
Abstract
Pulmonary hypertension (PH) initially results in compensatory right ventricular (RV) hypertrophy, but eventually in RV failure. This transition is poorly understood, but may be triggered by hypoxia. Measurements of RV oxygen tension (pO2) in PH are lacking. We hypothesized that RV hypoxia occurs in monocrotaline-induced PH in rats and that myo-inositol trispyrophosphate (ITPP), facilitating oxygen dissociation from hemoglobin, can relieve it. Rats received monocrotaline (PH) or saline (control) and 24 days later echocardiograms, pressure–volume loops were obtained and myocardial pO2 was measured using a fluorescent probe. In PH mean pulmonary artery pressure more than doubled (35 ± 5 vs. 15 ± 2 in control), RV was hypertrophied, though its contractility was augmented. RV and LV pO2 was 32 ± 5 and 15 ± 8 mmHg, respectively, in control rats. In PH RV pO2 was reduced to 18 ± 9 mmHg, while LV pO2 was unchanged. RV pO2 correlated with RV diastolic wall stress (negatively) and LV systolic pressure (positively). Acute ITPP administration did not affect RV or LV pO2 in control animals, but increased RV pO2 to 26 ± 5 mmHg without affecting LV pO2 in PH. RV oxygen balance is impaired in PH and as such can be an important target for PH therapy. ITPP may be one of such potential therapies.
Similar content being viewed by others

Introduction
Pulmonary hypertension (PH) is a condition involving increased pulmonary vascular resistance (PVR) that results in increased mean pulmonary artery pressure (mPAP) and afterload of the cardiac right ventricle (RV). This initially results in compensatory RV hypertrophy, but when RV is no longer able to compensate for the increased afterload, eventually RV failure occurs resulting in patient’s death1.
The cause of this transition from compensatory RV hypertrophy to decompensated RV failure is unclear2. Several lines of evidence indicate that impaired RV metabolism3 and in particular RV hypoxia, i.e. reduced myocardial oxygen tension, may underlie this phenomenon4,5. First, in PH oxygen delivery is impaired due to increased extravascular compression of RV coronary vessels (due to both RV hypertrophy and increased RV pressures) and reduced coronary perfusion pressure (due to reduced aortic pressure as a consequence of reduced LV cardiac output). Second, increased RV afterload results in proportionally increased energy demand. Third, capillary rarefaction was found in various animal PH models as well as in humans6. Fourth, these changes are accompanied by adverse metabolic reprogramming7. Fifth, increased activation of right ventricular hypoxia-inducible factor-1 (HIF-1) pathway, a marker of tissue hypoxia, was found in various models of PH8.
However, direct measurements of RV myocardial oxygen tension are lacking due to technical difficulties. Indirect methods, utilizing e.g. 1H NMR techniques which estimate the ratio of deoxygenated to oxygenated myoglobin or magnetic resonance imaging based on differences in magnetic properties between oxygenated and deoxygenated hemoglobin9 exhibit poor specificity and spatial resolution, but indeed suggest that RV oxygen partial pressure (pO2) may be reduced in humans with PH. However, some studies suggest normal RV oxygen availability10, while other studies indicate the presence of so called pseudohypoxia, i.e. activation of hypoxia-dependent molecular pathways without true tissue hypoxia11.
Recently myo-inositol tris-pyrophosphate (ITPP), a novel membrane-permeant allosteric effector of hemoglobin has been developed to enhance the oxygen release capacity of red blood cells specifically under hypoxic conditions12. ITPP lowers the affinity of hemoglobin for oxygen, thus counteracting the effects of hypoxia13. We have previously shown that ITPP shifts the oxygen dissociation curve downward, therefore increasing tissue oxygen delivery, increases oxygen delivery to hypoxic tumors14, and reduces HIF-1 expression by vascular endothelial cells15. This is accompanied by a potent anti-cancer effect in various tumor models in the mouse14,16. Moreover, we have recently shown that chronic ITPP treatment partially prevents post-myocardial infarction heart failure in the rat model17. However effects of ITPP on myocardial pO2 have never been studied.
Thus the aim of our study was to verify the hypothesis that RV is hypoxic in the rat model of monocrotaline (MCT)-induced PH and that acute ITPP administration is able to relieve it. For this purpose we used a specific fluorescence-based method to directly measure myocardial pO2 in situ in PH and assess the effects of ITPP treatment.
