
Abstract
The mammalian inner ear has a limited capacity to regenerate its mechanosensory hair cells. This lack of regenerative capacity underlies the high incidence of age-related hearing loss in humans. In contrast, non-mammalian vertebrates can form new hair cells when damage occurs, a mechanism that depends on re-activation of expression of the pro-hair cell transcription factor Atoh1. Here, we show that members of the E2F transcription factor family, known to play a key role in cell cycle progression, regulate the expression of Atoh1. E2F1 activates chicken Atoh1 by directly interacting with a cis-regulatory region distal to the avian Atoh1 gene. E2F does not activate mouse Atoh1 gene expression, since this regulatory element is absent in mammals. We also show that E2F1 expression changes dynamically in the chicken auditory epithelium during ototoxic damage and hair cell regeneration. Therefore, we propose a model in which the mitotic regeneration of non-mammalian hair cells is due to E2F1-mediated activation of Atoh1 expression, a mechanism which has been lost in mammals.
Similar content being viewed by others


Introduction
The inner ear consists of specialized sensory organs located in the cochlea and the vestibular system, which are responsible for sound detection and balance respectively. All of these sensory organs contain sensory hair cells (HCs), which are mechanoreceptors transducing sound and head movements into signals to the brain via sensory neurons1. The HCs are separated from one another and rest upon a basal layer of supporting cells (SCs), which play important roles in the homeostasis and function of the inner ear sensory epithelia1. The pro-neural gene Atoh1, a basic helix-loop-helix (bHLH) transcription factor, is essential for the formation of inner ear HCs2, neural cells in the spinal cord3, cerebellar neurons4, proprioceptive system neurons5,6 and secretory cells in the intestine7,8. Atoh1 mutants in both Drosophila and mice lack HCs in all sensory organs, demonstrating the importance of Atoh1 during neurogenesis and for the differentiation of HC progenitors2,9,10,11.
The loss of HCs is known to underlie deafness and balance disorders. In mammals, the inability to regenerate HCs when they are lost due to the effects of ototoxic agents, noise or ageing is a major cause of sensory hearing loss in humans. This includes the most common form, age-related hearing loss (ARHL) which is highly prevalent in humans with one in three people over the age of 65 having a disabling hearing impairment12. ARHL leads to social isolation and has been linked with both depression and dementia13. In contrast, non-mammalian vertebrates have a remarkable capacity to regenerate their vestibular and auditory HCs after damage. For example, in the avian auditory epithelium (the basilar papilla), SCs are normally quiescent but can either convert into new HCs or proliferate to form new HCs and SCs after tissue damage14. In both types of regenerative responses, Atoh1 expression is re-activated in SCs before new HCs are produced14,15.
The reasons for the limited capacity for HC regeneration in the vestibular organs of mammals and its complete absence in the organ of Corti are unknown, but could be due in part to the inability of adult mammalian SCs to re-activate Atoh1 expression after HC loss. In mammals, including humans, the damage-induced re-activation of ATOH116 and the formation of new HCs is very limited17,18,19,20. Several studies have shown that ectopic overexpression of Atoh1 resulted in the production of extra HCs at developmental stages21 and in young adults22. More recently, the overexpression of Atoh1 in adult human ear tissue has been demonstrated to generate a significant number of cells expressing HC markers23. Hence, the potential for a therapeutic approach to hearing restoration based on Atoh1 is currently being explored in translational research and clinical trials. However, it is likely that other key regulators are missing, since the overexpression of Atoh1 alone, which leads to the production of supernumerary HCs in the immature mammalian inner ear, is not sufficient to trigger the large-scale regeneration of fully differentiated and functional HCs at adult stages. The development of efficient HC regeneration therapies therefore requires a better understanding of the factors acting upstream of, or in conjunction with, Atoh1 to initiate a complete regenerative response in non-mammalian species.
Previous work has shown that the control of mammalian Atoh1 expression is largely dependent on two downstream cis-regulatory enhancers, known as enhancers A and B24. Here, we identified a third highly conserved region in avian species, enhancer C, which is not present in mammalian species. Using a combination of bioinformatics analysis, EMSA and reporter gene assays, we identified novel interactions between the Atoh1 locus and members of the E2F (adenovirus early gene 2 binding factor) transcription factor family, which are crucial regulators of the cell cycle25. We show that E2F1-3 factors have the capacity to up-regulate chick Atoh1 via direct binding to a novel regulatory element within enhancer C. Furthermore, we observe changes in the nuclear-cytoplasmic localisation of E2F1 during ototoxic-induced HC regeneration that coincides with the re-activation of ATOH1. Given the well-established role of E2F1 in controlling cell cycle re-entry, our findings suggest that enhancer C contributes to the reactivation of Atoh1 expression in mitotic SCs upon HC damage, thereby establishing a mechanistic link between two essential aspects of HC regeneration.
Results
Comparative alignment of the avian and mammalian Atoh1 3′ sequence and putative binding sites predictions
The regulation of mammalian ATOH1 expression has previously been shown to be controlled by two evolutionary conserved regions (ECRs) downstream of the Atoh1 locus which are sufficient to drive the expression of an Atoh1/lacZ reporter and replicate in most of the tissue specific expression of ATOH124,26. These two regions, defined as the Atoh1 enhancer A and B are highly evolutionary conserved between mouse and human. In order to identify similarities and differences between Atoh1 avian and mammalian regulatory regions, a comparative alignment of their sequences was performed. As shown in Fig. 1A, when the human, mouse and zebra finch sequences were aligned to the chicken Atoh1 3’ sequence, two highly conserved regions were detected in the four species corresponding to the Atoh1 enhancers A and B (Fig. 1A). The sequence homology for enhancers A and B across the four species was 81% and 66% respectively (Fig. 1B). Analysing the sequence downstream of these Atoh1 enhancers, an additional region was found in chicken and zebra finch that shares a similar degree of homology to that of enhancers AB. This is a 377 bp region and lies ~ 700 bp downstream of enhancer B in chick and shares 80% sequence homology between the chick and zebra finch genome. We designated this region as putative enhancer C (genomic location in chick chromosome, originally derived from Ensembl release 91, 4:37326061-37326437; in latest Ensembl release 102, 4:37047224-37047600; for sequence see Supplementary Figure S1).

