPs in PB1 domain (G34W, F66Y) (a) Radius of gyration of the protein back bone (compactness of protein), (b) Total number of Hydrogen bonds throughout simulations of wild and mutant structure, (c) RMSF values of Carbon alpha in the simulation, (d) RMSD values of Cα atoms of wild and mutants, (e) Solvent accessible surface of wild and variants.
For each residue of wild type and mutated protein fluctuations in RMSF were monitored to check the effect of mutation on dynamic behavior of protein residues. It is known from Fig. 6c that in G34W and F66Y the residue level fluctuations are quite high as compared to wild structure and other mutations. The wild protein has the highest fluctuation of 0.9 nm in residue 461. G34W has the highest fluctuation value of 1.4 nm in residue 576 while F66Y had the highest value of 0.9 nm in residue 580 residues (Fig. 6c). The effect of mutations on the structure of PKCɩ was analyzed by RMSD values. It is revealed from RMSD values that mutant structures are significantly unstable as compared to the wild structure (Fig. 6d).
It was showed that F66Y has higher SASA values followed by G34W. Both values are greater than wild structure. A higher SASA value indicates expansion of a protein, the results indicate that mutants are more unstable as compared to wild protein with F66Y being more unstable than G34W (Fig. 6e).
Molecular dynamic simulations analysis of (C1 and pseudo substrate) regulatory domain of protein
A total of 5 SNPs is observed in the C1 and pseudo substrate region. This region with PB1 and C1 domain make the regulatory region of the protein. In mutant R130C the radius of gyration has significantly reduced as compared to wild and other mutants, indicating a major change in the backbone of the protein structure, and altered compactness of the protein. Radius of gyration of other mutants is also changed (Fig. 7a). Maximum number of intramolecular hydrogen bonds in wild structure are around 400 while in mutants the number has been reduced. In Y169H lowest number of hydrogen bonds (an average of 360) are seen during 1–4 ns duration depicting decreased flexibility in its structure. Minor fluctuations in number of hydrogen bonds of other mutants are also observed (Fig. 7b).

Simulation analysis of the missense SNPs in C1 domain (R127K. R130K, R130C, G165E, R130H, Y169H), (a) Radius of gyration of the protein back bone, (b) Total number of Hydrogen bonds throughout simulations of wild and mutant structure, (c) RMSF values of Carbon alpha in the simulation, (d) RMSD values of Cα atoms of wild and mutants, (e) Solvent accessible surface of wild and variants.
Root mean square fluctuation (RMSF) values for each residue of native and mutant protein was examined. R127k had the highest RMSF of 1.3 nm at residues 561–596, followed by another fluctuation of 1 nm at residues 1–4. Y169H has the maximum fluctuation value of 0.7 nm from 441 to 481aminoacid residues. 0.9 nm fluctuation was recorded for R130C from 561 till last residues. G165E has a fluctuation of 0.5 nm from 161 to 201 residues. Maximum fluctuation of 0.6 nm was recorded for R130 from 281 to 290 residues (Fig. 7c). The effect of mutations on the structure of PKCι was analyzed by RMSD values. RMSD values showed that mutant structures are significantly unstable as compared to the wild structure (Fig. 7d). Maximum RMSD values were recorded for R130H, followed by G165E and then R130C. The difference between wild and R127K RMSD is not significant. It is demonstrated from the figure that mutation has considerable effect on the structure of PKCɩ (Fig. 7d).
From analysis solvent accessible surface area (SASA) it is exposed that Y169H has higher SASA values followed by G165E. After that R130H is found with higher values, with R127K values close to the wild structure. All mutant values are greater than the wild structure. A higher SASA value indicates expansion of a protein, the results indicate that mutants are more unstable as compared to wild protein with Y169H and G165E being more unstable than wild structure and other mutants (Fig. 7e).
Molecular dynamic simulation analysis of protein kinase domain of protein
SNP G398S is in the protein kinase domain of PKCɩ. This domain is the most conserved domain of the family. The SNP is observed to cause alterations to the protein. The compactness of the protein is predicted to be majorly affected by this mutation (Fig. 8a). Radius of gyration has reached to a maximum of 3 nm during 20 ns duration. This is a huge increase as compared to the gyration values of wild protein (Fig. 8b). Overall number of hydrogen bonds in G398S have been reduced when seen in comparison to wild structure, depicting a decreased flexibility of structure (Fig. 8c).

