
Abstract
The SnRK gene family is a key regulator that plays an important role in plant stress response by phosphorylating the target protein to regulate subsequent signaling pathways. This study was aimed to perform a genome-wide analysis of the SnRK gene family in wheat and the expression profiling of SnRKs in response to abiotic stresses. An in silico analysis identified 174 SnRK genes, which were then categorized into three subgroups (SnRK1/2/3) on the basis of phylogenetic analyses and domain types. The gene intron–exon structure and protein-motif composition of SnRKs were similar within each subgroup but different amongst the groups. Gene duplication and synteny between the wheat and Arabidopsis genomes was also investigated in order to get insight into the evolutionary aspects of the TaSnRK family genes. The result of cis-acting element analysis showed that there were abundant stress- and hormone-related cis-elements in the promoter regions of 129 SnRK genes. Furthermore, quantitative real-time PCR data revealed that heat, salt and drought treatments enhanced TaSnRK2.11 expression, suggesting that it might be a candidate gene for abiotic stress tolerance. We also identified eight microRNAs targeting 16 TaSnRK genes which are playing important role across abiotic stresses and regulation in different pathways. These findings will aid in the functional characterization of TaSnRK genes for further research.
Similar content being viewed by others
Introduction
Bread wheat (T. aestivum L.) is a major cereal crop and an important source of carbohydrates and protein in the human diet, accounting for 20% of daily calorie consumption. The most significant economic feature is grain yield, which is influenced by a variety of biotic and abiotic stressors. By 2050, it is expected that the demand for wheat will increase by 60%1. Plants use a variety of molecular defence mechanisms to deal with abiotic stresses such as salt, drought, and heat. Plants respond to environmental stresses in two ways: gene expression regulation and protein modification2. Protein kinase-mediated phosphorylation and dephosphorylation are important in protein modification3. SnRKs (Sucrose nonfermenting 1 (SNF1)-related protein kinases) are a group of protein kinase genes that have a role in a variety of physiological activities4. Based on their sequence similarities and gene architectures, plant SnRKs may be split into three subfamilies: SnRK1, SnRK2, and SnRK34,5. The SnRK1 subfamily has a highly conserved N-terminal protein kinase (Pkinase) domain, which is similar to SNF1 in yeast and AMPKs in mammals6. The SnRK2 and SnRK3 subfamilies are unique to plants, and both show more variability than the SnRK1 subfamily members in terms of plant diversity. A conserved P kinase domain and a C-terminal variable adjusting domain are found in members of the SnRK2 family7. SnRK3, known as CIPK (CBL-interacting protein kinases), also has conserved N-terminal protein kinase domains and NAF domains, as well as PPI domains at the C-terminus8,9.
Plant cells respond to starvation and metabolic stress through the SnRK1 family of genes. Catalytic components of heterotrimeric complexes, SnRK1 kinases interact with two additional subunits10. SnRK1 was shown to be involved in the stimulation of sucrose synthase expression and had a key function in carbohydrate metabolism control in S. tuberosum11. Low energy stress (e.g., darkness and hypoxia) may also cause SnRK1 nuclear translocation and subsequent induction of SnRK1 target genes, allowing for the replenishment of cellular energy for plant development12. SnRK1 genes were also shown to be hubs in a network of signalling pathways that included cell cycle control, pathogen responses, and meristem formation, as reported earlier13.
The SnRK2 genes, on the other hand, play a significant role in plants' responses to abiotic stressors, particularly osmotic and salt stress. In A. thaliana, for example, SnRK2.10 phosphorylates multiple target genes, including the dehydrins ERD10 and ERD14, to deal with osmotic stress14. In N. tabacum, SnRK2.1 regulates salt tolerance positively15,16. SnRK2 subfamily genes in A. thaliana may be divided into three groups: ABA-independent kinases, genes responsive to drought stress, and kinases that are substantially activated by ABA7,17. The most comprehensive study on ABA-dependent group 3 kinases is now underway. For example, one of the SnRK2 family genes, AtSnRK2.6 (OST1), is involved in the ABA signalling pathway in stomatal guard cells, and OST1 protein stability may be regulated by the E3-ubiquitin ligase HOS15 in Arabidopsis to reduce ABA signal sensitivity18,19.
CBL (calcium sensor calcineurin B-like proteins)-interacting protein kinases (CIPKs), also known as SnRK3 kinases, play an important role in plant stress tolerance4,20. The SOS (salt excessively sensitive) system, for example, was the first CBL-CIPK route found in A. thaliana. SOS3 (AtCBL4) on the cell membrane recognised the calcium signal created by salt stress, and then SOS3 joined with SOS2 (AtCIPK24) to phosphorylate SOS1 (Na+/H+ antiporter) to remove excess Na+ from root cells21,22. MdCIPK13 and MdCIPK22 also improved salt and drought tolerance in apple by phosphorylating the sucrose transporter MdSUT2.223,24. In B. napus, overexpression of BnCBL1-BnCIPK6 may improve high salinity tolerance and low potassium tolerance25. Finally, growing data has highlighted the relevance of SnRKs function in nutrient consumption and stress response, and researchers may be able to increase plant stress resistance by genetic modification of these genes.
The importance of SnRK genes function in nutrient consumption and stress response is becoming clearer, and researchers may be able to improve plant stress resistance by genetically modifying these genes. However, there is no information available on SnRK genes in wheat, one of the most important cereals. In the current study, we identified 174 SnRK gene family members in the genome of Triticum aestivum. We also evaluated their evolutionary relationships, gene architecture, protein motifs, chromosomal position, and cis-elements in promoter regions in a systematic way. The differential expression of the SnRK genes under abiotic stress was also investigated using qRT-PCR. The results obtained in this study provide important insights into the molecular pathways that underpin stress tolerance and molecular breeding.
Results
Identification of SnRK genes of wheat
A total of 174 SnRK genes were identified and further subdivided into three categories: SnRK 1, SnRK 2 and SnRK3 which have 14, 65 and 95 genes, respectively (Supplementary Table S1). The number of SnRK gene in wheat is higher than other plant species e.g. A. thaliana and rice. It may be due to hexaploid nature of wheat genome. Each protein's longest amino acid sequence was chosen for further investigation. The parameters of the gene characteristics including Gene IDs, amino acid length, molecular weights (MW), isoelectric points (pI), grand average of hydropathicity (GRAVY) and sub-cellular localization were analysed (Table 1). The molecular weight of all these TaSnRK proteins spans from 26.8 to 146.1 kDa, while the amino acid length spans from 239 to 1335. The isoelectric point of SnRKs family ranged from 4.3 to 9.4 indicating basic nature of proteins. The hydrophobicity and hydrophilicity are revealed by the GRAVY scores. SnRK proteins from T. aestivum, on the other hand, show a negative GRAVY score, implying that they are hydrophilic. The degree of hydrophilicity, on the other hand, is proportionate to the increased variability. According to the expected subcellular localization data, most of the TaSnRK genes are expressed in the nucleus and cytoplasm, followed by chloroplast and mitochondria (Table 1).