Alignment between the chick, human, mouse and zebra finch Atoh1 genomic sequences. (A) The chick Atoh1 sequence was used as the basis for comparison and conserved regions are highlighted in pink. Conservation levels are represented in the y axis (in percentages) and the length of the conserved regions is presented in the x axis (minimum length 100 bp). The diagram exported from mVista27 shows that some regions within the Atoh1 coding sequence are highly conserved among chick, human, mouse and zebra finch (Atoh1 coding sequences correspond to chromosome 6:64729125-64731245 in mouse, chromosome 4:93828753-93830964 in human and chromosome 4: 36493650-36494082 in chick; forward strand, all Ensembl release 91, accessed –December 2017). The two highly conserved regions previously characterized by Helms et al. 200024 and Ebert et al., 200328 and defined as the Atoh1 enhancer A and B are also conserved among the four species including zebra finch. An additional non-coding region (annotated as putative enhancer C; genomic location 4:37326061-37326437, release 91, updated to 4:37047224-37047600 in latest Ensembl release 102, November 2020; see Supplementary Figure S1 for sequence) shows a high degree of homology between chick and zebra finch but does not share homology with the 3’ region of the mouse and human Atoh1. During revision of this manuscript, it was identified that there is some reduced homology between a smaller 268 bp region of the putative chick enhancer C sequence and a sequence several kilobases further downstream of mouse and human Atoh1 genes; its position is shown below the main alignment using the human sequence as the reference. (B) Schematic representation (not to scale) of the position of Atoh1 enhancers A, B and putative enhancer C showing their respective homologies between chick, human, mouse and zebra finch using Clustal2.1. The predicted E2F transcription factor binding sites are marked along the chick, human, mouse and zebra finch Atoh1 locus.
In order to identify transcription factors potentially playing a role in the regulation of ATOH1 expression, we conducted a bioinformatic analysis using MatInspector software (https://www.genomatix.de/index.html version 9.0, Genomatix, Germany) to identify common transcription factor motifs within enhancers A, B and C from different species (Supplementary Figure S2). One putative transcriptional regulator of Atoh1 identified by this screen was the E2F transcription factor family. A large number of predicted E2F binding sites were present in the chick, human, mouse and zebra finch Atoh1 enhancers A and B and additionally within the putative avian enhancer C (Fig. 1B). This included a cluster of 5 E2F sites predicted within the putative enhancer C in chick, with 3 of them specific to the avian species (Fig. 1B). E2F proteins are known to be involved in controlling cell cycle re-entry25,29,30,31,32 therefore, the predicted E2F binding sites at the Atoh1 locus were prioritised for further investigation to determine their involvement in linking Atoh1 re-activation with cell cycle re-entry and cell proliferation.
E2F1-3 can induce transcription via elements within the chick Atoh1 but not the mouse Atoh1 enhancers
To confirm the validity of the predictions from the bioinformatic analysis, we first tested whether these putative Atoh1 regulatory elements could confer regulation by E2F1, the primary member of the E2F family, using reporter gene constructs. Distinct mouse and chick Atoh1 conserved regions containing combinations of the enhancers A, B, or C were cloned upstream of a minimal promoter in luciferase vectors (Fig. 2A). These were then co-transfected with increasing amounts of a human E2F1 expression vector in UB/OC-2 cells, a cell line derived from the immortomouse inner ear epithelium at embryonic day 1333. Both msAB-luc and chAB-luc constructs showed a dose-dependent increase in reporter activity in response to increasing E2F1 levels (Fig. 2B). However, the response of the chAB-luc was much larger showing an 8, 17 and 23-fold activation when co-transfected with increasing amounts of E2F1, whereas the maximal response of the msAB-luc construct was only threefold. Similarly, the construct containing the Atoh1 putative enhancer C (chC-luc) also showed strong dose dependent activation of 10, 18 and 34-fold difference suggesting that there are functional E2F1 regulatory elements within putative enhancer C. Furthermore, the construct containing all three chick Atoh1 conserved elements (chABC-luc) produced an even greater response: 27, 61 and 145-fold increases in reporter gene activity with increasing amounts of E2F1. To confirm that the activation of the Atoh1 reporter constructs was dependent on functional E2F1 protein, luciferase co-transfection assays were also performed with a construct expressing a mutant E2F1 protein34. When the mutant E2F1 construct was co-transfected, the large up-regulation that was previously observed in the chick Atoh1 constructs was almost completely abolished, confirming that the activation is dependent on a functional E2F1 protein (Fig. 2B). In contrast, the msAB-luc construct showed similar response upon transfection with both mutant or wild type E2F1 expression construct suggesting the small activation observed was not due to a direct interaction of E2F1 with the mouse enhancer. The ability of other members of the E2F family to regulate Atoh1 constructs was also tested in luciferase assays. As shown in Fig. 2C, E2F2 and E2F3 induced a less pronounced upregulation of the various reporter constructs than E2F1. E2F4, thought to act as a transcriptional repressor35,36,37,38 did not significantly regulate the mouse or chick Atoh1 enhancer AB. We did however observe a small, yet statistically significant repression of the putative enhancer C by E2F4 (Fig. 2C).

Effect of the E2F transcription factors on the mouse and chick Atoh1 enhancers and putative enhancer C. (A) Schematic representation of the luciferase constructs containing the mouse or chick Atoh1 conserved regions cloned into the luciferase vector pGL4.23. (B) Dose–response effects on the Atoh1 luciferase constructs of transfecting increasing amounts of an E2F1 expression construct or mutant E2F1 in the UB/OC-2 cell line. (C) Effect of E2F2, E2F3 and E2F4 on the activity of the Atoh1 luciferase constructs. Experiments were conducted in triplicate in two separate assays with different DNA preparations for each plasmid in each assay (n = 6). Relative luciferase values are expressed relative to activity of the reporters in the absence of E2F. Student t-test was conducted comparing each condition with 0 ng of the corresponding E2F expression construct. (*p < 0.05; **p < 0.01; #p < 10–5; ##p < 10–10). Error bars represent the s.e.m (n = 6).
Taken together, these results indicate that E2F1, E2F2 and E2F3 can strongly active the chick Atoh1 conserved elements but not the mouse Atoh1 enhancers. By contrast, E2F4 causes a small downregulation of the putative enhancer C. The greatest activation is observed when the chick Atoh1 enhancers A and B are combined with the putative enhancer C giving a 145-fold response upon the overexpression of E2F1.