Methods
Forty four male Wistar rats, weighing 180–220 g, were used. All study animals were used in compliance with local and institutional regulations. The study conformed to the Guide for the Care and Use of Laboratory Animals, US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and was approved by the local ethics committee (Second Warsaw Local Ethics Committee for Animal Experimentation). The manuscript follows the recommendations in the ARRIVE guidelines. ITPP was a kind gift from professor Jean-Marie Lehn, UNISTRA, University of Strasbourg to CK.
Study protocol
At the age of 4–5 weeks the rats received a subcutaneous injection of MCT (60 mg/kg, Sigma) to induce pulmonary hypertension (n = 26) or saline (n = 18) as a control group. Twenty four days later the animals from both control (n = 13) and MCT (n = 14) groups were anesthetized using inhaled 2% isoflurane, underwent echocardiographic examination, followed by hemodynamic evaluation and pO2 measurements. Thereafter they were euthanized and their lungs were processed for histological examination. This time point (day 24) was selected to provide approximately 25% mortality in MCT group, indicating severe PH.
The remaining animals from the control (n = 5) and MCT (n = 5) groups underwent echocardiographic examination, received a single intraperitoneal dose of ITPP (1.5 g/kg b.wt.) and 1 h later were re-anesthetized, underwent echocardiographic examination, followed by hemodynamic evaluation and pO2 measurements. Thereafter they were euthanized and their lungs were processed for histological examination. A total of 7 rats died in the MCT group, while there were no deaths in the control group.
Echocardiographic evaluation
Transthoracic echocardiography was performed using E-cube 15 Platinum (Alpinion Medical Systems) with 17 MHz linear transducer under a light isoflurane sedation. The rats were placed on a heating pad to sustain proper body temperature. Images of the parasternal short-axis view at the papillary muscle level, left and right parasternal long axis view to visualize LV and RV, respectively, and the apical 4-chamber view were recorded with careful attention to obtain high frame rate. LV and RV fractional shortening as well as LV and RV diastolic and systolic wall thickness were assessed using the M-mode in parasternal long axis view. All measurements were obtained by one observer blinded to the study groups.
Hemodynamic evaluation
As reported previously18, rats were put on a heating pad, anesthetized with 2% isoflurane (oxygen was used as a carrier gas, flow rate 0.5 l/min, providing fraction of inspired oxygen [FiO2] of 40%), intubated and put on an animal ventilator. The upper abdominal cavity was opened and the heart was exposed through cutting of the diaphragm. The LV and subsequently RV apex was punctured with a 25G needle and a microtip pressure–volume (PV) catheter (SPR-838, Millar Instruments; Houston, TX) was inserted into the LV, RV, and subsequently pulmonary artery. Its position was established based on pressure and volume signals. After stabilization for 5 min, the signals were continuously recorded at sampling rate of 1000/s using an ARIA P–V conductance system (Millar Instruments) coupled to a PowerLab/4SP A/D converter (AD Instruments; Mountain View, CA) and a personal computer. To characterize cardiac function, first the inferior vena cava was compressed for 10 s and then released to achieve reduction and augmentation of venous return and cardiac preload, respectively. Heart rate, maximal LV and RV systolic pressure (ESP), end-diastolic pressure (EDP), maximal slope of systolic pressure increment (+ dP/dt max) and diastolic pressure decrement (− dP/dt max), ejection fraction (EF), end-diastolic volume (EDV), end-systolic volume (ESV), stroke volume (SV), and cardiac output (CO) were computed using a cardiac P–V analysis program (PVAN3.2, Millar Instruments). Mean pulmonary artery pressure (mPAP) was calculated as 0.61 × systolic PAP + 2 mmHg. Pulmonary vascular resistance (PVR) was calculated as (mPAP—left atrial pressure)/CO. Indexes of contractility and stiffness [slope of end-systolic and end-diastolic P–V relations (ESPVR and EDPVR)] were also calculated using PVAN3.2. The volume signal was calibrated using serial dilutions of sodium chloride, as recommended by the manufacturer.
pO2 measurements
Myocardial oxygen tension (pO2) was measured using a fiberoptic oxygen-sensing device, the OxyLite Pro pO2 monitor (Oxford Optronics Ltd., Oxford, UK). This device measures pO2 by determining the oxygen-dependent fluorescent lifetime of ruthenium chloride. Probes (100 µm in diameter) are supplied precalibrated by the manufacturer. The tip of an optical fiber probe is covered with ruthenium chloride, which fluorescence lifetime is O2-dependent and is inversely proportional to the pO2 in the tissue at the circular area with a diameter of 230 µm around the tip. Following excitation by a flash of green light, the measured half-life of the phosphorescence signal can be quantitatively related to the oxygen tension.