Simulation analysis of the missense SNP in Protein kinase domain (G398S), (a) Radius of gyration of the protein back bone, (b) Total number of Hydrogen bonds throughout simulations of wild and mutant structure, (c) RMSF values of Carbon alpha in the simulation, (d) RMSD values of Cα atoms of wild and mutants, (e) Solvent accessible surface of wild and variants.
In the root mean square fluctuation values, a noticeable change at each domain was observed. The highest RMSF peak of G398S was observed at 0.9 nm in residue 200 of the protein. Overall RMSF values of mutant were noticed to be higher as compared to wild protein. Root mean square deviation values were compared for wild and G398S mutant, a major change in stability was under observation depicting a highly unstable state of protein (Fig. 8d). From analysis solvent accessible surface area (SASA) it was illustrated that mutant G398S has higher SASA values than the wild structure. Indicating that the mutant is unstable as compared to wild protein (Fig. 8e).
Molecular dynamic simulation analysis of AGC kinase domain of protein
Radius of gyration for mutant G581V is higher than the native protein and is increasing with the passage of time predicting a decrease in flexibility of the mutant structure (Fig. 9a). From hydrogen bond analysis wild structure was found to form more bonds than the mutant. The stability of mutant is therefore affected by fewer number of hydrogen bonds (Fig. 9b).

Simulation analysis of the missense SNP in AGC kinase domain (G581V), (a) Radius of gyration of the protein back bone, (b) Total number of Hydrogen bonds throughout simulations of wild and mutant structure, (c) RMSF values of Carbon alpha in the simulation. (d) RMSD values of Cα atoms of wild and mutants, (e) Solvent accessible surface of wild and variants.
A significant difference in the RMSF values of wild and G581V was noticed. The highest peak of wild structure observed was 0.9 nm, while that of mutant was 0.8 nm at residue 500 of the protein (Fig. 9c). Other than that, the mutant peaks are at increasing trend when compared with peaks of wild structure. This imparts significant deviations in both structures. Also, RMSD values of G581V are higher than the wild structure (Fig. 9d). SASA analysis indicated that mutant has greater values than mutant. The reason for this change could be effect of substitution of amino acids by change in size of surface of protein (Fig. 9e).
Association of pathogenic SNPs with cancer
The oncogenicity of selected SNPs were predicted through two tools. The FATHMM results for individual mutations are in the form of functional scores, SNP having score above 1 are considered as deleterious. CScape predicts the SNP as deleterious if the score is above 0.5. From FATHMM results, F66Y, G398S and G581V were predicted to be associated with cancer. CScape predicted all nine variants to be cancer drivers and oncogenic with a score greater than 0.6. Six variants F66Y, R127K, R130H, G165E, Y169H, G398S are categorized as high confidence oncogenic having score above 0.9.This suggests that all nine SNPs specifically F66Y, G398S and G581V can have possible role in protein dysregulation and causation of cancer (Table 5).
Connection of PKCɩ with cancer through Kaplan–Meier plotter
The effect of expression of PKCι on survival of cancer types like breast cancer, ovarian cancer, lung cancer and gastric cancer was determined through Kaplan–Meier Plotter. The red line is depicting the survival period of cancer patients having high expression levels of PKCι, while the black line illustrates survival period of cancer patients with low expression levels of the protein (Fig. 10). This representation is in the form of Kaplan–Meier curve that shows probability of survival of patients at a certain time period.

Probability survival curves for (a) Breast cancer, (b) Ovarian Cancer, (c) lung cancer, (d) Gastric cancer based on high and low expression of PKCɩ (Red line indicates higher expression of PKCɩ, while black line shows expression below the median line).
Kaplan–Meier plotter analysis revealed that high and low expression of PKCɩ was found to have no significant link on survival of breast cancer, lung cancer, gastric cancer and Ovarian cancer patients. (Fig. 10a–d).
Authentication of results through control study
For validation of our results, we performed a control analysis of the SNP, K274R which is proven as non-deleterious experimentally11. The score from Sift, Polyphen-2, CADD, MetaLR, PROVEAN, Mutation Assessor and REVEL predicted the SNP as non-deleterious, proving that these tools have a good accuracy level. The stability assessment of K274R was done through I-mutant, SDM, mCSM, MuPro and Dynamut. Except I-mutant & mCSM through which SNP is predicted as destabilizing, the other three tools prove it as stabilizing to the protein structure and function. Hope analysis also revealed that the SNP is possibly not damaging to the protein. Fathmm, and CScape both predicted the SNP to be benign. These results illustrated that all these tools have some accuracy level and can be used for filtration of deleterious SNPs that are to be tested experimentally (Supplementary file 3, Tables 1, 2, 3 and 4).