E2F binds to sequences within the chick Atoh1 enhancer AB and putative enhancer C
We next tested whether E2F binds directly to any of the nine putative binding sites predicted in the chick Atoh1 conserved regions (Fig. 3A) using an electrophoresis mobility-shift assay (EMSA). Nuclear extracts from UB/OC-2 cells were used for the analysis of molecular interactions between E2F protein and Atoh1 sequences since they endogenously express both SC and HC markers39 and E2F1 and ATOH1 proteins (Fig. 3B). EMSA experiments with nuclear proteins from UB/OC-2 cells bind to a radiolabelled E2F1 consensus sequence, causing several shifted band complexes (Fig. 3C; lane 2, labelled A–E). Incubating the reaction with a 500-fold excess of an unlabelled consensus E2F1-binding site or a mutated E2F1 consensus sequence identified that bandshift B in particular was attenuated by the E2F1 consensus but not the mutant sequence indicating this shift represents a specific interaction between E2F1 and its consensus binding site (Fig. 3C; lane 3, bandshift B). In agreement with this, EMSA assays using UB/OC-2 extracts transfected with E2F1 and its co-factor DP140 show an increase in intensity of shift B compared with other shifts suggesting this represents the binding to E2F1 protein. Supershift experiments using pre-incubation with an E2F1 antibody appear to attenuate binding of shift B, but also other shifts that are present. To investigate whether any of the chick Atoh1 E2F binding sites predicted by the bioinformatic analysis can compete with this known E2F binding site, competition was performed by adding a 500-fold excess of the nine unlabelled putative E2F probes predicted in chick Atoh1 (Fig. 3D; lanes 5–13). None of the nine competitors attenuated binding to the same degree as the consensus E2F1 binding site. However, probe S2 and to a lesser extent probe S6 show some competition in the E2F-specific shift B suggesting that of the nine predicted sites these two are the likeliest E2F binding sites (Fig. 3D; lanes 6 and 10, indicated by red asterisk).

Identification and verification of E2F recognition elements in the chick Atoh1 conserved elements. (A) Schematic diagram showing the location of nine putative E2F binding sites predicted in the chick Atoh1 conserved regions by MatInspector software (labelled as S1–S9) Numbers indicate the position of the enhancers in base pairs. (B) Immunofluoresence detection of endogenous expression of ATOH1 (green) and E2F1 (magenta) proteins in UB/OC-2 cells co-stained with DAPI (blue). (C) and (D): EMSA analysis performed using a radiolabelled E2F1 consensus sequence, containing a functional E2F1 binding site incubated alone (lane 1), with untransfected nuclear extracts or with nuclear extracts transfected with E2F1 and DP1 expression vectors as indicated. Competition assays were performed with 500 ng of non-radiolabelled competitors (consensus E2F1, lane 3 in both (C) and (D), and lane 9 in (C); mutant E2F1, lane 4 in (C) and (D), and lane 10 in (C); non-specific (N/S) probe, lane 5 and 11 in (C); sites S1 to S9 as described in (A), lanes 7–15 in (D). Lane 6 and 12 show a supershift assay with rabbit polyclonal anti-E2F1, pre-incubated with extracts before the binding assay.
Activation of the chick Atoh1 enhancer constructs by E2F1 is dependent on the predicted binding sites 2 and 6
To confirm that predicted E2F sites 2 and 6 identified in EMSA analyses are bound by E2F1 and investigate their role in the activation of the chick Atoh1 conserved regions we performed site-directed mutagenesis of these sites on the chABC-luc construct. Nucleotides to be mutated in site 2 and site 6 were identified by comparing their sequences to the experimentally derived consensus E2F1 binding site41 (Fig. 4A). Previous studies have shown that 4 core nucleotides 5’-GCGC-3’ within the E2F consensus sequence are critical for E2F binding activity42. We generated two point mutations within this core “GCGC” sequence of site 2 and site 6 in the chABC-luc construct (Fig. 4B) to produce chABC-s2mut and chABC-s6mut luciferase constructs respectively. In the presence of increasing amounts of co-transfected E2F1 the wild type construct (chABC-wt) generated a strong and highly significant activation. However, mutation of the putative E2F site 2 (chABC-s2mut), attenuated this activation by 37% compared to wild-type sequence. This reduction was observed only when 1200 ng of E2F1 was transfected in comparison to the response given by the wild type construct (Fig. 4C). Whereas, mutation of site 6 (chABC-s6mut) caused a greater attenuation, showing a 77% decrease in activation at the highest E2F1 level. The mutation in site 6 within the putative enhancer C also attenuated activation at lower levels of E2F1 transfection; 40% and 68% compared to the wild type construct when 400 and 800 ng of E2F1 were co-transfected respectively (Fig. 4C). These data suggest that both site 2 and site 6 in the chick Atoh1 flanking sequence play a role in conferring activation of the chick Atoh1 regulatory regions by E2F1 and that the effect is via a direct interaction between E2F1 and these regulatory elements.

The effect of site directed mutagenesis of the E2F1 putative sites 2 and 6 on E2F1 mediated activation of the Atoh1 regulatory region. (A) Consensus seuqeunce of the E2F1 binding site described by MatInspector, Genomatix. Cross species alignment of sites 2 and site 6 in the Atoh1 regulatory regions. Conserved nucleotides within the core sequence of site 2 and site 6 are shown in red. (B) Diagram showing the sequence and position of putative E2F sites 2 and 6 which both contain overlapping predicted binding sites for E2F1 and E2F4. Point mutations were introduced in the core GC nucleotides, replaced with TT (marked in bold and with asterisks). (C) Luciferase assays show a reduced response of the chABC-s2mut (*p < 0.05) and the chABC-s6mut (***p < 0.001) to E2F1 transfection in comparison with the wild type luciferase construct (chABC-wt). Each experiment was conducted in triplicate in two separate assays with different DNA preparations. Student t-test was conducted.
Profiling changes in E2F1 expression after HC injury in the chick basilar papilla
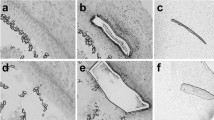
Damaged HCs in the avian inner ear can be replaced via direct conversion of SCs into HCs and through the re-entry of SCs into the cell cycle, which can then give rise to new HCs cells and SCs (reviewed in43,44). Given the proliferative functions of the E2F family, we investigated E2F1 expression during the process of HC regeneration using organotypic cultures of the E18 chick basilar papilla (BP) at different time points after HC damage (Fig. 5A). BPs were cultured for 2 days with the ototoxic antibiotic streptomycin, and as expected, a large reduction in the number of HCs was observed (determined by labelling actin-rich hair bundles with phalloidin, Fig. 5B; row I) in comparison to untreated cultures (Fig. 5B; row IV), suggesting that the majority of HCs have been lost. The BP cultures were then maintained for an additional 3 days and 6 days in streptomycin-free media. In cultures allowed to recover for 6 days after drug treatment, new HCs had re-emerged in the BP as shown by phalloidin labelling (Fig. 5B; see arrowheads in row III).