The rats were anesthetized using inhaled isoflurane (2%) and connected to a ventilator. The heart was exposed by left-sided thoracotomy, and a 20-gauge needle was used to pierce the epicardium and guide the probe into the myocardium immediately beneath the epicardium. A 100-µm diameter sensing tip was inserted into three locations in the cardiac RV and LV as well as in the liver and spleen. The pO2 signal was recorded until the signal was stable. Only good quality signals were analyzed (defined as at least 3 consistent non-zero pO2 measurements, differing by no more than 20% from the mean result). Subsequently data were averaged for each location. The measurements were performed at two FiO2 values: standard 40% and 100%.
Histological lung analysis
The lungs were inflated with OCT (Optimal Cutting Temperature) compound and then placed in plastic molds filled with OCT and snap-frozen in liquid nitrogen. All tissues were maintained at − 80 °C until further analysis.
The representative lung parts were cut into 5 µm-thick sections using Cryostat Microm HM550 (Thermo Fisher Scientific, Massachusetts, USA) and stained with the hematoxylin and eosin (H&E) stain for microscopic analysis. Arterioles with an external diameter of 30–90 µm were assessed and % wall thickness was expressed as (2 × wall thickness/external vessel diameter).
Statistical analysis
Shapiro–Wilk test was used to test normality of data distribution. Homogeneity of variances was tested by Bartlett’s. Normally distributed data were expressed as means ± SEM. One-way ANOVA was used to test differences between groups. Tukey post hoc test was used to compare data pairs. Non-normally distributed data were presented as median ± inter-quartile ranges + outliers. Subsequently statistical analysis of differences was tested using non-parametric methods (Mann–Whitney test to compare two groups or Kruskal–Wallis ANOVA to compare three or more groups followed by Dunn's post hoc test). Pearson correlation analysis was used to analyze the correlations. Differences were considered significant when P < 0.05. The primary end-point of our study was reduction of pO2 in RV in MCT vs. control rats. The study had 94% power to detect at least 25% reduction of pO2 in RV in MCT versus. control rats if 11 rats were included in each of the study groups. Statistical analyses were performed using SigmaPlot version 14.
Results
Pulmonary hypertension: the model
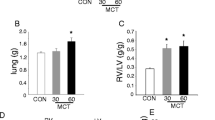
Twenty four days after MCT injection rats demonstrated significant hypertrophy of pulmonary arterioles (% wall thickness 23.4 ± 5.0% vs. 16.7 ± 4.5% in control rats, p < 0.05; Fig. 1A,B) and consequently increased pulmonary vascular resistance (PVR, Fig. 1C) and mean pulmonary artery pressure (mPAP, Fig. 1D), revealing typical chain of events for PH.

Histological and hemodynamic characterization of rats with monocrotaline-induced pulmonary hypertension and control rats. Representative hematoxylin and eosin stained images from control (A) and monocrotaline (MCT)-treated (B) rats, demonstrating small pulmonary arterioles (arrows). Yellow bars indicate arteriolar wall thickness. Clear wall thickening and luminal stenosis can be seen in a MCT rat. (C) demonstrates pulmonary vascular resistance (PVR), (D) mean pulmonary artery pressure (mPAP), and (E) left and right ventricular end-diastolic volumes. Representative right ventricular pressure–volume loops and tracings of right ventricular pressure and volumes are presented for control (F) and MCT (G) rats. (H) presents representative RV and LV PV loops from control and MCT rats. Horizontal lines represent mean (red) and median (black), the bottom and the top of the boxes represent the upper and the lower quartile, he whiskers represent 10th and 90th percentile, while the solid circles represent the individual data points. (C–E) control n = 9; MCT n = 14; since the data exhibited non-normal distribution, the groups were compared using Mann–Whitney test; * p < 0.05.
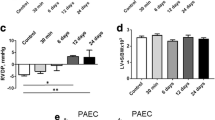
RV was dilated, which was reflected by increased RV end-diastolic volume (RVEDV, Figs. 1E–H, 2A,B), while LVEDV was reduced (Figs. 1E–H, 2A,B). Normalized right ventricular (RV) mass (Table 1) and RV diastolic wall thickness (Fig. 2C) were increased by 50%, while the corresponding parameters for LV were unchanged in PH rats.