Phylogenetic analysis of SnRK genes family
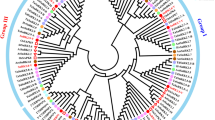
To study the evolutionary relationships among SnRK proteins and their classification, we performed unrooted phylogenetic tree analysis using the full-length amino acid sequences of 174 SnRKs genes of T. aestivum, 38 of A. thaliana and 50 of O. sativa (Fig. 1). The clustering of 38 AtSnRKs into three groups was reported earlier3. Based on the phylogenetic analysis and domains presence, 174 TaSnRKs were also divided into three groups in this study. Of these, 14 proteins in the TaSnRK1 subfamily have Pkinase (PF00069 of Pfam), UBA (PF00627), and KA1 (PF02149) domains, whereas 65 proteins in the TaSnRK2 subfamily have Pkinase domains with strong resemblance to AtSnRK2 subfamily, and 95 proteins in the SnRK3 subfamily have Pkinase and NAF (PF03822) domain (Fig. 1).

Phylogenetic tree of SnRK genes from T. aestivum, A. thaliana, and O. sativa. 174 SnRK genes from wheat, 38 from Arabidopsis and 50 from rice were clustered into three subgroups (SnRK1, SnRK2 and SnRK3). Wheat SnRK1, 2 and 3 subgroups are denoted as blue, green and red, respectively. The tree was generated using MEGA X software using the neighbor-joining method.
Motif composition and gene structural analysis of the SnRK gene family in T. aestivum
MEME analysis showed that 10 conserved motifs were identified in TaSnRK proteins (Fig. 2A). The conserved motif's sequence and length details have been listed in Supplementary Table S1. The conserved Pkinase domain including the pattern 1, 2, 3, 5, 6 was discovered in all TaSnRK genes in this investigation (Supplementary Fig. 1). Furthermore, TaSnRK genes from the same subfamily have comparable pattern compositions, but TaSnRK genes from different subfamilies have varied motif compositions. TaSnRK1 subfamily genes have 9 motif (motifs 1, 2, 3, 4, 5, 6, 7, 8 and 10) while TaSnRK2 subfamily genes had either motifs 1, 2, 4, 6, 8 or motifs 1, 2, 3, 4, 5 and 10. TaSnRK3 subfamily genes had 10 motifs while a few of them do not have any motif (Fig. 2A). In conclusion, the comparable gene architectures and conserved motif compositions of SnRK genes within the same subfamily substantially support the phylogenetic analysis based subfamily classifications.

(A) Arrangement of ten conserved motifs in the TaSnRK genes following MEME analysis. Different colored boxes represent different motifs and their position in each sequence of TaSnRK genes. (B) Gene structure of wheat SnRK. Exons are indicated in yellow rectangles and grey line connecting two exons represent introns.
The exon–intron structure of TaSnRK genes showed that the TaSnRK1 subfamily genes have more than 10 exons, while the TaSnRK2 subfamily genes have 2–34 exons followed by TaSnRK3 subfamily which have 1–33 exons (Fig. 2B). Notably, 14 genes are intron-less. In addition, the TaSnRK3 family is divided into two subgroups. The genes in SnRK3 subgroup 1 had more than 10 exons, while the genes in subgroup 2 had less than four exons.
Analysis of chromosomal location and orthologous genes in T. aestivum
The chromosomal distribution of all TaSnRK genes across the genome was investigated which provides useful information on the genomic regions (Fig. 3). The A sub-genome had 56 TaSnRK genes, comprising 2 genes from TaSnRK1 subfamily, 21 genes from TaSnRK2 subfamily, and 33 genes from TaSnRK3 subfamily, whereas the B sub-genome had 61 genes, comprising 8 genes from TaSnRK1 subfamily, 21 of TaSnRK2 subfamily, and 32 of TaSnRK3 subfamily. However, 4 genes of TaSnRK1 subfamily, 23 genes of TaSnRK2 subfamily, and 30 genes of TaSnRK3 subfamily were found on the D sub-genome (Supplementary Fig. S2A). These findings suggested that TaSnRK genes were distributed randomly throughout the A, B, and D chromosomes (Supplementary Fig. S2B).

Distribution of TaSnRK genes on the 21 chromosomes of wheat and within the three sub-genomes. Black lines representing the gene pairs. Physical map showing the chromosomal distribution, with position on the left side scale bar and the name of the gene on right side.
In this study, 57 orthologous pairings were identified between T. aestivum and H. valgare, while 166 between T. aestivum and A. thaliana. However, 53 orthologous genes were found within wheat species for instance between T. aestivum and T. urartu, while 102 orthologous genes between T. aestivum and Ae. dicoccoides and 63 between T. aestivum and Ae. tauschii (Supplementary Table S3).
We identified 11 segmental events across different chromosomes and 2 tandom duplication occurrences in the same chromosome using the BLAST and MCScanX techniques. The findings revealed that gene duplication may have produced some TaSnRK genes, and that segmental duplication events were important in the growth of TaSnRK genes in the wheat genome. We also looked into the frequency of tandem duplication occurrences. There were 45 TaSnRK gene pairs found in this area all of which were strongly related. (Supplementary Table S4). However, the identities of these were > 80%, indicating that they were included into tandem duplication occurrences.
We looked at the duplication events of the TaSnRK gene in the wheat genome since gene duplication has a big impact on the emergence of new functionalities and gene families. In addition, 55 gene pairs were found to be duplicated as shown in Fig. 4.

The synteny analysis of TaSnRK family in T. aestivum. Different colours represent SnRK subfamilies red lines indicate duplicated TaSnRK1 subfamily gene pairs, green lines indicated TaSnRK2 subfamily gene pairs and blue represented TaSnRK3 subfamily. The chromosome number is indicated at the bottom of each chromosom.
