
Abstract
Mikania micrantha is a notorious invasive weed that has caused huge economic loss and negative ecological consequences in invaded areas. This species can adapt well to invasive environments with various stress factors. The identification of gene families and functional pathways related to environmental adaptability is lack in M. micrantha at the multi-organ full-length transcriptome level. In this study, we sequenced the transcriptomes of five M. micrantha organs using PacBio single-molecule real-time sequencing and Illumina RNA sequencing technologies. Based on the transcriptome data, full-length transcripts were captured and gene expression patterns among the five organs were analyzed. KEGG enrichment analysis of genes with higher expression indicated their special roles in environmental stress response and adversity adaptation in the various five organs. The gene families and pathways related to biotic and abiotic factors, including terpene synthases, glutathione S-transferases, antioxidant defense system, and terpenoid biosynthesis pathway, were characterized. The expression levels of most differentially expressed genes in the antioxidant defense system and terpenoid biosynthesis pathway were higher in root, stem, and leaf than in the other two organs, suggesting that root, stem, and leaf have strong ability to respond to adverse stresses and form the important organs of terpenoid synthesis and accumulation. Additionally, a large number of transcription factors and alternative splicing events were predicted. This study provides a comprehensive transcriptome resource for M. micrantha, and our findings facilitate further research on the adaptive evolution and functional genomics of this species.
Similar content being viewed by others


Introduction
Mikania micrantha (Asteraceae), a perennial creeping herb or semi-woody vine originating from Central and south America, is ranked among the top ten worst weeds in the world1. The rapid expansion of this species throughout Pacific Islands and Southeast and South Asia regions, including south China, has caused huge economic loss and negative ecological consequences2. Mikania micrantha is widely found in agricultural and forest lands, open disturbed areas, and various climatic niches and biomes2,3. Strong adaptability of M. micrantha is its remarkable characteristic, and the reason behind it has become the focus of attention. High photosynthetic capacity is a representative feature of M. micrantha, which provides sufficient energy supply for rapid vegetative propagation and is conducive to invade ecosystems with different light intensities4. Strong allelopathic effect is another notable trait of M. micrantha. Its essential oil contains abundant terpenoids, especially monoterpenes and sesquiterpenes, which inhibit the growth of other plants and have a significant deterrent effect on oviposition of other insects5,6. In addition to high photosynthetic capacity and strong allelopathy, M. micrantha can adapt well to invasive environments with various abiotic factors. For example, M. micrantha can grow in various soil types, aquatic habitats, high salt conditions, and dry and cold areas7,8,9. These findings suggest that strong adaptability to environmental changes promotes the spread and invasion of M. micrantha. Therefore, it is necessary and important to study how M. micrantha adapts to the invasive environment for its invasion control.
To cope up with adverse environmental conditions, gene families related to stress, including terpene synthases (TPSs) and glutathione S-transferases (GSTs), have been induced and accumulated to protect invasive plants from stress damage and enhance their environmental adaptability. For example, the up-regulation of TPSs and GSTs in invasive plants Eupatorium adenophorum, Solanum elaeagnifolium, and Parthenium hysterophorus was conducive to enhancing tolerance to different light conditions, wounding stress, drought, and high-salinity environment10,11,12. In addition to gene families, some important functional pathways in invasive plants have also been shown to play roles in responding to stress stimuli. Stress stimuli are usually accompanied by an increase in the production of toxic reactive oxygen species (ROS). Excessive ROS may lead to oxidative stress, disrupt the normal metabolism, and affect plant growth and development13. Invasive plants have well-developed antioxidant defense systems to counter the deleterious effects of ROS14. Furthermore, terpenoids and their synthetic pathways (cytosolic mevalonate (MVA) and plastidial methylerythritol phosphate (MEP) pathway) are important secondary metabolites and functional pathways for plants to respond to various abiotic and biotic stresses15. The systematic analysis of these gene families and functional pathways can help us better understand the environmental adaptability of M. micrantha.
Transcriptome analysis is a cost-effective approach from which researchers can analyze gene expression patterns in specific organs and screen important gene families and functional pathways. Three previous studies using next-generation sequencing technology characterized the transcriptomes of M. micrantha's leaf, root and leaf mixtures, and young seedlings16,17,18. However, most of the assembled unigenes were not full-length transcript sequences, and these three studies did not generate gene expression profiles of the specific organs. In particular, the lack of transcriptional expression profiles of flower and seed may lead to the omission of potential genes and metabolic pathways related to environmental adaptation19. In addition, short reads generated by next-generation sequencing technology lead to difficulties with the explorations of alternative splicing (AS) events, and make the annotation of transcriptomes incomplete and error-prone. With the advancement of sequencing technology, PacBio single-molecule real-time (SMRT) sequencing can generate a single long read containing the entire transcript sequence without assembly20, helping overcome the limitations of next-generation sequencing technology. So far, PacBio SMRT sequencing has been successfully utilized to explore the adaptive mechanism of certain invasive plants21.
Here, we aimed to provide a comprehensive transcriptomic profile in different M. micrantha organs, namely the root, stem, leaf, flower, and seed, using both PacBio SMRT and Illumina RNA-Seq technology. Our objectives were to analyze the gene expression patterns among the five organs and to identify candidate genes and functional pathways related to abiotic and biotic stress factors. This work is the first comprehensive report on the full-length transcriptome of multiple organs of M. micrantha and it also provides valuable molecular resources for future research on the adaptation mechanisms and functional genomics of M. micrantha.
Results
The full-length sequences of PacBio SMRT sequencing
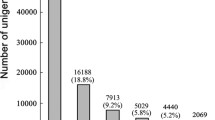
Based on PacBio SMRT sequencing, 3,751,089, 3,434,452, 3,900,180, 8,535,019, and 4,435,846 subreads were generated for root, stem, leaf, flower, and seed, with a N50 of 3040, 3367, 2611, 2198, and 4584 bp, respectively (Table S1; Fig. S1). Subreads were processed to generate circular consensus sequences (CCSs). By detecting the primers and poly(A) tail, 238,196, 232,290, 211,535, 257,905, and 231,877 full-length non-chimeric (FLNC) reads were identified for root, stem, leaf, flower, and seed, with a mean length of 2633, 3070, 2561, 1746, and 3762 bp, respectively (Table S2; Fig. S2). After Iterative Clustering for Error Correction (ICE) clustering, polishing, base correction, de-redundancy, and non-plant sequences filtering, 37,789, 34,034, 38,100, 54,937, and 53,906 unigenes were retained for root, stem, leaf, flower, and seed, respectively, with an average unigene length of 1802–3786 bp and N50 of 2238–4707 bp (Table S2). The length of most unigenes from five organs exceeded 2000 bp, accounting for 68.88% of the total number (Table S3; Fig. 1A). Based on Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment, about 88.1% (single-copy: 353; duplicated: 916) of the 1440 core embryophyte genes were found to be complete (90.6% were present when counting fragmented genes), suggesting the high integrity of the M. micrantha transcriptome (Fig. S3).