Echocardiographic characterization of rats with monocrotaline-induced pulmonary hypertension and normal rats. Short-axis view of the rat heart in control (A) and monocrotaline-treated (B) rats. Yellow line outlines contours of the right ventricular cavity, white line of the left ventricular cavity. Markedly dilated right ventricle and compressed left ventricle can be seen in monocrotaline-treated rats (B). Lower panels demonstrate right diastolic wall thickness (C) and ventricular fractional shortening (D). Horizontal lines represent mean (red) and median (black), the bottom and the top of the boxes represent the upper and the lower quartile, he whiskers represent 10th and 90th percentile, while the solid circles represent the individual data points. Control n = 12; MCT n = 12; since the data exhibited non-normal distribution, the groups were compared using Mann–Whitney test;* p < 0.05.
Ventricular function was characterized using echocardiography and pressure–volume (PV) loops. RV fractional shortening (FS, Fig. 2D) and ejection fraction (EF, Table 1) were impaired and RV PV loops were shifted to the right (Fig. 1E–H). Consequently cardiac output was reduced by 29% (Table 1).
However, maximum rate of RV pressure increase (RV dP/dt max) was slightly increased, while the most precise and load-independent index of contractility, slope of end-systolic pressure–volume relation (ESPVR) was increased by almost threefold (Table 1), indicating that RV contractility was actually augmented in our model and impairment of EF and FS was due to increased afterload. RV hypertrophy was not sufficient to prevent increase of RV wall stress: the diastolic wall stress was more than doubled, while the systolic wall stress—more than tripled (Table 1).
On the other hand, LV was unloaded and a result of reduced RV function, resulting in leftward and downward shift of LV PV loops (Fig. 1H) and a low grade reduction of LV diastolic and systolic wall stress (Table 1).
Myocardial oxygen tension
Mean myocardial pO2 was 32 ± 5 mmHg and 15 ± 8 mmHg in RV and LV (Fig. 3A–D), respectively, in the control animals, while that in the liver and spleen was 19 ± 11 and 15 ± 11 mmHg, respectively. Breathing with 100% oxygen did not affect these values (Fig. 3A–D).

Measurements of myocardial oxygen tension. (A) An image demonstrating measurement of right ventricular (RV) partial oxygen pressure (pO2) using a fiber-optic probe inserted into the RV, almost parallel to the epicardial surface. Representative recordings of pO2 measurements in control and monocrotaline-treated rats are presented on panels (B) and (C), respectively. The horizontal axis indicates pO2. Panel B presents pO2 values measured under 40% fraction of inspired oxygen (NORMAL O2), while panel C presents pO2 values measured under 40% fraction of inspired oxygen (NORMAL O2) and 100% fraction of inspired oxygen (HIGH O2). RV, right ventricle; LV, left ventricle. The results are summarized for the right and left ventricle on panels (D) and (E), respectively. Correlations between RV and LV pO2 and wall stress as well as LV systolic pressure are shown on panels (F) through (I). Horizontal lines represent mean (red) and median (black), the bottom and the top of the boxes represent the upper and the lower quartile, he whiskers represent 10th and 90th percentile, while the solid circles represent the individual data points. Control n = 11; MCT = 13. Kruskal–Wallis ANOVA was used to compare the groups followed by Dunn's post hoc test. Pearson correlation analysis was used to analyze the correlations.* p < 0.05.
In PH animals mean RV pO2 was reduced by 44% to 18 ± 9 mmHg, while LV pO2 was unchanged (11 ± 8 mmHg). pO2 values in the liver and spleen were not affected (14 ± 8 and 23 ± 11 mmHg, respectively). Breathing with 100% oxygen increased pO2 values in RV to 25 ± 5 mmHg without affecting LV pO2 values.
Myocardial oxygen delivery depends on 2 crucial factors, perfusion pressure and vascular resistance. Therefore, to gain insight into potential causes of this reduction of RV pO2 we correlated myocardial pO2 with LV systolic pressure (LVSP, as a measure of aortic pressure, supplying perfusion pressure to coronary arteries) and with RV wall stress (as a marker of external vascular compression). As Fig. 3F,H show, RV pO2 was highly negatively correlated with RV diastolic wall stress and positively correlated with LVSP. No such correlation was found for the LV pO2 values (Fig. 3G,I).