Discussion
SNPs in the human genome can considerably affect characteristics and complex diseases through their regulation and modifications12. The data from literature advocated the role of single nucleotide polymorphisms in progression of several diseases. The genetic variants in PKCι are still unexplored, It is therefore vital to unravel the pathogenic SNPs in PKCι as these can directly affect the structure and role of a protein13. As missense variants are directly involved in pathogenicity and treatment regimen of a disease, that’s why only missense variants were considered for in-dept study14. The use of bioinformatic tools is an effective and cost-efficient method to analyze a large set of SNPs that are functionally important in a disease and can investigate mechanism and bases for these mutations15.
The protein structure was predicted and then aligned through PYMOL. The PKCɩ being a potent and multi-functional protein was characterized for non-sense and missense SNPs mutational hotspots present in the protein. An average of 15 variations per exon were estimated. SNPs in the protein kinase and AGC kinase domain were the lowest in number. The study focused on distribution of missense and non-sense SNPs variations on different residues, exons, and domains of the protein along with mutational sensitivity of the protein PKCɩ for these variations. Overall, Exon 1 contained the highest number of pathogenic and non-pathogenic SNPs, followed by exon nine and exon fifteen had the lowest number of SNPs. Exon one encodes PB1 domain, the PB1 domain is responsible for protein–protein interaction of PKCɩ with other protein having a PB1 domain such as MEK5/ERK (MAPK mitogen activated kinase/Extracellular signal regulated kinase) and Par-6 (partitioning-defective 6)16,17. The region or domain in which SNP is located has a great impact on protein. Dysregulation in expression is mostly because of SNPs in the regulatory region of the protein. Variants in PKC beta were reported to elevate insulin resistance in diabetic patients18. The most deleterious 9 SNPs G34W, F66Y, R130C, R127K, G165E, Y169H, G398S, R130H and 581V. were scrutinized that were D > 75% tools. C1 and the pseudo substrate domain contain the highest number of deleterious SNPs. C1 domain of PKCɩ is not dependent on DAG and Ca+2, its activity is enhanced by phospholipid phosphatidylserine18. The SNPs might affect the activation, resulting in altered behavior of the protein. The radius of gyration of R130C has been noticeably decreased as compared to wild and other mutants, increasing compactness of the protein. The rate of folding has a direct relationship with compactness of the protein19.
According to Project HOPE analysis in almost all the domains of the protein, amino acid substitutions have changed the size of the residues making it smaller or larger than the wild ones affecting the hydrophobicity and charge. This will disturb domain wise interaction. The difference in mass and charge leads to difference in protein–protein spatiotemporal dynamics20. The most conserved domain of the protein is protein kinase domain. It contained only one SNP out of the selected list. Kinase domain is homologous in all members of the PKC family10. The degree of conservancy was further confirmed by ConSurf tool. Maximum conversed regions were found in the kinase domain, which has important role in stabilizing the structure of protein (Ono et al.10). A noticeable change to the protein might be caused by these substitutions primarily affecting its stability that can misfold and change its function.
Through molecular dynamic simulations an insight into structural and functional dynamics of protein and mutants is exposed21,22. Many factors affect the process of protein folding, including conformational and compositional stability. Those factors include accessible surface area, packing density and residue depth. A mutation, changing an amino acid with another, may alter the conformation of the protein. Subsequently, the new structural rearrangement must affect the specific physio-chemical properties of the residue which is mutated. For determination of structure and function of a protein solvent accessibility (SASA) is a crucial factor, which is computed from sequences and structures of proteins through different algorithms. If the residue that is mutated is internal, the probability of unfolding and major changes increase, much less if the residue is superficial23. The protein cores consist of densely packed residues having a certain depth that maintains its packing density. This packing fraction can be perturbed if there is a change in the SASA values, this will lead to malfunctioned protein–protein interactions and membrane embedded portion of trans-membrane protein24. The variant F66Y in the PB1 domain is observed to increase SASA of the protein structure more as compared to other SNPs and wild type of the protein (Fig. 6). Consequently, decreasing the overall compactness and stability of the protein. This might be since F66Y is in buried residues of the protein (Fig. 4), The change in amino acid from tyrosine to Phenylalanine can disturb the overall interactions most probably PB1 domain interactions of the protein because of difference in the hydrophobicity of amino acids. The change in RMSD, radius and SASA of the SNP 398S suggested that this is destabilize the protein more as compared to other mutants. The reason behind this could be that the SNP is in the regulatory region of the protein. The protein kinase domain is responsible for the phosphorylation function of the protein, so the mutation can possibly affect the phosphorylation function of the protein. In protein interactions of PKCɩ with Par6 and Par3, protein kinase domain remains in closed confirmation25 the variant in the domain can possibly alter the protein interactions of PKCɩ. Also, this is evident from Fig. 4 that G398S has a buried location in the protein, maybe that’s why its impact on protein functionality is substantial.