Length distribution of unigenes from PacBio SMRT sequencing (A) and Illumina RNA-Seq (B) across five organs.
De novo assembly of Illumina RNA-Seq data
Based on Illumina RNA-Seq, 43.23, 40.27, 41.01, 65.85, and 41.09 million clean reads were obtained for root, stem, leaf, flower, and seed, respectively, with Q20 exceeding 96.72%. Using Trinity software, clean reads were de novo assembled into 124,238, 60,232, 63,370, 93,229, and 66,411 unigenes for root, stem, leaf, flower, and seed. After filtering non-plant sequences, 124,233, 60,232, 63,370, 93,228, and 66,410 unigenes were finally retained for the five organs, respectively (Table S4). The length of most unigenes (84.70%) was shorter than 2000 bp (Table S3). In addition, the average length and N50 of unigenes generated by Illumina RNA-Seq were 1067–1312 bp and 1336–1685 bp, respectively, which were shorter than that from PacBio SMRT sequencing (Table S4; Fig. 1B).
Functional annotation
To obtain a comprehensive functional annotation of M. micrantha transcriptome, unigenes generated by PacBio SMRT sequencing were annotated in seven public databases, including NCBI non-redundant nucleotide sequences (NT), NCBI non-redundant protein sequences (NR), Gene Ontology (GO), Eukaryotic Orthologous Groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, and Pfam protein families. For root, stem, leaf, flower, and seed, 35,714 (94.51%), 32,614 (95.83%), 36,134 (94.84%), 49,197 (89.55%), and 50,962 (94.54%) unigenes were annotated to at least one database, respectively, suggesting that our transcriptome is well annotated and that most of unigenes may be functional (Table 1).
Based on NR database annotation, the top three homologous species for the five organs were Cynara cardunculus, Vitis vinifera, and Daucus carota (Fig. S4). The top homologous species was a plant of the Asteraceae family. For the GO function annotation, “binding”, “catalytic activities”, “metabolic process”, “cellular process”, “cell”, and “cell part” were functional categories with the most abundant unigenes (Fig. S5). In addition, numerous unigenes were assigned to “response to stimulus”, “response to biotic stimulus”, and “response to oxidative stress” category (Table S5). Positive response to stress stimuli is an important strategy for invasive plants to adapt to the environment. In the KEGG annotation, the top two pathways with the most abundant unigenes were “carbohydrate metabolism” and “translation”. Furthermore, “energy metabolism” and “environmental adaptation” were also worthy of attention, which are important pathways responsible for energy supply and stress responses (Fig. S6).
TFs identification and AS analysis
Using the iTAK pipeline, 1776 (root), 1293 (stem), 1627 (leaf), 2529 (flower), and 1733 (seed) unigenes were identified as TFs, which were classified into 68 families (Table S6). C3H (884), C2H2 (525), and bHLH (501) were the most abundant TF families (Fig. S7A). In addition, MYB (333) TFs were also found in the five organs. The differential expression levels of the top 15 TF families were further characterized. We found that the top 15 TF families had a certain amount of expression in the five organs of M. micrantha (Fig. S7B).
For root, stem, leaf, flower, and seed, 3300, 2324, 3219, 4730, and 3740 unique transcript models (UniTransModels) were constructed, among which the UniTransModels containing two isoforms were the most common (Fig. S8A). There were 329, 270, 358, 336, and 537 AS events identified in root, stem, leaf, flower, and seed, respectively. Retained introns (RIs) were detected as the most abundant AS event in all five organs, followed by alternative 3′ splice sites (A3) and alternative 5′ splice sites (A5). Mutually exclusive exons (MX) were the least frequent event (Fig. S8B).
Gene expression analysis
The number of unigenes in different expression level intervals was similar across the five organs (Fig. 2A). Using FPKM > 0.3 as the threshold for unigene expression, the total number of unigenes expressed in the five organs was 102,464 (Fig. 2B). Among them, 39,227 unigenes were co-expressed in all five organs. The information of differentially expressed genes (DEGs) identified in pairwise comparisons among the five organs is listed in Table S7. In total, 21,161 DEGs were identified among the five organs (Fig. S9). The number of DEGs between the five organs varied from 3469 (root vs stem) to 10,716 (leaf vs seed) (Fig. 2C). Notably, 933, 428, 1410, 1018, and 1292 DEGs showed significant higher expression in root, stem, leaf, flower, and seed, respectively (Figs. S10 and S11).

Gene expression patterns in five M. micrantha organs. (A) The FPKM interval distribution in the five organs. (B) Venn diagram of the number of unigenes expressed in five organs. (C) Number of differentially expressed genes in each pairwise comparison of five organs.
KEGG enrichment of unigenes with higher expression in each organ
According to the KEGG enrichment analysis results, there were obvious differences in enriched pathways in the five organs (Table S8; Fig. 3). The unigenes with higher expression in root were mainly enriched to defense response and protein processing pathways, such as “plant-pathogen interaction” and “protein processing in endoplasmic reticulum”. In stem, unigenes with higher expression were predominantly enriched to pathways related to the secondary metabolite, sugar, and terpenoid biosynthesis, such as “phenylpropanoid biosynthesis”, “starch and sucrose metabolism”, and “diterpenoid biosynthesis”. In flower, unigenes with higher expression were mainly related to “starch and sucrose metabolism”, “phenylpropanoid biosynthesis”, and “cutin, suberine, and wax biosynthesis”. The unigenes with higher expression in seed were mainly enriched in three fatty acid and sugar metabolism pathways, namely “biosynthesis of unsaturated fatty acids”, “galactose metabolism”, and “amino sugar and nucleotide sugar metabolism”. The unigenes with higher expression in leaf were significantly enriched in photosynthesis pathways, including “photosynthesis-antenna proteins”, “photosynthesis”, “porphyrin and chlorophyll metabolism”, and “carbon fixation in photosynthetic organisms”, which are important for the photosynthesis of M. micrantha.