Effect of ITPP
In additional experiments (n = 5) a single dose of ITPP was given intraperitoneally and 1 h later pO2 measurements were performed.
ITPP had no effect on pO2 values in the control animals: both LV and RV (Fig. 3D) as well as hepatic and splenic (not shown) oxygen tensions were not affected. 100% oxygen breathing had no effect in this setting, either. However, in PH animals, RV pO2 was significantly increased by acute ITPP administration by 44% to 26 ± 5 mmHg. ITPP did not affect pO2 values in the LV (Fig. 3E) or spleen or liver (not shown). 100% oxygen breathing did not affect RV pO2 values in MCT-treated rats that received ITPP. ITPP treatment had no effect on either echocardiographic or hemodynamic parameters (not shown).
Discussion
In this paper we reveal two new crucial facts regarding myocardial oxygen partial pressure in a rat model of monocrotaline-induced PH: (1) we demonstrate that PH is associated with reduced RV pO2 and that this reduction correlates with both reduced LV systolic pressure and increased RV diastolic wall stress and that (2) a new agent, ITPP, that increases oxygen dissociation from hemoglobin, improves RV pO2 in PH without affecting it in the healthy RV or LV.
RV function and structure in our pulmonary arterial hypertension model
In our model of MCT-induced PH, RV exhibited augmented contractile function as manifested by more than threefold increase of ESPVR and marked hypertrophy and dilation corresponding to a compensated stage of RV hypertrophy required to overcome more than fourfold increase of RV afterload. However, this was accompanied by reduction of cardiac output that in turn reduced LV preload and its output, resulting in decreased LV pressures and afterload. Thus, paradoxically RV wall stress increased, LV wall stress decreased and RV wall stresses were higher than those in LV, unlike in normal hearts.
RV myocardial oxygen tension
Here we showed that RV pO2 was more than twofold higher than LV pO2 (32 ± 5 mmHg and 15 ± 8 mmHg, respectively) in control rats. These values for LV are within the ranges reported by other authors and obtained using different methods: 45 mmHg in the LV of humans undergoing bypass surgery using voltammetric microelectrode technique19, 35 mmHg of mitochondrial O2 in the rat LV20, 10 mmHg in the rat LV21 and 8.6 mmHg in the swine LV using electron paramagnetic resonance oximetry22. These results indicate that LV pO2 values in the literature vary considerably and are probably method-dependent.
However, we are the first to report that RV pO2 is markedly higher than LV pO2 in control animals. It has long been known that the pO2 in the veins draining RV is higher than in those draining LV23 and that O2 extraction is higher in the LV than in the RV23. As RV oxygen consumption increases from rest to exercise, the initial effect is an increasing oxygen extraction (indicated by the falling venous oxygen tension). This increased oxygen extraction in the RV occurs with little change in flow when venous tension is above 10–12 mmHg. Conversely, in the LV increases in myocardial oxygen consumption result in negligible decreases in venous oxygen tension and large increases in coronary flow23. Our data nicely support these observations, indicating that indeed resting myocardial pO2 is much higher in RV than in LV. Higher extravascular compression in LV than in RV may be responsible for this phenomenon24.
Furthermore we showed here that RV pO2 was reduced by more than 40% in PH, while that in the LV was unchanged. This reduction correlated with both indices of reduced perfusion pressure (LVESP) as well as with indices of extravascular compression and oxygen utilization (RV wall stress). Thus it is difficult to determine, which of these factors was the main culprit here. Of note, no such correlation was found for LV pO2.
Effects of ITPP on myocardial oxygen tension
Affinity of hemoglobin for molecular oxygen is regulated in human red blood cells by 2,3-diphospho-D-glycerate II (DPG), an allosteric effector of hemoglobin. ITPP binds to the same allosteric pocket, resulting in rightward shift of hemoglobin-O2 dissociation curve, especially under low O2 conditions12, which could explain its specific effects found in hypoxic tumor tissues14. Moreover, ITPP enters red blood cells through the Band3 transporter, which distribution is mainly restricted to erythrocytes, which is responsible for its red blood cell specific affinity13,15.