E2F1 expression during HC regeneration in the chick BP. (A) Schematic representation of the experimental timeline of organotypic cultures of E18 chick BPs cultured in DMEM and 1% FBS for 2 days with 78 µM streptomycin (Strep) followed an additional 3 or 6 days in vitro (DIV) in the same media without streptomycin. The arrows indicate when the tissue was harvested. (B) Immunofluorescence of BPs showing the expression of E2F1 and ATOH1 in HCs (labelled with Phalloidin) and SCs (labelled with SOX2). Cultures maintained for 2 days in media with Strep show ATOH1 re-activation in some SCs whereas a downregulation of nuclear E2F1 expression is observed in comparison to control cultures maintained in DMEM (hollow arrowheads in zoom in areas in row (I) and (IV); scale bar: 10 µm. In BPs cultured for 3 additional days in streptomycin-free media (2d Strep + 3 DIV), nuclear E2F1 expression is observed in SCs (hollow arrowheads in zoom in areas in row II); scale bar: 10 µm). New HCs labelled with Phalloidin are formed in cultures maintained for 6 days in streptomycin-free media after drug treatment (2d Strep + 6 days DIV). In these cultures, ATOH1 is downregulated in SCs labelled with SOX2 whereas E2F1 is expressed in SCs and HCs (arrowheads in row III in E2F1 panel). Control experiments were conducted in parallel in DMEM and at the same timings plus additional days of in vitro (DIV). Technical replicates were three for each experiment with at least three biological samples.
Immediately after 2 days of streptomycin treatment and concurrent with HC loss, ATOH1 expression was up-regulated in SCs (labelled with the support cell marker SOX2 in Fig. 5B; row I) compared to untreated cultures (Fig. 5B, row IV). At this time point, a reduction of nuclear E2F1 as well as an increase in cytoplasmic E2F1 expression was apparent in SCs of treated cultures (Fig. 5B; comparing E2F1 in rows I and IV). After 3 days of recovery, a re-appearance of nuclear E2F1 expression was observed in SCs (Fig. 5B; row II compared to row I). In cultures allowed to recover for 6 days after drug treatment, when new HCs had re-emerged in the BP, ATOH1 expression was downregulated (Fig. 5B; row III). At this time-point, E2F1 was expressed in the nucleus of newly formed HCs and in SCs (Fig. 5; arrows in row III in E2F panel). In control BPs cultured for the same periods of time, but without streptomycin treatment, there were no significant changes in expression or nuclear/cytoplasmic localisation of E2F1 (Fig. 5B; rows IV-VI). However, some HC loss was apparent in BPs that were maintained for the longest period of culture (Fig. 5B; arrows in row VI).
Altogether, these results show that E2F1 is expressed in the HC and SC of the avian BP. During HC regeneration and at the time when ATOH1 expression is reactivated in the damaged BP, the subcellular localisation of E2F1 shifts transiently from the nucleus to the cytoplasm of SCs.
Enhancer C is well conserved in avian genomes but is not present in non-avian vertebrates
We investigated how widely the 377 bp chick enhancer sequence (for complete sequence see Supplementary Figure S1) is conserved across other avian species’ genomes and in non-avian vertebrates, some of which can also are able to regenerate hair cells. Figure 6 shows a comparative alignment in a number of species to the chick Atoh1 locus and reveals that while enhancer A, and to a lesser extent enhancer B, appear to be well conserved in mammals, avians and reptiles, enhancer C is absent from mammals and reptiles. Using Ensembl BLAST to screen all 50 avian, and 21 reptile and amphibian genomes in the Ensembl Release 102 (November 2020) revealed 45 of the 50 avian species’ genomes to have significant conservation of the enhancer C sequence (Supplementary Data S1) whereas no signifcant matches were found to the genomes of reptiles and amphibians. During revision of this manuscript it was identified that there is some homology between a smaller 268 bp region of the chick enhancer C sequence and a sequence several kilobases further downstream of mouse and human Atoh1 genes (see Supplementary Figure S4). Significantly, this sequence does not include the region of the enhancer C containing binding site S6 that confers E2F1 regulation.

Comparative analysis of the Atoh1 3′ sequence in mammalian, avian and non-mammalian non avian species. The comparative alignment was extended to non-avian vertebrate species (Ensembl release 101—August 2020). As shown, the putative enhancer C is not conserved in reptile species but is present in other avian species such as Golden Eagle. A complete analysis of all avian species can be found in Supplementary Data 1.
Discussion
In a comparative analysis of the avian and mammalian ATOH1 gene locus we found that the sequences of the previously known mammalian 3′ enhancers A and B, which have been shown to be largely responsible for controlling ATOH1 expression24, are also conserved in avian species. We also identified a third putative enhancer C, located approximately 700 bp 3′ of enhancer B, the sequence and position of which is highly conserved between chicken and zebra finch. Notably, this 377 bp sequence appears to be absent from both the mouse and human genome (Fig. 1). A subsequent bioinformatic analysis of putative transcription binding sites within these evolutionary conserved regions at the ATOH1 locus identified multiple putative E2F sites in both mammalian and avian sequences. Further investigation using reporter gene assays and site directed mutagenesis confirmed that sequences within the chicken locus can confer E2F1 regulation upon avian ATOH1 constructs whereas constructs with mouse enhancers A and B show a similar response to the base vector. All three chicken enhancers, A, B and C, were regulated by E2F1, but site directed mutagenesis indicated that this effect is primarily dependent on an E2F1 binding site within enhancer C (Fig. 4C). A second E2F1 site between chick enhancers A and B, also absent from mammalian sequences, also confers E2F1 activation. These E2F1 responsive sequences are not present in the mouse Atoh1 locus.
The avian specific regulation of ATOH1 by E2F1 is intriguing since it provides a potential mechanism by which ATOH1 activation, an event which is required for HC specification might be linked to cell cycle re-entry. In the chick BP, HCs can be regenerated by direct transdifferentiation which consists of the phenotypic conversion of SCs into HCs43,45,46. In addition, regeneration can also occur via cell division of SCs to form new SCs and HCs14,47,48,49. However, the mechanisms controlling cell cycle re-entry during HC regeneration are not well characterised. Hence, one possibility based on our results and the known roles of the E2F family in controlling proliferation is that E2F factors might be involved in the spontaneous regeneration of HCs via cell division and re-activation of ATOH1 observed in lower vertebrates after damage.