The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of unigenes with higher expression in each organ. The significantly enriched pathways with corrected p-value (q value) < 0.05 are shown. Number indicates the size of the dot, describing the number of unigenes enriched in the pathway. The color bar represents the q value and indicates significance of the enrichment. The blue represents higher value, and the red represents lower value.
Discovery of gene families related to stress response and adaptation
For root, stem, leaf, flower, and seed, 62, 26, 59, 37, and 12 unigenes were identified as putative TPSs, respectively. Among them, most TPSs belonged to TPS-a subfamily (61 unigenes) and TPS-b subfamily (65), and only a small amount of TPSs were classified as TPS-e (7), TPS-f (1), and TPS-g subfamily (5) (Tables S9 and S10). The neighbor-joining (NJ) tree showed that TPS sequences were classified into six clades, among which TPS sequences from M. micrantha were clustered in TPS-a, TPS-b, TPS-c, TPS-e, and TPS-g clade (Fig. 4). Most sequences of TPS-a and TPS-b subfamily had higher expression in root, leaf, or flower of M. micrantha, and the rest TPS subfamilies were expressed most highly in different organs of M. micrantha.

Phylogenetic analysis of terpene synthase (TPS) sequences from M. micrantha and other angiosperms.
A total of 172 unigenes annotated as GSTs were obtained from five organs, which were classified into Zeta (52 unigenes), Theta (37), Tau (18), Phi (19), glutathionyl-hydroquinone reductases (GHR, 15), dehydroascorbate reductase (DHAR, 11), Lambda (7), and Beta (1) subfamily and some unigenes with undetermined subfamily (Tables S9 and S11). The NJ tree showed that 23 GST sequences were divided into four groups, namely Zeta, GHR, Phi, and Theta subfamily (Fig. 5A). Ten conserved motifs were identified from 23 GST sequences, and motif distribution was obviously different among the four subfamilies (Fig. 5C). Sequences from GHR subfamily were mainly expressed in root, flower, and seed, while sequences distributed in Zeta, Phi, and Theta subfamilies had certain expression levels in the five organs of M. micrantha (Fig. 5B).

Neighbor-joining (NJ) phylogenetic tree, expression level, and conserved motif of glutathione S-transferase (GST). (A) NJ tree. (B) Expression heatmap. (C) Conserved motif.
Pathway related to environmental adaptation
Timely scavenging of ROS under high photosynthetic efficiency and potential stresses is critical for the normal growth and development of M. micrantha. A total of 419 unigenes involved in antioxidant defense system were identified in the five organs, which included superoxide dismutase (SOD, 40 unigenes), catalase (CAT, 155), ascorbate peroxidase (APX, 67), monodehydroascorbate reductase (MDHAR, 19), glutathione peroxidase (GPX, 44), glutathione reductase (GR, 22), glucose-6-phosphate 1-dehydrogenase (G6PDH, 46), and 6-phosphogluconate dehydrogenase (6-PGD, 26) (Table S12). Among them, 78 unigenes were identified as DEGs in the five organs (Fig. 6A). SOD can directly oxidize superoxide radicals (O2•−) to hydrogen peroxide (H2O2), which is further converted into H2O through CAT enzyme. The DEGs responsible for the synthesis of SOD and CAT exhibited higher expression levels in root, stem, leaf, and seed than that in flower. Consistently, the other antioxidant enzymes used for scavenging of toxic H2O2, such as APX, MDHAR, GPX, GR, G6PDH, and 6-PGD, were also be found to be predominantly expressed in root, stem, leaf, and seed (Fig. 6B).

Antioxidant defense system in M. micrantha and the differentially expressed genes (DEGs) involved in the antioxidant defense in five organs of M. micrantha. (A) A simplified diagram of the antioxidant defense system. Numbers in brackets represent DEG numbers. (B) A heatmap of the DEGs involved in the antioxidant defense in five organs.
Plants produce a variety of terpenoids in response to biotic and abiotic stresses. A total of 385 unigenes from the five organs were assigned to MVA and MEP pathways22 (Table S13; Fig. 7). In the MVA pathway, acetyl-CoA acetyltransferase (AACT, 20 unigenes), 3-Hydroxy-3-methylglutaryl-CoA synthase (HMGS, 16), 3-Hydroxy-3-methylglutaryl-CoA reductase (HMGR, 49), mevalonate kinase (MK, 4), phosphomevalonate kinase (PMK, 3), diphosphomevalonate decarboxylase (MPDS, 6), isopentenyl-diphosphate delta-isomerase (IPPI, 13), and farnesyl diphosphate synthase (FPPS, 18) were identified as important components in the synthesis of sesquiterpene precursors. Germacrene D synthase (GDS) acts as a key terpene synthase, which can catalyze the precursors into sesquiterpene germacrene D. In this study, we identified 52 GDSs in five organs (Table S13). In the MEP pathway, 66, 6, 2, 12, 8, 41, 36, 13, 10, and 23 unigenes were annotated as 1-Deoxy-D-xylulose-5-phosphate synthase (DXS), 1-Deoxy-D-xylulose-5-phosphate reductoisomerase (DXR), 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (MCT), 4-(cytidine 5-diphospho)-2-C-methyl-d-erythritol kinase (CMK), 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MDS), 4-Hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS), 4-Hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (HDR), IPPI, geranyl diphosphate synthase (GPPS), and (3S)-linalool synthase (TPS14), which were responsible for the biosynthesis of monoterpene linalool. Among the 385 unigenes identified in the MVA and MVP pathways, 80 unigenes were determined as DEGs in the five organs. Nearly half of the DEGs (38 DEGs, accounting for 47.50%) displayed lower expression levels in the seed than that in the other four organs, while the number of DEGs expressed most highly was similar in the root (18 DEGs), stem (22), leaf (19), and flower (18) (Fig. 7).

MVA and MEP pathways22 for terpenoid biosynthesis in M. micrantha and the differentially expressed genes (DEGs) involved in the terpenoid biosynthesis in five organs of M. micrantha. Numbers in brackets represent DEG numbers. The heatmap represents the expression level of DEGs in five organs. R: root; S: stem, L: leaf, F: flower, and Se: seed.