The pathogenicity and association of these 9 SNPs with cancer was confirmed through FATHMM and CScape. According to CScape all were oncogenic having 6 high confidence oncogenic SNPs with score above 0.9, the results from FATHMM demonstrated that F66Y, G398S and G581V are cancer related. These results are consistent with the result of MD simulations illustrating that these mutations can be significantly associated with cancer. The dysregulated expression of PKCɩ has been studied in various Ovarian Cancer26, Non-small lung carcinoma27, Colon Cancer28, Pancreatic Cancer29, Glioma30, Chronic myelogenous leukemia31 and Esophageal cancer32. But None of these variations were previously related with cancer. Variants in PKC beta were reported to elevate insulin resistance in diabetic patients18. Our studies of Kaplan–Meier plot illustrated that no significant association was found between expression of PKCɩ and breast, gastric, ovarian and lung cancer, however from literature it has been known that PKCɩ higher expression in gastric cancer can be linked with low overall survival33. PKCɩ expression in human non-small cell lung cancer (NSCLC) is over expressed and play an important role in altered growth of adenocarcinoma A549 human lung cancer cell line both in-vitro and in-vivo27. The study of control SNP revealed that in-silico tools can have some level of accuracy but as the computational tools used for scrutinization of SNPs are based on different algorithms, it is not necessary that highly conserved region variant always harvest noteworthy changes in the protein. Therefore, the confirmation of effects of these variants should be performed through genotype–phenotype based experiments. Generally, the study provides a starting point to investigate the deleterious variants in PKCι that can lead to altered structural dynamics mal function of the protein.
Conclusion
PKCɩ as an oncogenic gene plays essential role in control of cell cycle and regulatory activities. Alteration in the expression of this gene can be associated with various diseases specifically cancer. The first comprehensive and systemic in-silico investigation of missense SNPs in the protein PKCɩ was performed. A total of 9 SNPs (G34W, F66Y, R127K, R130C, R130H, G165E, Y169H, G398S, G581V) were reported as potentially deleterious due to their capability of affecting protein stability and conformational dynamics. Domain wise post translational modifications study revealed that phosphorylation sites are concentrated at the protein kinase domain, this suggests that variant in protein kinase domain will strongly affect the phosphorylation strategy of the protein. Kaplan Meier Plotter suggested that high expression of PKCι can be associated with low survival rates. A connection of protein and the mutants with cancer was predicted, highlighting the fact that these can be used as important candidates in the prognosis and therapeutics strategies of cancer and other metabolic diseases.
Methods
Prediction of protein structure and post translational modification
The protein sequence of PRKCI gene with transcript ID: PRKCI-201 ENST00000295797.5 was obtained from ENSEMBL database in FASTA format. ENSEMBL incorporates data from more than 25 databases for homo sapiens that includes COSMIC, gnomeAD, ExAC, and dbSNP34.The data consists of variant information, protein functional annotations, disease association, and sequence data. The data comprise of genetic and disease specific studies. As the complete structure of PKCι is not found in PDB bank therefore this sequence was then submitted to I-TASSER (Iterative Threading Assembly Refinement)35 which is an online tool for prediction of protein structures based on the threading approach of protein modelling and generates each predicted protein model with a confidence score ranging from − 5 to 27,36. The predicted models were then visualized with the help of PyMOL molecular visualization system. In addition, the predicted models by I-TASSER were cross-checked using InterPro database8 and other literature sources available, regarding the structural features of already studied and determined similar proteins. Validation of PKCι structure was performed by aligning kinase domain of the protein with crystal structure of PKCι (kinase domain, 38AX:ID from protein data base), similarly C1 and kinase domain of PKCɩ were aligned with PKC-theta ((1XJD, C1 and kinase domain).