Furthermore, the synteny of SnRK gene pairs between T. aestivum genome and A. thaliana genome was performed. The result showed that 166 SnRK genes of T. aestivum exhibiting syntenic relationship with AtSnRK genes (Fig. 5 and Supplementary Table S3), suggesting that these genes might have contributed to the evolution of TaSnRK gene family. Ks values, Ka values, Ka/Ks ratios and divergence time of paralogous and orthologous on SnRK family genes were estimated to assess the evolutionary constraints undertaking (Supplementary Table S4). The Ka/Ks ratios of the majority of segmentally duplicated TaSnRK gene pairs were < 1, the mean values of TaSnRK3 gene pairs (Ka/Ks = 0.30) and TaSnRK2 (Ka/Ks = 0.35) were lower than TaSnRK1 (Ka/Ks = 0.41). Furthermore, segmental gene divergence time spans from 18.76 to 34.97 Mya. These findings showed that the TaSnRK gene family may have been subjected to significant purifying selection during evolution.

Synteny analysis of SnRK genes between Triticum aestivum (Orange colour) and Arabidopsis thaliana (green colour). Gray lines in the background indicate the collinear blocks within T. aestivum and A. thaliana, while the red lines highlight the syntenic SnRK gene pairs.
Promoter analysis
PlantCARE was used to look at cis-elements (1.5 kb upstream from ATG) in order to better understand the function and regulatory processes of TaSnRK genes. We found 129 out of 174 TaSnRK genes had cis-elements (Supplementary Fig. S3, Supplementary Table S5). MyB, ABRE, and LTRE cis-elements were found to be involved in drought, ABA, and low-temperature responses. Auxin (9.77%), MeJA (51.72%), and Gibberallin (11.49%) cis-elements were found in phytohormones. It was also shown that most genes have many cis-element types. In addition, the TaSnRK1 (30.50) family had more cis-elements than the TaSnRK2 (19.29) or TaSnRK3 (23.81) families (Supplementary Table S5). Finally, the cis-elements study revealed that most TaSnRK genes can respond to a variety of environmental challenges, and that distinct subfamily genes can be regulated in various ways.
MicroRNAs targeting TaSnRK
The role of miRNAs in controlling the expression of TaSnRK genes have been investigated using the psRNATarget server. We predicted 16 TaSnRK genes as possible targets for eight different miRNAs (Table 2). Tae-miR319 implicated in the regulation of seven TaSnRK genes (TaSnRK3.33, TaSnrK3.36, TaSnRK2.37, TaSnRK2.35, TaSnRK2.36, TaSnRK3.38, TaSnRK2.39). Tae-miR408 accounted for regulating expression of three TaSnRK genes (TaSnRK3.63, TaSnRK3.64, TaSnRK3.62). The expression of TaSnRK genes (TaSnRK1.3, TaSnRK1.2, and TaSnRK2.39, TaSnRK2.38) may be influenced by the Tae-miR164 and Tae-miR167b (Table 2). Tae-miR1119 and Tae-miR160 were predicted to regulate the expression of TaSnRK3.38 and TaSnRK3.74, respectively.
Functional annotation of Hub genes and interacting network analysis
Based on their involvement in a biological and cellular process, we studied the function of potential hub genes. String database contains a collection of predicted and experimentally confirmed protein–protein interactions in wheat and other species. However, wheat SnRK found in string database is linked to them as well as numerous metabolic and regulatory processes. SnRK were defined based on the interaction observed. TaSnRK2.48 is included in the first, which serves as the network’s (Fig. 6). Here, the cluster is directly linked to TaSnRK3 subfamily (TaSnRK3.7, TaSnRK 3.14, TaSnRK 3.24, TaSnRK3.32, TaSnRK3.33, TaSnRK3.38, TaSnRK3.41, TaSnRK3.44, TaSnRK3.55, TaSnRK3.62 TaSnRK3.71, TaSnRK3.74, TaSnRK3.81, TaSnRK3.82, TaSnRK3.84 and TaSnRK3.93). These genes involved in Ca-dependent processes like as autophagy and maintenance. The second interaction with SnRK 2 subfamily (TaSnRK2.12, TaSnRK2.27 and TaSnRK2.36) was primarily linked to OST. Whereas the primary role of OST is to act as an activator of the abscisic acid (ABA) signalling pathway, which governs several ABA responses including stomata closure in response to dehydration, plant diseases, or changes in atmospheric relative humidity (RH). Pathogen-associated molecular patterns (PAMPs) (e.g. flg22 and LPS) of pathogenic bacteria such as P.syringae pv. tomato (Pst) and E. coli O157:H7 are required for stomata closer. TaSnRK 2.28 and TaSnRK1.9 have a direct connection that plays a crucial function in fatty acid synthesis. Further, there is no direct evidence of a direct interaction between TaSnRK2.61 and TaSnRK2.48 (Fig. 6). Apart from that, TaSnRK2.61 is the network’s second hub gene, which mainly interacts with SnRK2 subfamily, such as (TaSnRK2.16, TaSnRK2.28, TaSnRK2.31, TaSnRK2.38 and TaSnRK3.71), which are expressed preferentially in guard cells and appear to be involved ABA signalling mediating by reactive oxygen species.

The predicted protein–protein interactions network of SnRK in wheat based on the Arabidopsis orthologs using Cytoscape.
Expression profile of SnRK genes in different tissues under abiotic stress
To understand more about how TaSnRKs are involved in development and stress responses, we utilized the expVIP database to retrieve TPM values for all TaSnRKs from experiments including abiotic stress and various developmental stages. These TPM data were used to generate Heatmaps. The expression patterns of 174 TaSnRK genes in various wheat tissues were evaluated.