Discussion
In this study, we obtained a comprehensive overview of the transcriptomic profiles of the five M. micrantha organs based on the Illumina RNA-Seq and PacBio SMRT data. Previous transcriptome studies of M. micrantha used single or mixed vegetative organ samples and second-generation technology16,17,18. The average length of unigenes produced by these studies (585–1077 bp) were shorter than the 1802–3786 bp from the PacBio transcriptomic data of the five organs. This result indicates that PacBio SMRT sequencing has a better capability to capture long transcripts. In particular, PacBio SMRT sequencing captured more transcripts longer than 4000 bp in the seed transcriptome of M. micrantha, which may be partly related to the higher proportion of long transcripts in subread and FLNC read dataset. Moreover, our analysis contained vegetative organs and reproductive organs, which makes up for the lack of transcriptome data of reproductive organs and increases the transcript diversity of M. micrantha. The independent library construction of different organs provides putative opportunity for the discovery of organ-specific transcripts and gene expression profiles, and is conducive to exploring the potential role of different organs in the environmental adaptation of invasive plants. In addition, 89.55–95.83% of the PacBio unigenes from five M. micrantha organs had homology with sequences in seven public databases. The annotation rates were higher than that reported in previous next-generation transcriptome studies16,17,18, indicating that PacBio unigenes could promote annotation and provide more information for M. micrantha transcriptomes.
TFs are important regulatory proteins that control the gene expression, which play key roles in the regulation of physiological functions and the response of plants to various stresses. In this study, we found 1293–2529 unigenes encoding TFs in the five organs, which was comparable to those of model plant Arabidopsis thaliana23. C3H, C2H2, and bHLH were the most abundant TF families in the five organs. Consistently, these three types of TF families have also been found to be abundant in the transcriptome of other plants, such as invasive Phytolacca americana and non-invasive bermudagrass (Cynodon dactylon)24,25. Gene duplication event plays a significant role in the amplification of C3H, C2H2, and bHLH gene families26,27,28. The phenomenon of C3H, C2H2, and bHLH families being the most abundant may be the result of a higher number of gene duplication events in these gene families during the evolution of the M. micrantha genome. Furthermore, the existence of a large number of C3H, C2H2, and bHLH TFs may be related to their potential functions. C3H and C2H2 belong to zinc finger proteins, which can regulate the expression of stress-related genes and increase the resistance to salinity, drought, excessive light, and oxidative stress29,30. bHLH TFs are one of the most abundant regulatory protein families, involving in plant development, monoterpene linalool biosynthesis, and stress response31,32. In addition, MYB TFs were also found in the five organs. MYB TFs have been shown to involve in sesquiterpene germacrene D production33. The expression analysis showed that C3H, C2H2, bHLH, and MYB families were expressed in all five organs, suggesting that they may play important roles in organ development and defense response in M. micrantha. Our data provide a rich resource for further study on the potential regulatory roles of TFs in M. micrantha.
Increasing numbers of studies have shown that AS may occur in some genes under adverse environmental stress. For example, about 49% of intron-containing genes in Arabidopsis experienced AS events under salt stress34. The activation of AS promoted the adaptation of Spartina alterniflora to the high-salt environment21. The information of AS in M. micrantha is still unclear. In our study, we identified 1830 AS events in the five organs. RIs were identified as the most abundant AS events, which was in line with the patterns of AS reported in plants35. During the process of invasion, AS may participate in the adaptation of M. micrantha to invasive environment. In this sense, the roles of AS in the regulation of M. micrantha adaptation should be further studied.
Unigenes with higher expression in each organ were significantly enriched into distinct functional pathways. Unigenes with higher expression in root were enriched into “plant-pathogen interaction” pathway. Several studies have shown that “plant-pathogen interaction” participates in the response to pathogens attack in plant36. Unigenes with higher expression in stem were predominantly assigned to “diterpenoid biosynthesis” and “starch and sucrose metabolism” pathway. “Diterpenoid biosynthesis” is linked to gibberellins biosynthesis, which has a great effect on stem elongation37. Starch and sucrose are the main energy supplier in higher plants, which are implicated in the metabolism and growth of plants. The decrease of starch and sucrose content shortens the growth cycle and reduces vegetative growth in Lactuca sativa38. Photosynthesis-related pathways, such as “carbon fixation in photosynthetic organisms” and “photosynthesis”, were significantly enriched in leaf. These functional categories have significant implications for the photosynthesis of M. micrantha. Unigenes with higher expression in flower were enriched in “phenylpropanoid biosynthesis” and “cutin, suberine and wax biosynthesis” pathways. Phenylpropanoids are precursors of flavonoids that play vital roles in flower development and the defense against oxidative stress and abiotic stimuli39. “Cutin, suberine and wax biosynthesis” may be related to flower organ development in transgenic Arabidopsis plants40. Unigenes with higher expression in seed were mainly enriched in the “biosynthesis of unsaturated fatty acids” pathway. Unsaturated fatty acids play crucial roles in multiple biological processes, enabling plant to respond to biotic and abiotic stresses41. Taken all together, these results suggest that certain functional pathways may have crucial implications regarding the environmental adaptation and organ development of M. micrantha.
Plants possess a variety of TPSs, which are responsible for the biosynthesis of terpenoids that function in growth, development, and stress responses. In this study, 196 TPS unigenes were identified from M. micrantha transcriptome, which was comparable with those reported in some angiosperms42. The phylogenetic tree showed that M. micrantha TPS sequences were divided into TPS-a, TPS-b, TPS-c, TPS-f, and TPS-g subfamilies, which was congruent with the result of phylogenetic analysis of TPS in angiosperms43. Most TPSs in angiosperms are from the TPS-a and TPS-b subfamilies, while fewer TPSs belong to TPS-c, TPS-e/f, and TPS-g subfamilies44. TPS-c and TPS-e/f subfamilies contain sesqui-, mono-, and other diterpene synthases43. TPS-a, TPS-b, and TPS-g are angiosperm-specific subfamilies, comprising genes of specialized sesqui-, mono-, and diterpene biosynthesis, which participate in response to insect damage and abiotic stress42,45. Mikania micrantha TPS-a and TPS-b subfamilies were mainly highly expressed in the root, leaf, or flower, suggesting their significant implications for root, leaf, and flower. The function of the TPS gene family has been characterized in invasive plants. For example, EaTPS1 is likely involved in the biosynthesis of cadinene sesquiterpenes, which show potent antifeedant activity against insect Spodoptera exigua, indicating that TPS genes have important defensive roles in E. adenophorum46.