Phosphorylation sites for PKCɩ was predicted through Netphos-GPS (http://www.cbs.dtu.dk/services/NetPhos/)37 with a cut-off score of 0.5. Values equal to and greater than 0.5 were considered. Methylation sites were predicted by GPS-MSP (http://msp.biocuckoo.org/)38, while GPS (pail) (http://pail.biocuckoo.org/)39 was used for acetylation sites. Ubiquitination was analyzed through PDM-PUB (http://bdmpub.biocuckoo.org/)40.
Collection and processing of SNPs
Variations in the protein PKCɩ were identified from ENSEMBL (https://asia.ensembl.org/index.html)41. SNPs of PKCɩ excluding inframe and intronic were gathered and separated into regulatory variations (splice-site, 3′ and 5 UTRs) and coding SNPs (missense and non-sense SNPs). The data was retrieved in April 2021.The data base gave information about IDs of variants, amino acid coordinates, genomic coordinates, mutated base and amino acid residue information. Data about the protein was retrieved from Uniprot, InterPro and ENSEMBL. Residues of the protein were grouped into motifs, domains and loop regions. Within each domain and amino acid coordinate of the protein frequency of occurrence of SNPs was determined. Only missense variants were further subjected for prediction of pathogenicity. The scrutinized pathogenic SNPs were further mapped on exons and domains of the protein.
Analysis of coding SNP effect
SNPs were analyzed on 7 tools for sorting intolerant from tolerant (SIFT)42, Polymorphism Phenotyping v2 (PolyPhen-2)43, Protein variation effect Analyzer (PROVEAN)44, Mutation Accessor45, Rare exome variant ensemble learner (REVEL)46, meta LR47 and Annotation dependent depletion (CADD)48. Through these tools, rigorous screening of deleterious SNPs was performed. SNP was considered deleterious only if more than 75% tools predicted it to be deleterious. Mutationally, the most sensitive region of PKCɩ was determined by taking average and percentage of the deleterious SNPs and then a total of nine SNPs were selected for final analysis of the study.
Prediction of protein Stability
Stability change caused by the above mentioned mutations was determined through different tools such as mutation cut off scanning matrix (mCSM) (http://biosig.unimelb.edu.au/mcsm/)49, I-mutant (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi)50, Mu-Pro (http://mupro.proteomics.ics.uci.edu/)51 and Site directed Mutator (SDM) (http://marid.bioc.cam.ac.uk/sdm2)52. A variant was categorized as ‘Destabilizing’ SNP only when DDG values ≤ − 0.5 was given by at least 2 tools.
Functional and physio-chemical analysis of selected SNPs
Functional analysis of the selected SNPs in protein gave an understanding of biological consequences of stability, sub cellular localization and membrane binding. The other physiochemical changes like size and charge of amino acid, hydrophobicity and involvement in hydrogen and salt bridge formation was drawn from project HOPE (https://www3.cmbi.umcn.nl/hope/)53.
Evolutionary conservation study
Structure and function of the protein can be disrupted majorly by mutations that lie in the evolutionary conserved sections. The evolutionary conservation analysis of PKCι was performed through Consurf tool (https://consurf.tau.ac.il/) through conservation scores54.
Flexibility analysis of selected variants
Effect of mutation on dynamics of a protein were assessed computationally through DynaMut (http://biosig.unimelb.edu.au/dynamut/), an online tool for predicting fluctuations in proteins through normal mode analysis40. The Elastic Network Contact Model (ENCoM) was considered as destabilizing if the score was DDG < − 0.5. Molecular flexibility was predicted to be increased if delta-vibrational entropy (DDS) > 0.5 while with a DDS < − 0.5 molecular flexibility was considered as decreased.
Analysis of RMSD, RMSF, hydrogen bond and radius of gyration
The predicted model of PKCɩ was assessed for structural stability via GROMACS. Mutagenesis wizard tool of PyMOL55 was used to introduce point mutations. The obtained mutated structures were also examined for their influence on protein structure through GROMACS version 5.1. For stimulating the protein OPLS-AA force parameters were used56. The temperature was kept at 300 K while atmospheric pressure was maintained at 1. In a cubic box the system was solvated, neutralized and equilibrated for NVT and NPT simulation each. In detail ion steps are = 50,000, minim steps = 50,000, NPT steps = 50,000; 2 * 50,000 = 100 ps, NVT steps = 50,000; 2 * 50,000 = 100 ps and MD steps = 10,000,000; 2 * 10,000,000 = 20,000 ps (20 ns). MD simulation of 20 ns were performed on wild and mutated structures of the protein, the trajectory files were analyzed by Radius of gyration (Rg), Root mean Square Fluctuation (RMSF), Root mean square deviation (RMSD) and Solvent accessible surface (SASA).