Our results in ex vivo chick BP cultures treated with streptomycin showed that there are dynamic changes in the subcellular localization of E2F1 during HC loss, ATOH1 re-activation and HC regeneration. Soon after HC disappearance, E2F1 is downregulated in the nucleus of SCs and becomes enriched within the cytoplasm and perinuclear region (Fig. 5B-E2F1 expression in row I). Subsequently, a recovery of nuclear E2F1 occurs in SCs 3 days after the end of the streptomycin treatment (Fig. 5B-E2F1 expression in row II). Interestingly, the timing of the re-appearance of nuclear E2F1 expression in SCs is coincidental with the peak of division occurring in vivo during HC regeneration in post-hatch BPs14,50,51. A recent transcriptomic study has also demonstrated an up-regulation in E2F1 expression 3 days after ototoxic treatment in the chick BP in vivo52, which supports our in vitro results.
Several publications have reported nucleocytoplasmic shuttling of E2F1 and examined its effect on E2F1 target gene regulation and cell proliferation. In neurons, cytoplasmic E2F1 has been proposed to play a role in regulating apoptosis and/or necrosis53. Cytoplasmic E2F1 has also been suggested to control local protein translation as an RNA-binding protein54,55. The significance of the nucleo-cytoplasmic shuttling of E2F1 during HC loss and regeneration will require further investigation but it is likely that the cytoplasmic re-localisation of E2F1 prevents it from directly regulating gene expression and controlling cell cycle progression. Alternatively, nuclear E2F1 may be involved in the re-entry of post-mitotic quiescent SCs into the cell cycle. The availability of E2F to mediate gene regulation during the cell cycle is dependent on the Retinoblastoma tumor suppressor (Rb), a protein that belongs to the “pocket proteins” which are phosphorylated/dephosphorylated in response to cyclin-dependent kinases signals56,57,58. Hence, it is possible that nuclear E2F1, although present in mature HCs and SCs in chick, may be sequestered by Rb thereby preventing cell cycle re-entry, ATOH1 reactivation and HC proliferation. However, upon HC damage, cell signalling could trigger the phosphorylation of Rb and consequently release of E2F proteins leading to the reactivation of avian ATOH1 expression via interaction with enhancer C. Given the requirement for ATOH1 expression in HC specification, this may link SC proliferation with pro-HC gene expression in avians as proposed in Fig. 7. Further work is required to substantiate these hypotheses and to understand what role E2F re-localisation might play in this process.

Working hypothesis on the role of E2F1 on the control of Atoh1. Upon HC damage, cytoplasmic accumulation of E2F1 in SCs may occur in response to damage as an early response. Cytoplasmic E2F1 may be involved in apoptotic functions or controlling translation of proteins as an RNA-binding protein. A later response might be triggered to activate regeneration via cell division by shifting E2F1 protein into the nucleus of SCs. In the nucleus and upon phosphorylation of the Rb protein, E2F1 promotes re-entry of post-mitotic SCs into the cell cycle (from G0 phase to G1 phase). Nuclear E2F1 simultaneously induces transcription of chick Atoh1 via a regulatory element in putative enhancer C resulting in re-activation of the expression of chick ATOH1. This mechanism could be responsible for the spontaneous HC regeneration in avian species. In contrast, mouse Atoh1 gene is not responsive to E2F1 and therefore limits HC regeneration in mammalian species.
Studies with E2F knockout mice suggest unique tissue-specific roles for each E2F member during mouse development59. However, the phenotype in the inner ear in single and compound knockout animals has not yet been examined59,60. Studies with Rb mutant mouse models have examined the phenotype in the inner ear and demonstrated that there is an abnormal proliferation of vestibular and cochlear HCs during development61,62,63,64. This might be mediated by the effect of unbound E2Fs stimulating the cell cycle in the absence of Rb.
In conclusion, our data provide new insight into the comparative regulation of the key transcription factor ATOH1 in mammals and avians. Analysis of the genomes of all completed avian, reptile and amphibian species confirm the conservation of the 377 bp enhancer C sequence is avian specific whereas Atoh1 enhancers A and B is conserved across these species. Our results emphasize the potential role that a novel non-coding regulatory region, enhancer C, might play during avian HC regeneration to coordinate cell cycle re-entry with the re-activation of ATOH1 expression. Therefore, it may also have relevance to therapeutic approaches to preserve human hearing through potential strategies designed to modify the mammalian expression of ATOH1.
Material and methods
Bioinformatic analysis
Evolutionary conserved regions in the genomic sequence flanking the Atoh1 gene were identified using the comparative genomics browser mVista. Clustal2.1 was used to calculate the percentage homologies among the chick, human, mouse and zebra finch Atoh1 genomic sequences. MatInspector was used to predict transcription binding sites [Genomatix Platform; https://www.genomatix.de/index.html65,66]. Nine predicted E2F binding sites (S1–S9) were predicted within the enhancer regions and selected for functional analysis.
Plasmids and constructs
All luciferase constructs contain the Atoh1 conserved regions upstream of the minimal promoter and were made using pGL4.23 [luc2/minP] luciferase vector (Promega). The msAB-luc and chAB-luc were constructed by digesting a J2XnGFP containing the mouse Atoh1 enhancer (1408 bp) and a PE1-chick homology-AB vector containing the chick Atoh1 enhancer (1015 bp), kindly provided by Professor Jane Johnson (University of Texas Southwestern Medical School, USA) and subcloned into the pGL4.23[luc2/minP] luciferase vector. The chC-luc (genomic location in chick chromosome genomic location in chick chromosome 4:37047224-37047600; Ensembl release 102 –November 2020) was made by amplifying chicken genomic DNA using the following primers: Chick C-forward, 5′-TAAGCAGTCGACTGTCCTCTC-GCCCGCCCTG-3′; Chick C-reverse, 5′-ATTCGTGTCGACGGTTACAGTGTCGGTGAGCTGC-3′. The amplified PCR product (383 bp) was cloned into the pGL4.23 [luc2/minP] luciferase vector. The chABC-luc was generated by amplifying chick genomic DNA with the primer Chick ABC-forward: 5′-TAGAAGTCGACGCAGCGCATTTCCATGTTGAG-3′ and the Chick C-reverse listed above. After PCR amplification, the ABC fragment (1905 bp) was cloned into the pGL4.23 [luc2/minP] luciferase vector. The following expression constructs used in transfections have been previously reported: a human E2F1 expression construct cloned in pcDNA3 (kindly obtained from Professor Kristian Helin, University of Copenhagen, Denmark67, mutant E2F1 containing a change in the DNA binding domain located in exon 3 in the E2F1 cDNA34, E2F2 is a pCMV-Neo-Bam construct68, E2F3 is a pcDNA3 construct69, E2F4 is a pcDNA3 construct70 and human DP1 cDNA (accession number: L23959) is a pCMV-Neo-Bam1 (a gift from Dr Tony Kouzarides, The Gurdon Institute, University of Cambridge, UK) subcloned into the pcDNA3 expression construct.