GSTs are one of the largest gene families and have functions in protecting plants from abiotic and biotic stress. GSTs are classified into distinct subfamilies, among which Zeta and Theta subfamilies exist in both plants and animals, whereas Tau and Phi subfamilies are specific to plants47. The overexpression of Tau and Phi GSTs contributes to plant tolerance to a variety of abiotic stresses, such as dehydration, high salinity, heavy metals, and oxygen stress48,49. Zeta GSTs enhance plant growth and germination at low temperature, and Theta GSTs confer tolerance to abiotic and oxidative stresses in tobacco plants50,51. The GHR and DHAR subfamilies are also important components of the GST gene family, whose members are up-regulated or differentially regulated under high-temperature, drought, and other abiotic stresses52,53. In our study, a total of 172 unigenes annotated as GSTs were identified in the five organs of M. micrantha, mainly including members of the Zeta, Theta, Tau, Phi, GHR, and DHAR subfamilies. Moreover, the members of Zeta, Theta, Phi, and GHR subfamilies were found to highly express in the five organs. Given the extensive functions of these GST subfamilies, it can be inferred that they may play an important role in M. micrantha.
Alien plants are expected to be exposed to multiple biotic and abiotic stresses when adapting to invasive environments. Stresses are often accompanied by an increase in production of ROS, leading to an imbalance of intracellular redox homeostasis and disrupting normal metabolism13. Plants have evolved antioxidant defense system to protect them from ROS damage. The protective roles of antioxidant enzymes in enhancing plant tolerance to various adverse environments have been widely proved. The SOD and CAT activities of Ailanthus altissima were significantly inducted under drought and salt stress54. Stress stimulus may induce the increase of glutathione and ascorbate content, and these two non-enzymatic antioxidants are important components for responding to different abiotic stresses13. Antioxidant enzyme APX, MDHAR, GPX, GR, G6PDH, and 6-PGD have been involved in the regeneration of glutathione and ascorbate, enhancing the ROS scavenging capacity and stress adaptability of plants55,56. Here, we reported unigenes encoding antioxidant enzyme SOD, CAT, APX, MDHAR, GPX, GR, G6PDH, and 6-PGD, indicating that antioxidant defense system is applied in M. micrantha, and that these enzymes may be conducive to improve the tolerance to drought stress, high light, and biotic infestation of M. micrantha57,58. Most DEGs in the antioxidant defense system exhibited higher expression levels in root, stem, leaf, and seed than in flower, suggesting that root, stem, leaf, and seed have strong ability to respond to adverse stresses.
Mikania micrantha plants produce rich terpenoids, especially monoterpenes and sesquiterpenes, which play an important role in plant growth and development and stress response59. In our transcriptome data, we found unigene sequences of key genes in the MVA and MEP pathways. Eighty unigenes were differentially expressed in five organs, and the majority of them (77 unigenes) were predominantly expressed in root, stem, leaf, and flower, indicating that root, stem, leaf, and flower form the main organs of terpenoid synthesis and accumulation in M. micrantha5. In addition, the enzymes in these two synthetic pathways and their roles in growth, development, and stress response in plants have been well established. Gene encoding DXS was demonstrated to respond to wounding stress by increasing terpene biosynthesis in invasive weed S. elaeagnifolium11. Genes encoding AACT, HMGS, HMGR, MPDS, IPPI, FPPS, DXS, DXR, MCT, CMK, MDS, HDS, HDR, and GPPS were found to involve in plant development and growth, pathogen resistance, and abiotic stress tolerance59. Germacrene D and linalool are important constituents of sesquiterpenes and monoterpenes in plants. Germacrene D possesses significant allelopathic activity, which can inhibit the seed germination and the growth of other plants60. Meanwhile, germacrene D is an important response factor to insect attack and salt stress61,62. Linalool shows significant phytotoxic and antimicrobial activities63. In our study, we identified terpene synthases for the synthesis of germacrene D and linalool, which is of great significance for protecting M. micrantha from herbivore damage and insect infestation and enhancing its stress adaptability.
In summary, we reported the full-length transcriptomes of five M. micrantha organs by applying the PacBio SMRT and Illumina RNA-Seq technologies. A total of 218,766 unigenes were obtained from five organs and were functionally annotated in seven public databases. Furthermore, TFs and AS events were identified. Gene expression patterns among the five organs and genes with higher expression in each organ were also analyzed. KEGG enrichment analysis of unigenes with higher expression indicated their special roles in environmental stress response and adversity adaptation in the various five organs. In addition, we identified gene families and pathways related to biotic and abiotic stress factors, including TPS and GST families, terpenoid biosynthesis pathway and antioxidant defense system. This study not only provides a large number of transcriptome resources for understanding the adaptation to invasive environment but will also contribute to further research on the adaptive evolution and functional genomics and offer a cue for promoting better prevention and control of this species.
Materials and methods
Plant materials and RNA extraction
The sampling work was approved by Sun Yat-sen University, and our research complies with the laws of the People’s Republic of China. The voucher specimen was identified by Xiaoxian Ruan and stored at the Herbarium of Sun Yat-sen University (voucher number: Ruan201705).
The M. micrantha plant was selected from the Neilingding Island, Shenzhen, Guangdong Province, China (22°24′35.13"N, 113°48′37.09"E). Its five organs (root, stem, leaf, flower, and seed) were collected from the same plant and stored in RNAfixer (Bioteke Corporation). Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. RNA integrity was assessed using standard 1% agarose gel electrophoresis and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and its concentration was determined using a Nanophotometer (Implen, Munich, Germany). High-quality RNA was used for cDNA synthesis and library construction.
Illumina library preparation, sequencing, and de novo assembly
Illumina-based cDNA library for each organ was constructed using NEBNext Ultra RNA Library Prep Kit (NEB, USA), according to the manufacturer’s recommendations. The libraries were sequenced on an Illumina NovaSeq platform (Illumina, San Diego, CA, USA), generating 150 bp paired-end reads. The raw reads were quality-filtered using in-house Perl scripts to eliminate reads that contain adapters, more than 10% ambiguous bases (N), or more than 50% low-quality bases (Qphered ≤ 20). Clean reads were de novo assembled into unigenes using Trinity v2.4.064. To improve the sequence quality, the unigene sequences were assessed by using BLAST searching against the NR database with an E-value cutoff of 1 × 10–5. The unigenes with top hits to non-plant organisms were excluded.