Here we showed that ITTP increased RV pO2 in the PH rats. What is interesting, RV pO2 in PH rats (18 mmHg) was still higher than LV pO2 both in control rats (15 mmHg) and PH rats (14 mmHg). However, acute administration of a single dose of ITPP improved only RV pO2 in PH rats (to 26 mmHg), without affecting LV pO2. This again stresses the fact that oxygen supply and utilization is differently regulated in RV and LV and what is acceptable for LV, is hypoxic for RV. This may be due to different density of mitochondria and capillaries between these two ventricles10, but factors responsible for these differences remain unknown.
Limitations of the study
The study was performed solely in a model of MCT-induced PH, which is a limitation of the study. MCT is known to cause PH through injury of pulmonary arterial endothelium, but also causes local inflammation and fibrosis. Therefore our results require confirmation in other models of PH. We used only acute ITPP therapy, since the concept of chronic ITPP therapy was complicated by the fact that ITPP was also a potent stimulator of PTEN (phosphatase and tensin homolog deleted on chromosome ten), which could also contribute to its effects in the chronic setting14.
Conclusions
We show that RV pO2 is markedly reduced in the rat model of MCT-induced PH and that a new agent, ITPP, that facilitates oxygen release from hemoglobin pecifically under hypoxic conditions, is able to restore it upon acute administration. This indicates that RV is indeed hypoxic in PH and oxygen supply and more broadly RV energetics can be an important target for PH therapy; ITPP may be one of such potential therapies. Future studies are needed to verify this hypothesis.
Data availability
All data are available at the Department of Clinical Physiology, Centre of Postgraduate Medical Education, Warsaw, Poland.
References
Thenappan, T., Ormiston, M. L., Ryan, J. J. & Archer, S. L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 360, j5492–j5492. https://doi.org/10.1136/bmj.j5492 (2018).
Ryan, J. J. & Archer, S. L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 115, 176–188. https://doi.org/10.1161/CIRCRESAHA.113.301129 (2014).
Koop, A. M. C. et al. Metabolic remodeling in the pressure-loaded right ventricle: Shifts in glucose and fatty acid metabolism—A systematic review and meta-analysis. J. Am. Heart Assoc. 8, e012086. https://doi.org/10.1161/JAHA.119.012086 (2019).
Asosingh, K. & Erzurum, S. Mechanisms of right heart disease in pulmonary hypertension (2017 Grover Conference Series). Pulm. Circ. 8, 2045893217753121. https://doi.org/10.1177/2045893217753121 (2018).
Gómez, A. et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J. Am. Coll. Cardiol. 38, 1137–1142. https://doi.org/10.1016/S0735-1097(01)01496-6 (2001).
Ruiter, G. et al. Right ventricular oxygen supply parameters are decreased in human and experimental pulmonary hypertension. J. Heart Lung Transplant 32, 231–240. https://doi.org/10.1016/j.healun.2012.09.025 (2013).
Oikawa, M. et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J. Am. Coll. Cardiol. 45, 1849–1855. https://doi.org/10.1016/j.jacc.2005.02.065 (2005).
Pullamsetti, S. S., Mamazhakypov, A., Weissmann, N., Seeger, W. & Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 130, 5638–5651. https://doi.org/10.1172/JCI137558 (2020).
Sree Raman, K. et al. Right ventricular myocardial deoxygenation in patients with pulmonary artery hypertension. J. Cardiovasc. Magn. Reson. 23, 22. https://doi.org/10.1186/s12968-020-00694-0 (2021).
Balestra, G. M. et al. Increased in vivo mitochondrial oxygenation with right ventricular failure induced by pulmonary arterial hypertension: mitochondrial inhibition as driver of cardiac failure?. Respir. Res. 16, 6–6. https://doi.org/10.1186/s12931-015-0178-6 (2015).
Ryan, J. J. & Archer, S. L. Emerging concepts in the molecular basis of pulmonary arterial hypertension. Circulation 131, 1691–1702. https://doi.org/10.1161/CIRCULATIONAHA.114.006979 (2015).
Fylaktakidou, K. C., Lehn, J.-M., Greferath, R. & Nicolau, C. Inositol tripyrophosphate: A new membrane permeant allosteric effector of haemoglobin. Bioorg. Med. Chem. Lett. 15, 1605–1608. https://doi.org/10.1016/j.bmcl.2005.01.064 (2005).
Duarte, C. D., Greferath, R., Nicolau, C. & Lehn, J.-M. myo-Inositol trispyrophosphate: A novel allosteric effector of hemoglobin with high permeation selectivity across the red blood cell plasma membrane. ChemBioChem 11, 2543–2548. https://doi.org/10.1002/cbic.201000499 (2010).