Association of PKCɩ and mutants with cancer
Pathogenicity of selected variations could have role in causation of different cancers. Association of pathogenic SNPs with cancer was predicted through tools CSCAPE (http://www.cscape.biocompute.org.uk/cgi-bin/submitcancer.cgi)57 and FATHMM (http://fathmm.biocompute.org.uk/)58. Through FATHMM the coding and non-coding variants were analyzed for its functional impact, while CScape was used for the prediction of oncogenic status of deleterious variants.
The effect of expression of PKCɩ on probability survival of different types of cancers such as breast cancer, ovarian cancer, lung cancer and gastric patients was determined through Kaplan–Meier Plotter, which is a software for integration of gene expression data with clinical data59. The data base contains information of over 22,277 genes and their impact on survival of breast, ovarian, lung and gastric cancer. The plot was generated and compared for survival of patients in low and high expression cohort.
Authentication of results through control study
For assessment of our results through in-silico tools we took a control SNP, K274K from Uniprot. The SNP is in the kinase domain of the protein and has been proved to be non-deleterious to the structure and function of the domain and protein11. We applied Sift, PROVEAN, metaLR, CADD, Polyphen-2, REVEL and Mutation Assessor to K274K for pathogenicity test. Stability assessment was performed through SDM, MuPro, mCSM, Dynamut and I-mutant. Project Hope analysis for the SNP was also done. Prediction of cancer driver/passenger was checked through Fathmm and CScape.
References
Martiny-Baron, G. & Fabbro, D. Classical PKC isoforms in cancer. Pharmacol. Res. 55(6), 477–486. https://doi.org/10.1016/j.phrs.2007.04.001 (2007).
Garg, R. et al. Protein kinase C and cancer: What we know and what we do not. Oncogene 33, 5225–5237. https://doi.org/10.1038/onc.2013.524 (2014).
Steinberg, S. F. Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378. https://doi.org/10.1152/physrev.00034.2007 (2008).
Murray, N. R. et al. Protein kinase Cι is required for Ras transformation and colon carcinogenesis in vivo. J. Cell Biol. 164, 797–802. https://doi.org/10.1083/jcb.200311011 (2004).
Griner, E. M. & Kazanietz, M. G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. 7, 281–294. https://doi.org/10.1038/nrc2110 (2007).
RoyA, A. K. & Zhang, Y. I. TASSER: Aunifiedplatform forautomated proteinstructureandfunction prediction. NatureProtocols. 5, 725 (2010).
Yang, J. & Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354 (2021).
Blumberg, P. et al. Wealth of opportunity-the C1 domain as a target for drug development. Curr. Drug Targets 9, 641–652. https://doi.org/10.2174/138945008785132376 (2008).
Ono, Y. et al. Protein kinase C zeta subspecies from rat brain: Its structure, expression, and properties. Proc. Natl. Acad. Sci. 86, 3099–3103. https://doi.org/10.1073/pnas.86.9.3099 (1989).
Spitaler, M., Villunger, A., Grunicke, H. & Uberall, F. Unique structural and functional properties of the ATP-binding domain of atypical protein kinase C-ι. J. Biol. Chem. 275, 33289–33296 (2000).
Zhang, S. et al. Whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. bioRxiv. https://doi.org/10.1101/583237 (2019).
Wall, S. M. In Novartis Foundation Symposium. 231 (Wiley, 1999).
Ferrer-Costa, C., Orozco, M. & de la Cruz, X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J. Mol. Biol. 315, 771–786 (2002).
Chitrala, K. N., Nagarkatti, M., Nagarkatti, P. & Yeguvapalli, S. Analysis of the TP53 deleterious single nucleotide polymorphisms impact on estrogen receptor alpha-p53 interaction: A machine learning approach. Int. J. Mol. Sci. 20, 2962 (2019).
Hirano, Y. et al. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J. Biol. Chem. 279, 31883–31890. https://doi.org/10.1074/jbc.M403092200 (2004).
Diaz-Meco, M. T. & Moscat, J. MEK5, a new target of the atypical protein kinase C isoforms in mitogenic signaling. Mol. Cell. Biol. 21, 1218–1227. https://doi.org/10.1128/MCB.21.4.1218-1227.2001 (2001).