PacBio library construction and SMRT sequencing
Total RNA for each of the five organs was used to construct the library separately following the PacBio SMRT sequencing experimental protocol. For each organ, three fractionated libraries (1–2 kb, 2–3 kb, and 3–6 kb) were constructed to eliminate loading bias. The libraries were sequenced on the PacBio Sequel platform (Pacific Biosciences, Menlo Park, CA, USA). Subreads were generated from the PacBio raw data by using SMRTlink v5.1 software (http://www.pacb.com/products-and-services/analytical-sofware/smrt-analysis/). The CCSs were obtained from subreads, and then classified into FLNC and non-full-length (nFL) reads based on the presence of 5′-primer, 3′-primer, and poly(A) tail. FLNC reads were clustered into consensus sequences by using ICE algorithm65. The consensus sequences were polished by nFL reads using the Arrow algorithm to obtain polished consensus sequences. The polished consensus sequences were further corrected by Illumina clean reads using LoRDEC software66, and redundant sequences were removed by using CD-HIT software (c = 0.95)67. Finally, the non-redundant transcripts (referred as unigenes) with top hits to non-plant organisms were excluded based on the results of NR annotation. The retained unigenes from five organs were merged and de-redundant to generate the reference transcriptome sequences of M. micrantha. The integrity of the reference transcriptome sequences was evaluated by BUSCO analysis with the core conserved gene set (embryophyta_odb9)68.
Functional annotation
The unigenes of the five organs were functionally annotated in seven public databases. NT database annotation was conducted using BLAST 2.7.1 + with an E-value cutoff of 1 × 10−5. Searches against Pfam were performed by using the Hmmscan function of HMMER69. GO annotation was performed based on Blast2GO (http://www.blast2go.com)70 and in-house scripts. The NR, KOG, and Swiss-Prot database annotations were conducted using Diamond v0.8.36 software71 with an E-value cutoff of 1 × 10−5. KEGG pathway analysis was conducted using the KAAS (KEGG Automatic Annotation Server)72 and KEGG database22 with an E-value cutoff of 1 × 10−10.
AS analysis and TFs identification
Coding GENome reconstruction Tool (Cogent v3.1, https://github.com/Magdoll/Cogent) was used to process non-redundant PacBio transcripts to create the k-mer profile. Based on the k-mer similarity, the De Bruijn graph method was used to reconstruct the transcripts into UniTransModels. GMAP-2017-06-2073 was applied to map transcripts to UniTransModels. Transcripts mapped to the same UniTransModels were examined to detect splicing junctions, and transcripts with the same splicing junctions were collapsed. Collapsed transcripts with different splicing junctions were defined as transcription isoforms. AS events were identified using SUPPA74 with default parameters. In addition, iTAK software75 was used to identify plant TFs in the five organs of M. micrantha.
Gene expression quantification, DEGs identification, and enrichment analysis
The Illumina clean reads for each organ were mapped to the reference transcriptome sequences using Bowtie276. The raw readcount value of each unigene in each organ was counted using RSEM software76, and then normalized to the FPKM (Fragments Per Kilobase of transcript per Million mapped reads). FPKM value was used to quantify the expression level of each unigene in each organ. Differentially expressed genes (DEGs) among the five organs were identified using the DEGSeq R package77. Benjamini–Hochberg multiple-testing correction was applied to adjust p-value for reducing the false discovery rate. Unigenes were assigned as DEGs with the criteria of corrected p-value < 0.005 and |log2(fold change)| > 1. KEGG pathways related to unigenes with higher expression in each organ were enriched using KOBAS v2.078. Corrected p-values (q values) were used to determine the significance of KEGG enrichment, with corrected p-values < 0.05 considered significant.
Analysis of gene families related to stress response and adaptation
Based on the functional annotations from NR, Swiss-Prot, KOG, and Pfam databases, the environmental stress-related gene families (TPSs and GSTs) were identified in M. micrantha. Furthermore, TPS and GST sequences were used to construct NJ phylogenetic trees using MEGA v5.1079. First, the TPS protein-coding sequences with complete domains and more than 500 amino acids were screened from the M. micrantha transcriptome. Second, sequences with more than 95% similarity were removed using CD-HIT67. Third, 30 TPS protein sequences of different angiosperms derived from GenBank were obtained (Table S14), which include all TPS subfamily in angiosperms. Finally, 16 TPS protein-coding sequences from M. micrantha transcriptome and 30 TPS protein sequences from GenBank were used to construct the TPS phylogenetic tree. The filtering of GST protein-coding sequences was consistent with that of TPS except for two conditions. The GST protein-coding sequences were more than 100 amino acids and had potential conserved motifs predicted by MEME Suite. MEME analysis parameters were set to -nmotifs 10, -minw 10, and -maxw 50. We finally used 23 GST protein-coding sequences from M. micrantha transcriptome to construct a GST phylogenetic tree. The family of GST protein-coding sequences was determined based on the classification in UniProt database (https://www.uniprot.org/). Using MAFFT v780 with the default parameters, the protein sequences of these two gene families were separately aligned. 1000 bootstrap replications were performed to ensure the reliability of phylogenetic trees. Phylogenetic trees were visualized using FigTree v1.4.281.
Ethics statements
This study has not involved humans and animals.
Data availability
The Illumina raw data have been deposited in NCBI Sequence Read Archive (SRA) database as follows: root: SRR10596657; stem: SRR10596656; leaf: SRR10596655; flower: SRR10596654; and seed: SRR10596653. The PacBio transcriptome data were available under the Bioproject PRJNA592884 on the NCBI SRA database.
References
Holm, L. G., Plucknett, D. L., Pancho, J. V. & Herberger, J. P. The world’s worst weeds (University Press of Hawaii, 1977).
Zhang, L. Y., Ye, W. H., Cao, H. L. & Feng, H. L. Mikania micrantha H.B.K. in China–an overview. Weed Res. 44, 42–49 (2004).
Banerjee, A. K., Mukherjee, A. & Dewanji, A. Potential distribution of Mikania micrantha Kunth in India−evidence of climatic niche and biome shifts. Flora 234, 215–223 (2017).
Cui, C., Wang, Z., Su, Y. J. & Wang, T. New insight into the rapid growth of the Mikania micrantha stem based on DIA proteomic and RNA-Seq analysis. J. Proteomics 236, 104126 (2021).
Sun, M. et al. GC-MS analysis of essential oil from different organs of Mikania micrantha. Guangdong Agr. Sci. 40, 111–115 (2013).
Zhang, M., Ling, B., Kong, C., Pang, X. & Liang, G. Chemical components of volatile oil from Mikania micrantha and its biological activity on insects. Chin. J. Appl. Ecol. 14, 93–96 (2003).
Yue, M. F. et al. Flooding with shallow water promotes the invasiveness of Mikania micrantha. Ecol. Evol. 9, 9177–9184 (2019).
Hu, L., Deng, T., Zhang, Q. & Li, M. The adaptation of Mikania micrantha to acid soi. Ecol. Environ. Sci. 25, 1439–1445 (2016).