Kieda, C. et al. Stable tumor vessel normalization with pO(2) increase and endothelial PTEN activation by inositol trispyrophosphate brings novel tumor treatment. J. Mol. Med. (Berl.) 91, 883–899. https://doi.org/10.1007/s00109-013-0992-6 (2013).
Kieda, C. et al. Suppression of hypoxia-induced HIF-1alpha and of angiogenesis in endothelial cells by myo-inositol trispyrophosphate-treated erythrocytes. Proc. Natl. Acad. Sci. USA 103, 15576–15581. https://doi.org/10.1073/pnas.0607109103 (2006).
El Hafny-Rahbi, B. et al. Tumour angiogenesis normalized by myo-inositol trispyrophosphate alleviates hypoxia in the microenvironment and promotes antitumor immune response. J. Cell Mol. Med. 25, 3284–3299. https://doi.org/10.1111/jcmm.16399 (2021).
Oknińska, M. et al. Treatment of hypoxia-dependent cardiovascular diseases by myo-inositol tris pyrophosphate (ITPP) through enhancement of oxygen delivery by red blood cells. J. Cell Mol. Med. 24, 2272–2283. https://doi.org/10.1111/jcmm.14909 (2020).
Paterek, A. et al. Acute heart rate-dependent hemodynamic function of the heart in the post-myocardial infarction rat model: Change over time. Can. J. Cardiol. 34, 1341–1349. https://doi.org/10.1016/j.cjca.2018.08.009 (2018).
Al-Obaidi, M. K., Etherington, P. J., Barron, D. J., Winlove, C. P. & Pepper, J. R. Myocardial tissue oxygen supply and utilization during coronary artery bypass surgery: Evidence of microvascular no-reflow. Clin. Sci. (Lond. Engl. 1979) 98, 321–328 (2000).
Mik, E. G. et al. Mitochondrial oxygen tension within the heart. J. Mol. Cell Cardiol. 46, 943–951. https://doi.org/10.1016/j.yjmcc.2009.02.002 (2009).
Tokunaga, C. et al. Albumin resuscitation improves ventricular contractility and myocardial tissue oxygenation in rat endotoxemia*. Crit. Care Med. 35, 1341–1347. https://doi.org/10.1097/01.ccm.0000260242.77637.57 (2007).
Zhao, X. et al. Endothelium-derived nitric oxide regulates postischemic myocardial oxygenation and oxygen consumption by modulation of mitochondrial electron transport. Circulation 111, 2966–2972. https://doi.org/10.1161/circulationaha.104.527226 (2005).
Tune, J. D., Gorman, M. W. & Feigl, E. O. Matching coronary blood flow to myocardial oxygen consumption. J. Appl. Physiol. 97, 404–415. https://doi.org/10.1152/japplphysiol.01345.2003 (2004).
Cai, Z. et al. Right ventricular oxygen delivery as a determinant of right ventricular functional reserve during exercise in juvenile swine with chronic pulmonary hypertension. Am. J. Physiol. Heart Circul. Physiol. 317, H840–H850. https://doi.org/10.1152/ajpheart.00130.2019 (2019).
Acknowledgements
The authors thank Professor Jean-Marie Lehn, ISIS, University of Strasbourg, 8 Allée Gaspard Monge, 67083 Strasbourg, France and Professor Claude Nicolau, Friedman School of Nutrition Science and Policy, Tufts University Boston, MA 02115, USA, for their constant collaboration, interest and support.
Funding
The study was supported by the National Science Centre, Poland Grant No. (2019/35/B/NZ5/04432).
Author information
Authors and Affiliations
Contributions
MM wrote the main manuscript text, UM, CS, AT, CK and MM contributed to the study concept, MO, ZZ, KZ, AP and KB performed the experiments, summarized and interpreted the data. All authors reviewed and accepted the final manuscript.
Corresponding author
Ethics declarations
Competing interests
CK is a shareholder of Normoxys Inc., manufacturer of ITPP. The other authors have no conflicts of interest to disclose.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oknińska, M., Zambrowska, Z., Zajda, K. et al. Right ventricular myocardial oxygen tension is reduced in monocrotaline-induced pulmonary hypertension in the rat and restored by myo-inositol trispyrophosphate. Sci Rep 11, 18002 (2021). https://doi.org/10.1038/s41598-021-97470-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97470-6
- Springer Nature Limited