Five different tissues at three different developmental stages were taken for this study. The time points are the Zadoks scale (Z-scale). Different TaSnRK sub-families show differential induction in different tissues (Fig. 7A). TaSnRKs such as TaSnRK1.1, TaSnRK1.2, TaSnRK1.3, TaSnRK1.4, TaSnRK1.5, TaSnRK1.6, TaSnRK1.8, TaSnRK1.9 and TaSnRK1.10, TaSnRK2.1, TaSnRK2.2, TaSnRK2.3, TaSnRK2.4, TaSnRK2.5, TaSnRK2.6, TaSnRK2.7, TaSnRK2.8, TaSnRK2.9, TaSnRK2.10, TaSnRK2.11, TaSnRK2.12, TaSnRK2.16, TaSnRK2.17, TaSnRK2.18, TaSnRK2.25, TaSnRK2.26, TaSnRK2.27, TaSnRK2.50, TaSnRK2.51, TaSnRK2.55, TaSnRK2.56, TaSnRK2.57, TaSnRK2.62, TaSnRK2.64, TaSnRK3.16, TaSnRK3.17, TaSnRK3.18, TaSnRK3.19, TaSnRK3.30, TaSnRK3.31, TaSnRK3.43, TaSnRK3.44, TaSnRK3.50, TaSnRK3.53, TaSnRK3.54, TaSnRK3.55, TaSnRK3.68, TaSnRK3.69, TaSnRK3.70, TaSnRK3.71, TaSnRK3.72, TaSnRK3.86, TaSnRK3.87, TaSnRK3.89, TaSnRK3.90 and TaSnRK3.95 showed induction in spike tissues at Z39 stage. Whereas, TaSnRK1.1, TaSnRK1.2, TaSnRK1.3, TaSnRK1.4, TaSnRK1.5, TaSnRK1.6, TaSnRK1.8, TaSnRK1.9 and TaSnRK1.10 etc. were up regulated in leaves tissues at Z75 stage. Multiple TaSnRKs were induced at different spike stages especially at Z39 stage. Forty genes of TaSnRK3 subfamily were induced in root tissues at Z10 stage. This suggests different TaSnRKs might be involved in development of different tissues at different stages. Many TaSnRKs showed induction at the dough development stage in wheat such as TaSnRK1.1, TaSnRK1.2, TaSnRK1.3, TaSnRK1.8 to, TaSnRK1.9 and TaSnRK1.10, TaSnRK2.1, TaSnRK2.2, TaSnRK2.3, TaSnRK2.7, TaSnRK2.8, TaSnRK2.9, TaSnRK2.10, TaSnRK2.11, TaSnRK2.12, TaSnRK2.14, TaSnRK2.15, TaSnRK2.16, TaSnRK2.17, TaSnRK2.18, TaSnRK2.25, TaSnRK2.26, TaSnRK2.27, TaSnRK2.35, TaSnRK2.36, TaSnRK2.37, TaSnRK2.38, TaSnRK2.39, TaSnRK2.49, TaSnRK2.50, TaSnRK2.51, TaSnRK2.55, TaSnRK2.56, and TaSnRK2.57. However, the expression of TaSnRK3 subfamily was down regulated in Z85 stage as the grain matures (Fig. 7A). Very few members of the TaSnRKs sub-family showed no induction during any of the different developmental stages e.g., TaSnRK1.7, TaSnRK1.11, TaSnRK1.12, TaSnRK1.13, TaSnRK1.14, TaSnRK2.43, TaSnRK2.44, TaSnRK2.45, TaSnRK2.46, TaSnRK3.37, TaSnRK3.40, TaSnRK3.42, TaSnRK3.51, TaSnRK3.82, and TaSnRK3.83. The SnRK subfamily 1, 2 and 3 were induced in leaf tissue at Z10 stages.

Heatmap of TaSnRK genes (A) tissue specific expression in leaf, root, shoot and spike at seedling, vegetative and reproductive stages (B) abiotic stress specific expression under drought, heat and osmotic stress at seedling stage. The color scale represents log2 expression values. Ck-control; HS1-heat stress at 1hr; HS2-heat stress at 6hr; DS1- drought stress at 1hr; DS2- drought stress at 6hr; PEG1- osmotic stress at 2hr; PGE2- osmotic stress at 12 hr.
The expression patterns of TaSnRK family genes in wheat tissues under abiotic stresses were evaluated (Fig. 7B). A total of 16 genes with high expression levels in all studied tissues under abiotic stresses (log2-based values > 5) were assigned to sub-group1. SnRK3.40, for example, was shown to be highly expressed in all vegetative organs, with log2-based average values. In sub-group2, the expression levels of 55 TaSnRK genes were significantly lower across all detected tissues (log2-based values > 2). In sub-group3, 103 TaSnRK genes were involved with the lowest expression levels in diverse tissues at various stages. Meanwhile, sub-group1 had one TaSnRK1, 6 TaSnRK2 genes and 9 TaSnRK3 genes; Sub-group2 had 8 TaSnRK1, 21 TaSnRK2, and 26 TaSnRK3 genes and Sub-group3 had 5 TaSnRK1, 38 TaSnRK2, and 69 TaSnRK3 genes (Fig. 7A). The expression pattern of TaSnRK genes was nonetheless studied using drought, salinity and heat stresses26. The expression levels of TaSnRK genes have been significantly changed under various abiotic stresses (Fig. 7B). TaSnRK3.17 and TaSnRK3.18, for example, were highly stimulated by all treatments, whereas TaSnRK3.15 and TaSnRK3.16 responded to dehydration by increasing their levels of expression. But, TaSnRK 3.37, TaSnRK3.45, and TaSnRK3.46 have demonstrated high expression levels in response to heat and drought stress (Fig. 7B). These results demonstrated that TaSnRKs have a large variety of patterns of expression and that even genes within the same subfamily had different patterns of expression.
Validation of TaSnRK for abiotic stresses using qRT-PCR
Members of the SnRK family serve critical roles in plant abiotic stress response. In plants, however, the specific mechanism underpinning SnRK function is not fully understood. The expression analysis of SnRK2s and SnRK3s in leaf tissue was identified by qRT-PCR following different stress treatments at different time intervals. This was to study the function of TaSnRK2 and TaSnRK3 genes in responding to salt, heat, and drought stress (Fig. 8).

qRT-PCR based differential expression analysis of 10 TaSnRK genes under abiotic stress. Heat stress treatment at 37 °C (A) HS240, & (B) RAJ3765; Heat stress treatment at 42 °C (C) HS240, & (D) RAJ3765; Salt stress treatment at 150 mM NaCl (E) Kharchia65, (F) HD2687 at 12 h and (G) Kharchia 65, (H) HD 2687 for 24 h; drought stress at 25% (v/v) PEG treatment (I) C306 and (J) Wl711 for 24 h. The error bars indicate the standard deviation.