Banerjee, A. K., Mukherjee, A., Guo, W. X., Liu, Y. & Huang, Y. L. Spatio-temporal patterns of climatic niche dynamics of an invasive plant Mikania micrantha Kunth and its potential distribution under projected climate change. Front. Ecol. Evol. 7, 291 (2019).
Ahmad, J. et al. Drought and salinity induced changes in ecophysiology and proteomic profile of Parthenium hysterophorus. PLoS One 12, e0185118 (2017).
Tsaballa, A. et al. Use of the de novo transcriptome analysis of silver-leaf nightshade (Solanum elaeagnifolium) to identify gene expression changes associated with wounding and terpene biosynthesis. BMC Genom. 16, 504 (2015).
Zhang, S. et al. EalspF1, essential enzyme in isoprenoid biosynthesis from Eupatorium adenophorum, reveals a novel role in light acclimation. J. Integr. Agr. 13, 1024–1035 (2014).
Sharma, P., Jha, A. B., Dubey, R. S. & Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 1–26 (2012).
Wu, M., Li, Z. Y. & Wang, J. B. Transcriptional analyses reveal the molecular mechanism governing shade tolerance in the invasive plant Solidago canadensis. Ecol. Evol. 10, 4391–4406 (2020).
Pichersky, E. & Raguso, R. A. Why do plants produce so many terpenoid compounds?. New Phytol. 220, 692–702 (2018).
Huang, Y. L. et al. Transcriptome analysis of an invasive weed Mikania micrantha. Biol. Plantarum 56, 111–116 (2012).
Yang, M. et al. The emergence of the hyperinvasive vine, Mikania micrantha (Asteraceae), via admixture and founder events inferred from population transcriptomics. Mol. Ecol. 26, 3405–3423 (2017).
Guo, W. X. et al. Comparative transcriptome analysis of the invasive weed Mikania micrantha with its native congeners provides insights into genetic basis underlying successful invasion. BMC Genomics 19, 392 (2018).
Mátyás, K. K. et al. Different expression pattern of flowering pathway genes contribute to male or female organ development during floral transition in the monoecious weed Ambrosia artemisiifolia L. (Asteraceae). PeerJ 7, e7421 (2019).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 13, 278–289 (2015).
Ye, W. B. et al. The full-length transcriptome of Spartina alterniflora reveals the complexity of high salt tolerance in monocotyledonous halophyte. Plant Cell Physiol. 61, 882–896 (2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Gliwicka, M., Nowak, K., Balazadeh, S., Mueller-Roeber, B. & Gaj, M. D. Extensive modulation of the transcription factor transcriptome during somatic embryogenesis in Arabidopsis thaliana. PLoS One 8, e69261 (2013).
Liu, D. F. et al. Full-length transcriptome analysis of Phytolacca americana and its congener P. icosandra and gene expression normalization in three Phytolaccaceae species. BMC Plant Biol. 20, 396 (2020).
Zhang, B., Liu, J. X., Wang, X. S. & Wei, Z. W. Full-length RNA sequencing reveals unique transcriptome composition in bermudagrass. Plant Physiol. Bioch. 132, 95–103 (2018).
Pi, B. Y., He, X. H., Ruan, Y., Jang, J. C. & Huang, Y. Genome-wide analysis and stress-responsive expression of CCCH zinc finger family genes in Brassica rapa. BMC Plant Biol. 18, 373 (2018).
Jiao, Z. C. et al. Genome-wide study of C2H2 zinc finger gene family in Medicago truncatula. BMC Plant Biol. 20, 401 (2020).
Lu, R. et al. Characterization of bHLH/HLH genes that are involved in brassinosteroid (BR) signaling in fiber development of cotton (Gossypium hirsutum). BMC Plant Biol. 18, 304 (2018).
Wang, K., Ding, Y. F., Cai, C., Chen, Z. X. & Zhu, C. The role of C2H2 zinc finger proteins in plant responses to abiotic stresses. Physiol. Plantarum 165, 690–700 (2019).
Jiang, A. L. et al. Genome-wide analysis of the C3H zinc finger transcription factor family and drought responses of members in Aegilops tauschii. Plant Mol. Biol. Rep. 32, 1241–1256 (2014).
Aslam, M. Z., Lin, X., Li, X., Yang, N. & Chen, L. Q. Molecular cloning and functional characterization of CpMYC2 and CpBHLH13 transcription factors from Wintersweet (Chimonanthus praecox L.). Plants 9, 785 (2020).
Sun, X., Wang, Y. & Sui, N. Transcriptional regulation of bHLH during plant response to stress. Biochem. Bioph. Res. Co. 503, 397–401 (2018).
Zvi, M. M. B. et al. PAP1 transcription factor enhances production of phenylpropanoid and terpenoid scent compounds in rose flowers. New Phytol. 195, 335–345 (2012).
Ding, F. et al. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genomics 15, 431 (2014).
Marquez, Y., Brown, J. W. S., Simpson, C., Barta, A. & Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 22, 1184–1195 (2012).
Hou, S., Zhang, C., Yang, Y. & Wu, D. Recent advances in plant immunity: recognition, signaling, response, and evolution. Biol. Plantarum 57, 11–25 (2013).
Kuroha, T. et al. Ethylene-gibberellin signaling underlies adaptation of rice to periodic flooding. Science 361, 181–186 (2018).
Qin, X. X. et al. Phosphoproteomic analysis of lettuce (Lactuca sativa L.) reveals starch and sucrose metabolism functions during bolting induced by high temperature. PLoS One 15, e0244198 (2020).
Nakabayashi, R., Mori, T. & Saito, K. Alternation of flavonoid accumulation under drought stress in Arabidopsis thaliana. Plant Signal. Behav. 9, e29518 (2014).
Wang, L. L. et al. GbFT, a FLOWERING LOCUS T homolog from Ginkgo biloba, promotes flowering in transgenic Arabidopsis. Sci. Hortic. 247, 205–215 (2019).
He, M., Qin, C. X., Wang, X. & Ding, N. Z. Plant unsaturated fatty acids: biosynthesis and regulation. Front. Plant Sci. 11, 390 (2020).
Zhao, L., Zhao, X. J., Francis, F. & Liu, Y. Genome-wide identification and characterization of the TPS gene family in wheat (Triticum aestivum L.) and expression analysis in response to aphid damage. Acta Physiol. Plant. 43, 64 (2021).
Chen, F., Tholl, D., Bohlmann, J. & Pichersky, E. The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229 (2011).
Jiang, S. Y., Jin, J. J., Sarojam, R. & Ramachandran, S. A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 11, 2078–2098 (2019).