Transient transfections and luciferase activity assays
For Dual Luciferase Assays, UB/OC-2 cells were cultured in MEM media supplemented with 10% FBS and 50 U/ml γ-IFN at 33 °C39. Prior to transfection, cells were plated in 6 well plates (2 × 105 cells per well) at 37 °C and transfected using the calcium chloride method71 with 200 ng of the Atoh1 reporter constructs with increasing amounts of E2F expression constructs (as indicated in Fig. 2) and 10 ng of phRL-null (Promega). The total amount of transfected DNA was maintained constant using the empty pSI mammalian expression vector (Promega). A glycerol shock was conducted 24 h after transfecting the DNA reporter constructs and then cells were cultured for 16 h in complete media at 33 °C. Following this, cells were harvested and enhancer activity was quantified using the Dual Luciferase Reporter Assay System (Promega). Each assay was conducted in triplicate in two separate experiments using different DNA preparations for each plasmid.
Electrophoretic mobility-shift assays (EMSA)
EMSA experiments were performed with nuclear extracts from UB/OC-2 cells transfected with E2F1 and DP1 (the co-factor for E2F) since a significant improvement in the E2F-DP1 complex interacting with a consensus E2F binding site was found (Fig. 3C). Double stranded oligonucleotides probes (for sequences see Supplementary Table S1) were labelled in a standard T4 kinase reaction with γ32P γATP (GE Healthcare). Binding reactions were conducted at room temperature for 10 min with 10 µl of 2 × Parker buffer (16% Ficoll, 40 mM HEPES at pH 7.9, 100 mM KCl, 2 mM EDTA, 1 mM DTT, 4 mM MgCl2), 1.5 µg poly (dI·dC), 10ug of nuclear extracts protein and 50 ng of the radiolabelled oligonucleotide. For competition experiments, an excess of 500-fold molar of cold consensus or mutant oligonucleotide competitors were added to the reaction for 2 h at room temperature before adding the radiolabelled oligonucleotide. For supershift experiments, 1 µg of antibody was added to the binding reaction 1 h before adding the radiolabelled probe. Reaction mixtures were resolved on a 4% poly-acrylamide gel (29:1) 0.25 × TBE acrylamide gel and electrophoresed in 0.25 × TBE at 200 V for 2 to 3 h at 4 °C. The polyacrylamide gel was dried under a vacuum system for 1–2 h at 80 °C and exposed to an X-ray film at − 80 °C for between 10 h and 5 days. Following film exposure, film was developed using Kodak film reagents according to the manufacturer’s instructions.
Site-directed mutagenesis
The chABC-s2mut and chABC-s6mut luciferase constructs were generated by QuickChange II Site-Directed Mutagenesis (Agilent Technologies). Oligonucleotides were designed using the primer design tool provided on the manufacture’s website (http://www.genomics.agilent.com/primerDesignProgram.jsp) to introduce two point mutations in the predicted E2F site 2 and site 6 in the chABC-luc construct. Primers designed for this technique were (mutated bases underlined): P2_Sense_mutagenic primer, 5′-CGCTTTAAAGAAA-TGCCTCAAAAAAAGATAAAAAATGGCACAAAGCAAAGC-3′, P2_Antisense_mutagenic primer, 5′-GCTTTGCTTTGTGCCATTTTTTATCTTTTTTTGAGGCATTTCTTTAAAGCG-3′, P6_Sense_ mutagenic primer, 5′-TCCCGCGCCCAACTTGGGACAGCGACGC-3′, P6_Antisense_mutagenic primer, GCGTCGCTGTCCCAAGTTGGGCGCGGGA.
Immunohistochemistry
Immunohistochemistry on chick tissues was performed as described previously72 with the following antibodies: chicken anti-peptide polyclonal against mouse Atoh1 (used at 1:5000) kindly supplied by Matthew Kelley73; rabbit polyclonal E2F1 (against the N terminal of E2F1; P100821_P050) from Aviva Systems Biology used at 1:200; mouse monoclonal anti-SOX2 (BD Pharmingen) used at 1:200. Secondary antibodies (Invitrogen) were: goat anti-(rabbit Ig) conjugated to Alexa Fluor 633 (1:1000), goat anti-(mouse Ig) conjugated to Alexa 550 (1:1000) and goat anti-(chick IgY) conjugated to Alexa 488 (1:1000). Samples were counterstained with Phalloidin 405 (1:1000) and imaged on a Zeiss LSM Meta 510 and 880 confocal microscopes (Zeiss). For cell line immunohistochemisty, UB/OC-2 cells were seeded in 6 well plates containing 13 mm diameter uncoated glass coverslips (VWR). Cells were grown until 80% confluency and fixed in 4% paraformaldehyde for 2 min. Following fixation, cells were washed three times in PBS for 5 min before proceeding for immunohistochemistry with ATOH1 and E2F1 antibodies as described above.
Explant cultures
Basilar papillae (BPs) were dissected from embryonic chicks at E18 as described previously51 and maintained in chilled Leibovitz’s L-15 media (Gibco, Invitrogen) during the dissecting procedure. Isolated BPs were cultured onto Millicell cell culture inserts (Millipore) placed into 35 mm culture dishes containing 1.5 ml of Dulbecco's Modified Eagle Medium (DMEM) supplemented with 1% fetal bovine serum and 1 mM HEPES buffer. For the drug experiment, 78 μM of streptomycin (Sigma) was added to the previous culture medium. Cultures were maintained for 2 days in vitro (DIV) at 37 °C and 5% CO2. After 2 days of incubation with streptomycin, all BPs were either harvested or were rinsed and maintained in culture for 3 or 6 days in streptomycin-free media which was replenished at 2 days intervals (see Fig. 5A). Untreated cultures were maintained in parallel and served as matched controls. For each experiment, three biological replicates were performed with a minimum of 3 samples per run. All animal procedures were carried out in accordance with the UK Home Office guidelines and approved by University College London and by the UK Home Office.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Pickles, J. O. & Corey, D. P. Mechanoelectrical transduction by hair cells. Trends Neurosci. 15, 254–259 (1992).