Osterhoff, M. et al. Association of polymorphisms within the protein kinase C β promoter with insulin-resistance in non-obese subjects. Exp. Clin. Endocrinol. Diabetes. 114, OR5_25 (2006).
Mlu, L., Bogatyreva, N. & Galzitskaia, O. Radius of gyration is indicator of compactness of protein structure. Mol. Biol. 42, 701–706 (2008).
Islam, M. J., Khan, A. M., Parves, M. R., Hossain, M. N. & Halim, M. A. Prediction of deleterious non-synonymous SNPs of human STK11 gene by combining algorithms, molecular docking, and molecular dynamics simulation. Sci. Rep. 9, 1–16 (2019).
Hu, X. et al. The dynamics of single protein molecules is non-equilibrium and self-similar over thirteen decades in time. Nat. Phys. 12, 171–174. https://doi.org/10.1038/nphys3553 (2016).
Chitrala, K. N. & Yeguvapalli, S. Computational screening and molecular dynamic simulation of breast cancer associated deleterious non-synonymous single nucleotide polymorphisms in TP53 gene. PLoS ONE 9, e104242. https://doi.org/10.1371/journal.pone.0104242 (2014).
Savojardo, C., Manfredi, M., Martelli, P. L. & Casadio, R. Solvent accessibility of residues undergoing pathogenic variations in humans: From protein structures to protein sequences. Front. Mol. Biosci. 7, 460 (2021).
Chakravarty, S. & Varadarajan, R. Residue depth: A novel parameter for the analysis of protein structure and stability. Structure 7, 723–732 (1999).
Wang, C., Shang, Y., Yu, J. & Zhang, M. Substrate recognition mechanism of atypical protein kinase Cs revealed by the structure of PKCι in complex with a substrate peptide from Par-3. Structure. 20, 791–801 (2012).
Jatoi, A. et al. A mixed-methods feasibility trial of protein kinase C iota inhibition with auranofin in asymptomatic ovarian cancer patients. Oncology 88, 208–213 (2015).
Liu, L. et al. Protein kinase C-iota-mediated glycolysis promotes non-small-cell lung cancer progression. OncoTargets Ther. 12, 5835. https://doi.org/10.2147/OTT.S207211 (2019).
Guo, W., Wu, S., Liu, J. & Fang, B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 68, 7403–7408 (2008).
Evans, J. D., Cornford, P. A., Dodson, A., Neoptolemos, J. P. & Foster, C. S. Expression patterns of protein kinase C isoenzymes are characteristically modulated in chronic pancreatitis and pancreatic cancer. Am. J. Clin. Pathol. 119, 392–402 (2003).
Patel, R. et al. Involvement of PKC-ι in glioma proliferation. Cell Prolif. 41, 122–135 (2008).
Fatkullina, S. The Role of Atypical Protein Kinase C Iota in Cancer Invasion (McGill University, 2015).
Wang, B.-S. et al. PKCι counteracts oxidative stress by regulating Hsc70 in an esophageal cancer cell line. Cell Stress Chaperones 18, 359–366 (2013).
Hashimoto, I. et al. Clinical significance of PRKCI gene expression in cancerous tissue in patients with gastric cancer. Anticancer Res. 39, 5715–5720. https://doi.org/10.21873/anticanres.13771 (2019).
Cunningham, F. et al. Ensembl. Nucleic Acid Res. 2019(47), D745–D751 (2019).
Roy, A., Kucukural, A. & Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. https://doi.org/10.1038/nprot.2010.5 (2010).
Zhang, C., Freddolino, P. L. & Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 45, W291–W299 (2017).
Singh, H. B. & Borbora, D. J. M. G. In silico assessment of human CD14 gene revealed high-risk single nucleotide polymorphisms and their impact on innate immune response against microbial pathogens. Meta Gene 17, 136–140. https://doi.org/10.1016/j.mgene.2018.05.010 (2018).
Deng, W. et al. Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins. Brief. Bioinform. 18, 647–658. https://doi.org/10.1093/bib/bbw041 (2017).
Deng, W. et al. GPS-PAIL: Prediction of lysine acetyltransferase-specific modification sites from protein sequences. Sci. Rep. 6, 1–10. https://doi.org/10.1038/srep39787 (2016).
Qiu, W., Xu, C., Xiao, X. & Xu, D. Computational prediction of ubiquitination proteins using evolutionary profiles and functional domain annotation. Curr. Genom. 20, 389–399. https://doi.org/10.2174/1389202919666191014091250 (2019).