Zhou, H. C., Shamala, L. F., Yi, X. K., Yan, Z. & Wei, S. Analysis of terpene synthase family genes in Camellia sinensis with an emphasis on abiotic stress conditions. Sci. Rep. 10, 933 (2020).
Liu, Y. et al. Characterization of defensive cadinenes and a novel sesquiterpene synthase responsible for their biosynthesis from the invasive Eupatorium adenophorum. New Phytol. 229, 1740–1754 (2021).
Frova, C. The plant glutathione transferase gene family genomic structure, functions, expression and evolution. Physiol. Plantarum 119, 469–479 (2003).
Yang, Q., Liu, Y. J. & Zeng, Q. Y. Overexpression of three orthologous glutathione S-transferases from Populus increased salt and drought resistance in Arabidopsis. Biochem. Syst. Ecol. 83, 57–61 (2019).
Srivastava, D., Verma, G., Chauhan, A. S., Pande, V. & Chakrabarty, D. Rice (Oryza sativa L.) tau class glutathione S-transferase (OsGSTU30) overexpression in Arabidopsis thaliana modulates a regulatory network leading to heavy metal and drought stress tolerance. Metallomics 11, 375–389 (2019).
Stavridou, E. et al. Tolerance of transplastomic tobacco plants overexpressing a Theta class glutathione transferase to abiotic and oxidative stresses. Front. Plant Sci. 9, 1861 (2018).
Takesawa, T., Ito, M., Kanzaki, H., Kameya, N. & Nakamura, I. Over-expression of zeta glutathione S-transferase in transgenic rice enhances germination and growth at low temperature. Mol. Breeding 9, 93–101 (2002).
Wang, W. B. et al. Molecular cloning and functional characterization of a sweetpotato chloroplast IbDHAR3 gene in response to abiotic stress. Plant Biotechnol. Rep. 14, 9–19 (2020).
Lallement, P. A., Brouwer, B., Keech, O., Hecker, A. & Rouhier, N. The still mysterious roles of cysteine-containing glutathione transferases in plants. Front. Pharmacol. 5, 192 (2014).
Filippou, P., Bouchagier, P., Skotti, E. & Fotopoulos, V. Proline and reactive oxygen/nitrogen species metabolism is involved in the tolerant response of the invasive plant species Ailanthus altissima to drought and salinity. Environ. Exp. Bot. 97, 1–10 (2014).
Hossain, M. A., Piyatida, P., da Silva, J. A. T. & Fujita, M. Molecular mechanism of heavy metal toxicity and tolerance in plants: central role of glutathione in detoxification of reactive oxygen species and methylglyoxal and in heavy metal chelation. J. Bot. 2012, 1–37 (2012).
Zhang, L. W. et al. Differentially expressed genes related to oxidoreductase activity and glutathione metabolism underlying the adaptation of Phragmites australis from the salt marsh in the Yellow River Delta, China. PeerJ 8, e10024 (2020).
Zhang, L. L., Sun, F. F. & Wen, D. Z. Effects on irradiance and soil moisture on antioxidants and membrane lipid peroxidation products in Mikania micrantha and Chromolaena odoratum. Chin. J. Plant Ecol. 33, 974–983 (2009).
Zhang, L. L. & Wen, D. Z. Photosynthesis, chlorophyll fluorescence, and antioxidant enzyme responses of invasive weed Mikania micrantha to Bemisia tabaci infestation. Photosynthetica 46, 457–462 (2008).
Pu, X. J., Dong, X. M., Li, Q., Chen, Z. X. & Liu, L. An update on the function and regulation of methylerythritol phosphate and mevalonate pathways and their evolutionary dynamics. J. Integr. Plant Biol. 63, 1211–1226 (2021).
Scrivanti, L. R. & Anton, A. M. Allelopathic effect of endemic South American Bothriochloa species (Poaceae: Andropogoneae). J. Essent. Oil Res. 31, 247–254 (2018).
Lamine, M., Gargouri, M. & Mliki, A. Identification of the NaCl-responsive metabolites in Citrus roots: a lipidomic and volatomic signature. Plant Signal. Behav. 15, 1777376 (2020).
Fu, J. Y. Molecular cloning and expression analysis of a putative sesquiterpene synthase gene from tea plant (Camellia sinensis). Acta Physiol. Plant. 35, 289–293 (2012).
Kim, J. Phytotoxic and antimicrobial activities and chemical analysis of leaf essential oil from Agastache rugosa. J. Plant Biol. 51, 276–283 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Gordon, S. P. et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One 10, e0132628 (2015).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514 (2014).
Fu, L. M., Niu, B. F., Zhu, Z. W., Wu, S. T. & Li, W. Z. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Eddy, S. R. Profile hidden Markov models. Bioinformatics 14, 755–763 (1998).
Conesa, A. et al. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Wu, T. D., Reeder, J., Lawrence, M., Becker, G. & Brauer, M. J. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. In Statistical Genomics: Methods and Protocols, (Mathé, E., Davis, S., Eds.) 1418, 283–334 (Springer: New York, NY, USA 2016).
Alamancos, G. P., Pagès, A., Trincado, J. L., Bellora, N. & Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 21, 1521–1531 (2015).
Zheng, Y. et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670 (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Wang, L. K., Feng, Z. X., Wang, X., Wang, X. W. & Zhang, X. G. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Mao, X. Z., Cai, T., Olyarchuk, J. G. & Wei, L. P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793 (2005).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Rambaut, A. FigTree v1.4.2: Tree Figure Drawing Tool. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 5 March 2019) (2014).
Acknowledgements
We thank Qiqi Huang, Ning Li, Ruixiang Xu, and Shufeng Li for sampling.
Funding
This work was supported by the National Natural Science Foundation of China [31670200, 31770587, 31872670, and 32071781], Guangdong Basic and Applied Basic Research Foundation [2021A1515010911], and Project of Department of Science and Technology of Shenzhen City, Guangdong, China [JCYJ20190813172001780 and JCYJ20210324141000001].
Author information
Authors and Affiliations
Contributions
X.R. performed the data analysis and wrote the manuscript; Z.W. checked English grammar; Y.S. designed the experiments and wrote the manuscript; T.W. wrote and approved the final manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruan, X., Wang, Z., Su, Y. et al. Full-length transcriptome analysis of multiple organs and identification of adaptive genes and pathways in Mikania micrantha. Sci Rep 12, 3272 (2022). https://doi.org/10.1038/s41598-022-07198-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07198-0
- Springer Nature Limited