Bermingham, N. A. et al. Math1: An essential gene for the generation of inner ear hair cells. Science 284, 1837 (1999).
Lai, H. C., Klisch, T. J., Roberts, R., Zoghbi, H. Y. & Johnson, J. E. In vivo neuronal subtype-specific targets of Atoh1 (Math1) in dorsal spinal cord. J. Neurosci. 31, 10859 (2011).
Klisch, T. J. et al. In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proc. Natl. Acad. Sci. 108, 3288 (2011).
Helms, A. W. & Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development 125, 919 (1998).
Rose, M. F. et al. Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64, 341–354 (2009).
Yang, Q., Bermingham, N. A., Finegold, M. J. & Zoghbi, H. Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294, 2155 (2001).
Gerbe, F. et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol. 192, 767–780 (2011).
Jarman, A. P., Grau, Y., Jan, L. Y. & Jan, Y. N. Atonal is a proneural gene that directs chordotonal organ formation in the Drosophila peripheral nervous system. Cell 73, 1307–1321 (1993).
Jarman, A. P., Grell, E. H., Ackerman, L., Jan, L. Y. & Jan, Y. N. Atonal is the proneural gene for Drosophila photoreceptors. Nature 369, 398–400 (1994).
Groves, A. K., Zhang, K. D. & Fekete, D. M. The genetics of hair cell development and regeneration. Annu. Rev. Neurosci. 36, 361–381 (2013).
Bowl, M. R. & Dawson, S. J. Age-related hearing loss. Cold Spring Harb. Perspect. Med. 9, a033217 (2019).
Livingston, G. et al. Dementia prevention, intervention, and care. Lancet 390, 2673–2734 (2017).
Cafaro, J., Lee, G. S. & Stone, J. S. Atoh1 expression defines activated progenitors and differentiating hair cells during avian hair cell regeneration. Dev. Dyn. 236, 156–170 (2007).
Lewis, R. M., Hume, C. R. & Stone, J. S. Atoh1 expression and function during auditory hair cell regeneration in post-hatch chickens. Hear. Res. 289, 74–85 (2012).
Wang, G.-P. et al. Notch signaling and Atoh1 expression during hair cell regeneration in the mouse utricle. Hear. Res. 267, 61–70 (2010).
Forge, A., Li, L., Corwin, J. & Nevill, G. Ultrastructural evidence for hair cell regeneration in the mammalian inner ear. Science 259, 1616 (1993).
Rubel, E. W., Dew, L. & Roberson, D. Mammalian vestibular hair cell regeneration. Science 267, 701 (1995).
López, I., Honrubia, V., Lee, S.-C., Li, G. & Beykirch, K. Hair cell recovery in the chinchilla crista ampullaris after gentamicin treatment: A quantitative approach. Otolaryngol. Head Neck Surg. 119, 255–262 (1998).
Kawamoto, K., Izumikawa, M., Beyer, L. A., Atkin, G. M. & Raphael, Y. Spontaneous hair cell regeneration in the mouse utricle following gentamicin ototoxicity. Hear. Res. 247, 17–26 (2009).
Gubbels, S. P., Woessner, D. W., Mitchell, J. C., Ricci, A. J. & Brigande, J. V. Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455, 537–541 (2008).
Zheng, J. L. & Gao, W.-Q. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 3, 580–586 (2000).
Taylor, R. R. et al. Regenerating hair cells in vestibular sensory epithelia from humans. Elife 7, e34817 (2018).
Helms, A. W., Abney, A. L., Ben-Arie, N., Zoghbi, H. Y. & Johnson, J. E. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 127, 1185 (2000).
Cam, H. & Dynlacht, B. D. Emerging roles for E2F: Beyond the G1/S transition and DNA replication. Cancer Cell 3, 311–316 (2003).
Gálvez, H., Tena, J. J., Giraldez, F. & Abelló, G. The repression of Atoh1 by Neurogenin1 during inner ear development. Front. Mol. Neurosci. 10, 321–321 (2017).
Mayor, C. et al. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047 (2000).
Ebert, P. J. et al. Zic1 represses Math1 expression via interactions with the Math1 enhancer and modulation of Math1 autoregulation. Development 130, 1949 (2003).
Johnson, D. G., Schwarz, J. K., Cress, W. D. & Nevins, J. R. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 365, 349–352 (1993).
Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 (1998).
Helin, K. Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet. Dev. 8, 28–35 (1998).
Bertoli, C., Skotheim, J. M. & de Bruin, R. A. M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518–528 (2013).
Rivolta, M. N. et al. Auditory hair cell precursors immortalized from the mammalian inner ear. Proc. Biol. Sci. 265, 1595–1603 (1998).
Yu, W. et al. First somatic mutation of E2F1 in a critical DNA binding residue discovered in well-differentiated papillary mesothelioma of the peritoneum. Genome Biol. 12, R96 (2011).
Ginsberg, D. et al. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 8, 2665–2679 (1994).
Vairo, G., Livingston, D. M. & Ginsberg, D. Functional interaction between E2F-4 and p130: Evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 9, 869–881 (1995).
Popov, B., Chang, L.-S. & Serikov, V. Cell cycle-related transformation of the E2F4–p130 repressor complex. Biochem. Biophys. Res. Commun. 336, 762–769 (2005).
Litovchick, L., Florens, L. A., Swanson, S. K., Washburn, M. P. & DeCaprio, J. A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 25, 801–813 (2011).
Rivolta, M. N. & Holley, M. C. Cell lines in inner ear research. J. Neurobiol. 53, 306–318 (2002).
Girling, R. et al. A new component of the transcription factor DRTF1/E2F. Nature 362, 83–87 (1993).
Tao, Y., Kassatly, R. F., Cress, W. D. & Horowitz, J. M. Subunit composition determines E2F DNA-binding site specificity. Mol. Cell. Biol. 17, 6994–7007 (1997).
Lu, Z. et al. E2F–HDAC complexes negatively regulate the tumor suppressor gene ARHI in breast cancer. Oncogene 25, 230 (2005).
Stone, J. S. & Cotanche, D. A. Hair cell regeneration in the avian auditory epithelium. Int. J. Dev. Biol. 51, 633–647 (2007).