TaSnRK2.16 expression was fivefold higher in HS240 after a 37 °C heat stress treatment (Fig. 8A), followed by TaSnRK3.74 (~ twofold) and TaSnRK2.16 (~ three fold) after a 42 °C heat stress (Fig. 8C). At 37 °C, the expression levels of four genes, TaSnRK2.16, TaSnRK2.11, TaSnRK3.23, and TaSnRK2.8 were up-regulated (Fig. 8B) compared to 42 °C heat stressed Raj3765 genotype (Fig. 8D). Furthermore, expressions of SnRK genes were carried out at different time intervals to investigate the role of SnRK during salt stress treatment. Three genes, TaSnRK2.11, TaSnRK2.12, and TaSnRK3.23 had up to eightfold higher expression levels in Kharchia65 (Fig. 8E) whereas four SnRK gens in HD2687 had upto 5.5-fold higher expression levels at 12 h (Fig. 8G). While at 24 h after salt stress, expression levels of two genes, TaSnRK2.11 and TaSnRK2.12 rose 32- and 20-folds in wheat genotype Kharchia 65 (Fig. 8F) respectively, followed by TaSnRK2.11 (five fold) in HD2687 (Fig. 8H) at. We chose two genotypes i.e. C306 and WL711, to test under drought stress. In wheat genotype C306, TaSnRK2.11 expression was tenfold up-regulated, but TaSnRK3.87 expression was fourfold up-regulated (Fig. 8I), whereas TaSnRK2.3, TaSnRK2.11, TaSnRK3.18, and TaSnRK2.8 was more than fourfolds (Fig. 8J) in WL711. Abiotic tolerance in wheat might be improved by utilizing a higher expression level of TaSnRK2.11 which has been shown to increase under salinity, drought, and heat stress conditions. We analysed the RNA-seq and qRT-PCR data of SnRK genes to determine the magnitude of RNA transcription level (Fig. 9). A total of ten transcripts were chosen for this. The results revealed that the log fold-change values of these selected transcripts were found comparable in correspondence with RNA-seq results (Fig. 9A,B).

Comparison between magnitude of gene expression of RNA-seq and qRT-PCR data of heat stress (A) and drought stress (B).
Discussion
This study identified 174 TaSnRK genes in the T. aestivum genome, which were classified as TaSnRK1, TaSnRK2, or TaSnRK 3 based on their subfamily classification. The TaSnRK gene family was carefully searched, including evolutionary linkages, protein patterns, gene architectures, gene duplication, distributions of chromosomal, and the promoter cis-elements. This research progresses towards the future functionality of SnRK genes in order to improve abiotic stress adaption of plant. SnRK genes were already reported in A. thaliana4, O. sativa27, B. distachyon28, and E. grandis29 having 39, 48, 44, and 34, respectively. The number of TaSnRK genes in the genome of T. aestivum is substantially higher than in diploid plants. There were 14 TaSnRK1 genes, 65 TaSnRK2 genes , and 95 TaSnRK3 genes were discovered and classified into three subfamilies. According to more detailed description, T. aestivum and other species have similar member proportions for each subfamily. T. aestivum is a naturally occurring amphidiploid evolved from T. urartu (AA), A. speltoides (BB), and A. tauschii (DD). There were 56, 61, and 57 TaSnRK genes discovered in the A, B, and D sub-genomes, respectively demonstrating that SnRK genes had a similar functional role in progenitor species.
Although the conserved domains of the SnRK subfamily genes differ, the N-terminal protein kinase domain is retained. It has been discovered that SnRK3 subfamily genes interact with CBLs in a calcium-dependent manner due to the NAF domain. Furthermore, the NAF domain identifies a set of heterologous kinases that CBL calcium sensor protein targets and participate in a range of signalling cascades8. According to this study, distinct TaSnRKs subfamily genes shared different types of conserved domains. This might imply that the TaSnRK genes family is functionally diverse based on the domains contained.
In the AtSnRK and TaSnRK genes, certain subfamily genes showed substantial structural exon–intron divergences and gene length differences. In genes with fewer introns, increased expression in plants has been previously reported30,31. A compact gene structure with few introns has enabled rapid activation and responsiveness to different environmental conditions31. However, when the transcriptome data used in this study was combined, we found no evidence that TaSnRK gene with fewer introns had shown higher expression levels than the other TaSnRKs genes.
According to accumulating data, gene activity was often linked to discrepancies in the promoter region32. In gene promoter regions, cis-elements played a major role in controlling gene expression during development and environmental changes33,34. TaSnRKs had several cis-elements, including growth hormones, MyB, ABRE, and LTR according to promoter analysis in this investigation. Most gene promoters contained at least one of these components, demonstrating that many TaSnRKs were capable of responding to a variety of abiotic stresses while also promoting growth. When gene expression profiles from TaSnRKs with MyB and ABRE cis-elements were merged under drought stress, TaSnRKs with MyB and ABRE cis-elements increased by an average of 6.3-fold, but TaSnRKs without cis-elements only increased by 3.5-fold. As a result, cis-elements analysis can help with gene function studies, particularly gene expression patterns under different stress conditions.
TaSnRK gene expression levels were analysed using transcriptome data in different tissues and organs of T. aestivum26. The research results showed that expression patterns of these genes are divided into three categories (Fig. 7). In this study, it we found that subgroup-2 TaSnRKs contain fewer cis-elements than TaSnRKs in subgroups-1 and -3 in their promoters. Every gene in subgroup-1 has an average of 5.08 Auxin, 13.55 MeJA, and 8.47 ABRE, 1.69 MYB, 3.38 LTR compared with each gene in subgroup3 and averages of 2.05 in Auxin, 11.08 of MeJA, 1.43 of Gibberellins and 6.67 ABRE, 1.43 MYB, 0.61 LTR, each gene in subgroup 2 with a total of 0.35 Auxin, 9.12 MeJA, 4.56 Gibberellin, 3.50 ABRE, 1.40 MYB, 0.35 LTRE. These data showed that TaSnRK activity is linked to promoter region differences.
The roles and functions of several TaSnRKs in response to different abiotic stresses were also determined. Drought stress findings showed that ABA production and A. thaliana signals in response to drought were orthologous to AtSnRK2.339 and imply that TaSnRK2.11 and AtSnRK2.3 play the same role in response to drought stress. The extreme expression changes in T. aestivum against effects of drought, salt, thermal stress and ABA induction were reported in TaSnRK2.8, TaSnRK2.12, TaSnRK3.11, TaSnRK2.16, TaSnRK3.23 and TaSnRK3.83 whereas its orthologs, AtSnRK2.2, could also respond to osmotic stress and ABA induction in A. thaliana. This shows that under different conditions the TaSnRK2.11 gene may be activated substantially.
Previous research has shown that ABA-independent regulation of SnRKs occurs in terrestrial plants, such as Arabidopsis SnRK2.1, SnRK 2.4, SnRK 2.5, SnRK 2.9, and SnRK 2.10, which are activated by osmotic stress following an ABA-independent pathway7. Arabidopsis ABA-independent SnRKs control stress related gene/transcripts under hyperosmotic conditions, complementing the action of ABA-dependent SnRK2s function35. As reported in other plants, ABA-independent SnRK2s in wheat also showed sensitive reaction to osmotic stress. Specific responses to low nitrogen or sulfur deprivation36, however, appear to be initiated in an ABA-independent manner, as in other plants. These findings support the hypothesis that the plant-specific SnRK2 subfamily is important in stress response signalling both in Arabidopsis and wheat. These pathways are not solely responsible for energy-saving decisions, but they do resulting complex remodelling of cell metabolism, as evidenced by the interactions with DNA repair and maintenance pathways and TOR in Arabidopsis37 network as indicated by STRING studies. The fact that a single stress induces the expression of a large number of SnRK2 genes implies that there is a significant compensating impact or pleiotropy within this family in wheat. It is well known that most stresses result in oxidative damage38,39. The third SnRK subfamily, the other hand, played an important role in response to osmotic, salt, and heat stresses, because it consists of proteins kinases interacting with calcineurin B-like calcium binding domains7 which are mostly involved in drought and salt resistance, being the SOS (salt overly sensitive) mechanism being the best-known40.