Hubbard, T. et al. The ensembl genome database project. Nucleic Acids Res. 30, 38–41 (2002).
Sim, N.-L. et al. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. https://doi.org/10.1093/nar/gks539 (2012).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. https://doi.org/10.1038/nmeth0410-248 (2010).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE https://doi.org/10.1371/journal.pone.0046688 (2012).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 39, e118–e118. https://doi.org/10.1093/nar/gkr407 (2011).
Ioannidis, N. M. et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. https://doi.org/10.1016/j.ajhg.2016.08.016 (2016).
Mahfuz, A. & Khan, M. A. Identification of deleterious single nucleotide polymorphism (SNP) s in the human TBX5 gene & prediction of their structural & functional consequences: An in silico approach. Biochem. Biophys. Rep. https://doi.org/10.1101/2020.05.16.099648 (2020).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. https://doi.org/10.1038/ng.2892 (2014).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342. https://doi.org/10.1093/bioinformatics/btt691 (2014).
Wang, L. et al. Construction of a genomewide RNA i mutant library in rice. Plant Biotechnol. J. 11, 997–1005. https://doi.org/10.1111/pbi.12093 (2013).
Abbas, R. A. A., Albakry, A. M. S., Khaier, M. A. M. & Elnasri, H. A. Computational analysis of single nucleotide polymorphism (SNPs) in humanSLC5A1 gene. Int. J. Biomed. Sci. Eng. 7, 85. https://doi.org/10.11648/j.ijbse.20190704.12 (2019).
Pandurangan, A. P., Ochoa-Montaño, B., Ascher, D. B. & Blundell, T. L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 45, W229–W235. https://doi.org/10.1093/nar/gkx439 (2017).
Mustafa, H. A. et al. Computational determination of human PPARG gene: SNPs and prediction of their effect on protein functions of diabetic patients. Clin. Transl. Med. 9, 1–10. https://doi.org/10.1186/s40169-020-0258-1 (2020).
Ashkenazy, H. et al. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350. https://doi.org/10.1093/nar/gkw408 (2016).
DeLano, W. L. Pymol: An open-source molecular graphics tool. Protein Crystallogr 40, 82–92 (2002).
Siu, S. W., Pluhackova, K. & Böckmann, R. A. Optimization of the OPLS-AA force field for long hydrocarbons. J. Chem. Theory Comput. 8, 1459–1470 (2012).
Rogers, M. F., Shihab, H. A., Gaunt, T. R. & Campbell, C. CScape: A tool for predicting oncogenic single-point mutations in the cancer genome. Sci. Rep. 7, 1–10. https://doi.org/10.1038/s41598-017-11746-4 (2017).
Kumar, R. D., Swamidass, S. J. & Bose, R. Unsupervised detection of cancer driver mutations with parsimony-guided learning. Nat. Genet. 48, 1288–1294. https://doi.org/10.1038/ng.3658 (2016).
Rosen, K., Prasad, V. & Chen, E. Y. Censored patients in Kaplan–Meier plots of cancer drugs: An empirical analysis of data sharing. Eur. J. Cancer. 141, 152–161. https://doi.org/10.1016/j.ejca.2020.09.031 (2020).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.S., N.M.A. and Y.S.; methodology, M.S. and N.M.A.; experimentation, H.S., K.K.; validation H.S.; formal analysis, H.S. and K.K.; investigation,N.K., K.K. and H.S.; resources, M.S.; data curation, N.M.A.; writing—original draft preparation, H.S. and N.K.; writing—review and editing, Y.S. and M.S.; visualization, M.S. and N.M.A.; supervision, M.S.; project administration, M.S.; funding acquisition, M.S. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shah, H., Khan, K., Khan, N. et al. Impact of deleterious missense PRKCI variants on structural and functional dynamics of protein. Sci Rep 12, 3781 (2022). https://doi.org/10.1038/s41598-022-07526-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07526-4
- Springer Nature Limited
This article is cited by
-
Possible prognostic impact of PKCι genetic variants in prostate cancer
Cancer Cell International (2024)
-
Multiplex Cas9-based excision of CLCuV betasatellite and DNA-A revealed reduction of viral load with asymptomatic cotton plants
Planta (2023)
-
Insilico prediction and functional analysis of nonsynonymous SNPs in human CTLA4 gene
Scientific Reports (2022)