Warchol, M. E. Sensory regeneration in the vertebrate inner ear: Differences at the levels of cells and species. Comp. Stud. Ear 273, 72–79 (2011).
Morest, D. K. & Cotanche, D. A. Regeneration of the inner ear as a model of neural plasticity. J. Neurosci. Res. 78, 455–460 (2004).
Taylor, R. R. & Forge, A. Hair cell regeneration in sensory epithelia from the inner ear of a urodele amphibian. J. Comp. Neurol. 484, 105–120 (2005).
Collado, M. S., Burns, J. C., Hu, Z. & Corwin, J. T. Recent advances in hair cell regeneration research. Curr. Opin. Otolaryngol. Head Neck Surg. 16, 465–471 (2008).
Rubel, E. W., Furrer, S. A. & Stone, J. S. A brief history of hair cell regeneration research and speculations on the future. Inn. Ear Dev. Regen. 297, 42–51 (2013).
Burns, J. C. & Stone, J. S. Development and regeneration of vestibular hair cells in mammals. Semin. Cell Dev. Biol. 65, 96–105 (2017).
Roberson, D. W., Alosi, J. A. & Cotanche, D. A. Direct transdifferentiation gives rise to the earliest new hair cells in regenerating avian auditory epithelium. J. Neurosci. Res. 78, 461–471 (2004).
Shang, J., Cafaro, J., Nehmer, R. & Stone, J. Supporting cell division is not required for regeneration of auditory hair cells after ototoxic injury in vitro. J. Assoc. Res. Otolaryngol. JARO 11, 203–222 (2010).
Jiang, L. et al. Transcriptomic analysis of chicken cochleae after gentamicin damage and the involvement of four signaling pathways (Notch, FGF, Wnt and BMP) in hair cell regeneration. Hear. Res. 361, 66–79 (2018).
Strachan, G. D., Koike, M. A., Siman, R., Hall, D. J. & Jordan-Sciutto, K. L. E2F1 induces cell death, calpain activation, and MDMX degradation in a transcription independent manner implicating a novel role for E2F1 in neuronal loss in SIV encephalitis. J. Cell. Biochem. 96, 728–740 (2005).
Ishizaki, J., Nevins, J. R. & Sullenger, B. A. Inhibition of cell proliferation by an RNA ligand that selectively blocks E2F function. Nat. Med. 2, 1386 (1996).
Darbinian, N., White, M. K. & Khalili, K. Regulation of the Pur-alpha promoter by E2F-1. J. Cell. Biochem. 99, 1052–1063 (2006).
Bagchi, S., Weinmann, R. & Raychaudhuri, P. The retinoblastoma protein copurifies with E2F-I, an E1A-regulated inhibitor of the transcription factor E2F. Cell 65, 1063–1072 (1991).
Bandara, L. R. & La Thangue, N. B. Adenovirus E1a prevents the retinoblastoma gene product from complexing with a cellular transcription factor. Nature 351, 494–497 (1991).
Chellappan, S. P., Hiebert, S., Mudryj, M., Horowitz, J. M. & Nevins, J. R. The E2F transcription factor is a cellular target for the RB protein. Cell 65, 1053–1061 (1991).
Swiss, V. A. & Casaccia, P. Cell-context specific role of the E2F/Rb pathway in development and disease. Glia 58, 377–390 (2010).
Rocha-Sanchez, S. M. & Beisel, K. W. Pocket proteins and cell cycle regulation in inner ear development. Int. J. Dev. Biol. 51, 585–595 (2007).
Clarke, A. R. et al. Requirement for a functional Rb-1 gene in murine development. Nature 359, 328–330 (1992).
Sage, C. et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14, 3037–3050 (2000).
Mantela, J. et al. The retinoblastoma gene pathway regulates the postmitotic state of hair cells of the mouse inner ear. Development 132, 2377 (2005).
Sage, C. et al. Essential role of retinoblastoma protein in mammalian hair cell development and hearing. Proc. Natl. Acad. Sci. 103, 7345 (2006).
Quandt, K., Frech, K., Karas, H., Wingender, E. & Werner, T. Matlnd and Matlnspector: New fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucl. Acids Res. 23, 4878–4884 (1995).
Cartharius, K. et al. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics 21, 2933–2942 (2005).
Helin, K. et al. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell 70, 337–350 (1992).
Wu, C. L., Zukerberg, L. R., Ngwu, C., Harlow, E. & Lees, J. A. In vivo association of E2F and DP family proteins. Mol. Cell. Biol. 15, 2536–2546 (1995).
Lui, J. C. & Baron, J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc. Natl. Acad. Sci. 110, 6181 (2013).
Beijersbergen, R. L. et al. E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo. Genes Dev. 8, 2680–2690 (1994).
Kingston, R. E., Chen, C. A. & Okayama, H. Calcium phosphate transfection. Curr. Protoc. Immunol. 31, 10.13.1-10.13.9 (1999).
Daudet, N. & Lewis, J. Two contrasting roles for Notch activity in chick inner ear development: Specification of prosensory patches and lateral inhibition of hair-cell differentiation. Development 132, 541 (2005).
Driver, E. C., Sillers, L., Coate, T. M., Rose, M. F. & Kelley, M. W. The Atoh1-lineage gives rise to hair cells and supporting cells within the mammalian cochlea. Dev. Biol. 376, 86–98 (2013).
Acknowledgements
We would like to thank Professor Jane Johnson (Southwestern Texas Medical University) for providing chick and mouse Atoh1 constructs and Dr. Matthew Kelley (National Institute on Deafness and Other Communication Disorders) for providing the ATOH1 antibody. We would also thank Dr. Lisa S. Nolan for the advice provided for experiments and revision of the manuscript. This work was supported by the following funding: Wellcome Trust [091092/Z/09/Z to SD and JG], Medical Research Council [MR/N004329/1 to SD and JG] & [MR/S003029/1; ND].
Author information
Authors and Affiliations
Contributions
M.G.D., S.J.D., N.D., and J.E.G. designed the research studies. M.G.D. performed the experiments and analyzed the data. M.G.D., S.J.D., N.D., and J.E.G. interpreted the data. M.G.D. wrote the manuscript and all authors contributed to revising and reviewing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gómez-Dorado, M., Daudet, N., Gale, J.E. et al. Differential regulation of mammalian and avian ATOH1 by E2F1 and its implication for hair cell regeneration in the inner ear. Sci Rep 11, 19368 (2021). https://doi.org/10.1038/s41598-021-98816-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98816-w
- Springer Nature Limited