The findings of this study provide a thorough description of the SnRK gene family in T. aestivum. It helps us to better understand the biological role of specific TaSnRK genes in T. aestivum. The study presented just a fundamental characterisation of the TaSnRK genes and a comprehensive functional validation would be required to hold the importance of the SnRK family.
Conclusions
SnRK genes are involved in a variety of signalling pathways, including responses to biotic and abiotic stresses. In this study, SnRK gene family has been intensively investigated in wheat. A total of 174 TaSnRK genes were discovered and categorized into three subgroups based on motif composition and gene structural similarity within each subfamily. Phylogenetic analyses of SnRK genes in A. thaliana and O. sativa can also be used to derive the evolutionary characteristics of the TaSnRK genes. Furthermore, the TaSnRK family's microRNA targeting, cis-acting elements, and gene expressions were investigated in order to better understanding the biological role of TaSnRK genes in T. aestivum.
Materials and methods
Identification and characterization of SnRKs
Protein sequences of SnRKs identified in related plant species were obtained from the Phytozome database (http://www.phytozome.net/) and the Rice Annotation Project (RAP) (https://rapdb.dna.affrc.go.jp/). BLASTP searches were performed against the bread wheat protein sequences (ftp:/ftp.ensemblgenomes.org/pub/plants/release-51/fasta/triticum aestivum/pep/) using an e-value cut-off of 0.0001 and bit-score > 100. Potential SnRK candidates were discovered using the methods described above. Following the removal of duplicate results, the final sequences were checked for the existence of SnRK related domains using HMMscan (https://www.ebi.ac.uk/Tools/hmmer/search/hmmscan), the SMART database (http://smart.embl-heidelberg.de/)41 PFAM42 NCBI CDD (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)43. Additionally, tools from ExPASy (http://www.expasy.ch/tools/pitool.html) were used to compute the number of amino acids, molecular weights (MW), and isoelectric point (pI) of each SnRK protein.
Phylogenetic analysis of SnRK genes family
A multiple sequence alignment was performed with ClustalW with default settings44,45 of 174 non-redundant TaSnRK amino acid. Using MEGA X and the Neighbor-Joining (NJ) technique45, a phylogenetic tree was created utilizing the poisson model, pairwise deletion, and 1000 replications of bootstrap. MEGA X has been used to create an unrooted NJ tree that includes all A. thaliana and O. sativa SnRKs protein sequences.
Motif composition and gene structural analysis of the SnRK gene family in T. aestivum
The Multiple Expectation Maximization for Motif Elicitation (MEME) online tool46 (http://meme.sdsc.edu/meme/itro.html) was used to find conserved motifs in TaSnRK proteins with the following parameters: The number of repetitions is unlimited, the maximum number of motifs is ten, and the ideal motif length is six to one hundred residues. The exon–intron structures of TaSnRK family genes were analysed using the Gene Structure Display Server online application (GSDS v.2.0: http://gsds.cbi.pku.edu.ch) based on the gff3 data file47.
Analysis of chromosomal location and orthologous genes in T. aestivum
Using MapChart version 3.048, the chromosomal coordinates of all TaSnRK genes were mapped to 21 chromosomes of the wheat genome based on physical location from the Plant ensemble database. All T. aestivum gene sequences were aligned using BLASTP with an e value of 1e−10 to find gene duplication. The pattern of duplicated SnRK were classified as segmental, tandem duplications using MCScanX with default parameters49. A tandem duplication is defined as a chromosomal area of less than 200 kb that contains two or more genes50. TBtools were used to exhibit synteny relationships of the orthologous SnRK genes between T. aestivum and A. thaliana, the syntenic analysis maps and synonymous (Ks) and non-synonymous (Ka) substitution of each duplicated TaSnRK gene51.
cis-Elements in promoter regions of TaSnRKs
The wheat genome database was used to extract upstream sequences (1500 bp) from the start codon of each TaSnRK gene, which were subsequently utilised for cis-element distributions in promoter regions using PlantCARE software (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)52.
Prediction of MIR genes targeting TaSnRK
The TaSnRK gene transcript sequences were obtained from the wheat genome database. The psRNATarget service was used to examine the matured miRNA sequences53,54 and the TaSnRK transcript sequences with default settings55,56.
Analysis of protein–protein interactions
The STRING v1054 databases were used to identify protein–protein functional interactions.
SnRK protein sequences were uploaded to the STRING57 application, and the database was searched with A. thaliana as the reference organism. All identified interaction partners were gathered and searched against the A. thaliana genome using blast software at e-value 1−e10.
Using Cytoscape58, the one best-hit for each gene was selected for the creation of a PPI network. Finally, the top hub gene from the interaction network was predicted using Cytoscape's cytoHubba plugin (Cytoscape Consortium 2016).
RNA-seq derived gene expression profiling
The value of TPM (transcripts per million) for each TaSnRK was obtained from the expVIP database (http://www.wheat-expression.com/). Clustvis (https://biit.cs.ut.ee/clustvis/) was used to create heatmaps59.
Plant material and growth conditions
In this experiment, seeds of bread wheat genotypes (C-306, WL-711, RAJ3765, HS240, Kharchia 65, and HD2687) were procured from Germplasm Unit, ICAR-Indian Institute of Wheat and Barley Research, Karnal, India. Seeds were sterilised with 1% sodium hypochloride for 10 min and then rinsed with distilled water three times and germinated in petri plates at 22 °C under controlled conditions. Seedlings were moved to a culture bottle filled with full-strength Hoagland's solution after 5 days of germination and allowed to grow for seven days. Each genotype was seeded in two sets of three biological replications. For drought stress, two contrasting wheat genotypes, C306-drought tolerant and WL711, drought susceptible were used. After 14 days of growth in Hoagland's solution, one set of seedlings was given osmotic shock using 25% (v/v) polyethylene glycol (PEG) 6000 for 24 h, while untreated used as control60. Leaf samples from control and stressed seedlings were harvested at above mentioned time intervals for expression analysis. For heat stress, two contrasting wheat genotypes were chosen: RAJ3765-heat resistant and HS240-heat sensitive. These plants were kept at 42 °C for 3 h (at Basal), followed by at 37 °C for 3 h (at Normal), and finally at 42 °C for 3 h (at Acquired). At the time interval stated above, leaf samples from the basal and acquired stress levels were collected. Kharchia 65-salt resistant and HD2687-salt sensitive wheat genotypes were utilised to study salt stress. Two week old seedlings of both genotypes were subjected to 150 mM NaCl treatment. The leaf samples were collected 12 h and 24 h after the treatment. All obtained samples were wrapped in foil and immediately frozen in liquid nitrogen at − 80 °C for total RNA isolation.
RNA isolation and real time PCR analysis
RNA was extracted using TRIzol Reagent according to the manufacturer's instructions. To get DNA-free RNA, the extracted RNA was treated with DNase I (NEB, USA). The first strand cDNA synthesized from 1 g of total RNA by using Superscript-III reverse transcriptase (Invitrogen, USA). The cDNA was diluted to 1:2 for real-time qRT-PCR analysis, and 1.5 µL of the diluted cDNA was utilised as a template in a 20 µL reaction volume according to the manufacturer's instructions. Real time quantitative RT-PCR analysis was carried out by using the BIO-RAD CFX96 using SYBR Green (Bio-Rad). The endogenous control β-actin was used to standardise the expression data61. Using the 2−ΔΔCt method, the expression was measured as a relative fold change62. The error bars show the standard deviation of the three biological replicates' expression.
Research statement
Experimental research and field studies on plants complies with relevant institutional, national, and international guidelines and legislation.
References
FAO. Crop Prospects and Food Situation - Quarterly Global Report No.1. https://doi.org/10.4060/ca8032en (2020).
Bohnert, H. J., Gong, Q., Li, P. & Ma, S. Unraveling abiotic stress tolerance mechanisms—Getting genomics going. Curr. Opin. Plant Biol. 9, 180–188 (2006).
Hunter, T. Protein kinases and phosphatases: The Yin and Yang of protein phosphorylation and signaling. Cell 80, 225–236 (1995).
Hrabak, E. M. et al. The Arabidopsis CDPK-SnRK superfamily of protein kinases. Plant Physiol. 132, 666–680 (2003).
Halford, N. G. & Hardie, D. G. SNF1-related protein kinases: Global regulators of carbon metabolism in plants?. Plant Mol. Biol. https://doi.org/10.1023/A:1006024231305 (1998).
Celenza, J. L. & Carlson, M. A yeast gene that is essential for release from glucose repression encodes a protein kinase. Science (80-). 233, 1175–1180 (1986).
Kulik, A., Wawer, I., Krzywińska, E., Bucholc, M. & Dobrowolska, G. SnRK2 protein kinases—Key regulators of plant response to abiotic stresses. OMICS J. Integr. Biol. 15, 859–872 (2011).
Albrecht, V., Ritz, O., Linder, S., Harter, K. & Kudla, J. The NAF domain defines a novel protein–protein interaction module conserved in Ca+-regulated kinases. EMBO J. 20,1051–1063 (2001).
Ohta, M., Guo, Y., Halfter, U. & Zhu, J. K. A novel domain in the protein kinase SOS2 mediates interaction with the protein phosphatase 2C ABI2. Proc. Natl. Acad. Sci. U. S. A. 100, 11771–11776 (2003).
Hardie, D. G., Carling, D. & Carlson, M. The AMP-activated/SNF1 protein kinase subfamily: Metabolic sensors of the eukaryotic cell?. Annu. Rev. Biochem. 67, 821–855 (1998).
Purcell, P. C., Smith, A. M. & Halford, N. G. Antisense expression of a sucrose non-fermenting-1-related protein kinase sequence in potato results in decreased expression of sucrose synthase in tubers and loss of sucrose-inducibility of sucrose synthase transcripts in leaves. Plant J. 14, 195–202 (1998).
Ramon, M. et al. Default activation and nuclear translocation of the plant cellular energy sensor SnRK1 regulate metabolic stress responses and development. Plant Cell 31, 1614–1632 (2019).
Halford, N. G. & Hey, S. J. Snf1-related protein kinases (SnRKs) act within an intricate network that links metabolic and stress signalling in plants. Biochem. J. 419, 247–259 (2009).
Maszkowska, J. et al. Phosphoproteomic analysis reveals that dehydrins ERD10 and ERD14 are phosphorylated by SNF1-related protein kinase 2.10 in response to osmotic stress. Plant Cell Environ. 42, 931–946 (2019).
Diédhiou, C. J., Popova, O. V., Dietz, K. J. & Golldack, D. The SNF1-type serine-threonine protein kinase SAPK4 regulates stress-responsive gene expression in rice. BMC Plant Biol. 8, 1–13 (2008).
Zhang, H. et al. Cloning and characterization of SnRK2 subfamily II genes from Nicotiana tabacum. Mol. Biol. Rep. https://doi.org/10.1007/s11033-014-3440-y (2014).
Kawa, D. et al. SnRK2 protein kinases and mRNA decapping machinery control root development and response to salt. Plant Physiol. 182, 361–371 (2020).
Mustilli, A. C., Merlot, S., Vavasseur, A., Fenzi, F. & Giraudat, J. Arabidopsis OST1 protein kinase mediates the regulation of stomatal aperture by abscisic acid and acts upstream of reactive oxygen species production. Plant Cell 14(12), 3089–3099 (2002).
Ali, A. et al. Rheostatic control of ABA signaling through HOS15-mediated OST1 degradation. Mol. Plant 12, 1447–1462 (2019).
Kim, K. N. et al. Isolation and characterization of a novel rice Ca2+-regulated protein kinase gene involved in responses to diverse signals including cold, light, cytokinins, sugars and salts. Plant Mol. Biol. 52, 1191–1202 (2003).
Guo, Y. et al. A calcium sensor and its interacting protein kinase are global regulators of abscisic acid signaling in Arabidopsis. Dev. Cell https://doi.org/10.1016/S1534-5807(02)00229-0 (2002).
Liu, J., Ishitani, M., Halfter, U., Kim, C. S. & Zhu, J. K. The Arabidopsis thaliana SOS2 gene encodes a protein kinase that is required for salt tolerance. Proc. Natl. Acad. Sci. U. S. A. https://doi.org/10.1073/pnas.97.7.3730 (2000).
Ma, Q. J. et al. A CIPK protein kinase targets sucrose transporter MdSUT2.2 at Ser 254 for phosphorylation to enhance salt tolerance. Plant Cell Environ. 42, 918–930 (2019).
Ma, Q. J. et al. An apple sucrose transporter MdSUT2.2 is a phosphorylation target for protein kinase MdCIPK22 in response to drought. Plant Biotechnol. J. 17, 625–637 (2019).
Greco, M., Chiappetta, A., Bruno, L. & Bitonti, M. B. In Posidonia oceanica cadmium induces changes in DNA methylation and chromatin patterning. J. Exp. Bot. 63, 695–709 (2012).
Coello, P. et al. Evidence that abscisic acid promotes degradation of SNF1-related protein kinase (SnRK) 1 in wheat and activation of a putative calcium-dependent SnRK2. J. Exp. Bot. 63, 913–924 (2012).
Kobayashi, Y., Yamamoto, S., Minami, H., Kagaya, Y. & Hattori, T. Differential activation of the rice sucrose nonfermenting1-related protein kinase2 family by hyperosmotic stress and abscisic acid. Plant Cell 16, 1163–1177 (2004).
Wang, L. et al. Genome-wide analysis of SnRK gene family in Brachypodium distachyon and functional characterization of BdSnRK29. Plant Sci. https://doi.org/10.1016/j.plantsci.2015.05.008 (2015).
Wang, Y. et al. Comprehensive analysis of SNRK gene family and their responses to salt stress in eucalyptus grandis. Int. J. Mol. Sci. 20, 1–21 (2019).
Chung, B. Y. W., Simons, C., Firth, A. E., Brown, C. M. & Hellens, R. P. Effect of 5′UTR introns on gene expression in Arabidopsis thaliana. BMC Genom. https://doi.org/10.1186/1471-2164-7-120 (2006).
Jeffares, D. C., Penkett, C. J. & Bähler, J. Rapidly regulated genes are intron poor. Trends Genet. 24, 375–378 (2008).
Xue, T. et al. Genome-wide and expression analysis of protein phosphatase 2C in rice and Arabidopsis. BMC Genom. 9, 1–21 (2008).
Ali, G. M. & Komatsu, S. Proteomic analysis of rice leaf sheath during drought stress. J. Proteome Res. 5, 396–403 (2006).
Sarda, X. et al. Two TIP-like genes encoding aquaporins are expressed in sunflower guard cells. Plant J. 12, 1103–1111 (1997).
Soma, F. et al. ABA-unresponsive SnRK2 protein kinases regulate mRNA decay under osmotic stress in plants. Nat. Plants 3, 1–8 (2017).
Gonzalez-Ballester, D., Pollock, S. V., Pootakham, W. & Grossman, A. R. The central role of a SNRK2 kinase in sulfur deprivation responses. Plant Physiol. 147, 216–227 (2008).
Shen, C. et al. TOR signaling is a determinant of cell survival in response to DNA damage. Mol. Cell. Biol. 27, 7007–7017 (2007).
Kobayashi, Y. et al. Abscisic acid-activated SNRK2 protein kinases function in the gene-regulation pathway of ABA signal transduction by phosphorylating ABA response element-binding factors. Plant J. 44, 939–949 (2005).
Shin, R., Alvarez, S., Burch, A. Y., Jez, J. M. & Schachtman, D. P. Phosphoproteomic identification of targets of the Arabidopsis sucrose nonfermenting-like kinase SnRK2.8 reveals a connection to metabolic processes. Proc. Natl. Acad. Sci. U. S. A. 104, 6460–6465 (2007).
Ji, H. et al. The salt overly sensitive (SOS) pathway: Established and emerging roles. Mol. Plant 6, 275–286 (2013).
Letunic, I., Doerks, T. & Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, 302–305 (2012).
Finn, R. D. et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Marchler-Bauer, A. et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, 225–229 (2011).
Saitou, N. & Nei, M. ESCALA CIWA-AR Escala CIWA-Ar(Clinical Institute Withdrawal Assesment for Alcohol) Evaluación del Síndrome de Abstinencia Alcohólica. Mol. Biol. Evol. 4, 406–425 (1987).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Bailey, T. L. et al. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 37, 1–7 (2009).
Hu, B. et al. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 31, 1296–1297 (2015).
Voorrips, R. E. Mapchart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 1–14 (2012).
Houb, E. B. The arms race is ancient history in Arabidopsis, the wildflower. Nat. Rev. Genet. 2, 516–527 (2001).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327 (2002).
Kozomara, A. & Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1181 (2014).
Sun, F. et al. Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol. https://doi.org/10.1186/1471-2229-14-142 (2014).
Dai, X. & Zhao, P. X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr319 (2011).
Katiyar, A. et al. Identification of novel drought-responsive microRNAs and trans-acting siRNAs from Sorghum bicolor (L.) Moench by high-throughput sequencing analysis. Front. Plant Sci. https://doi.org/10.3389/fpls.2015.00506 (2015).
Szklarczyk, D. et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Paul, S. et al. Cytoscape: A software environment for integrated models. Genome Res. 13, 426 (1971).
Metsalu, T. & Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570 (2015).
Kaur, A., Gupta, O. P., Meena, N. L., Grewal, A. & Sharma, P. Comparative temporal expression analysis of MicroRNAs and their target genes in contrasting wheat genotypes during osmotic stress. Appl. Biochem. Biotechnol. 181, 613–626 (2017).
Muthusamy, S. K., Dalal, M., Chinnusamy, V. & Bansal, K. C. Differential regulation of genes coding for organelle and cytosolic ClpATPases under biotic and abiotic stresses in Wheat. Front. Plant Sci. https://doi.org/10.3389/fpls.2016.00929 (2016).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods https://doi.org/10.1006/meth.2001.1262 (2001).
Acknowledgements
This work was supported by Indian Council of Agricultural Research, New Delhi, India-core grant. We are thankful to ICAR-Indian Institute of Wheat and Barley Research for providing the facility and Dr SK Muthusamy and Stacy Dris for helpful discussion. This paper is IIWBR contribution No. 329.
Author information
Authors and Affiliations
Contributions
P.S. conceived the study. S.M. and P.S. analysed the data, S.M., R.S., P.S. wrote the first draft. P.S., R.S., R.T., G.P.S. edited the manuscript. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mishra, S., Sharma, P., Singh, R. et al. Genome-wide identification and expression analysis of sucrose nonfermenting-1-related protein kinase (SnRK) genes in Triticum aestivum in response to abiotic stress. Sci Rep 11, 22477 (2021). https://doi.org/10.1038/s41598-021-99639-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-99639-5
- Springer Nature Limited