
Abstract
Emphysema is an important feature of chronic obstructive pulmonary disease (COPD). Genetic factors likely affect emphysema pathogenesis, but this question has predominantly been studied in those of European ancestry. In this study, we sought to determine genetic components of emphysema severity and characterize the potential function of the associated loci in Korean population. We performed a genome-wide association study (GWAS) on quantitative emphysema in subjects with or without COPD from two Korean COPD cohorts. We investigated the functional consequences of the loci using epigenetic annotation and gene expression data. We also compared our GWAS results with an epigenome-wide association study and previous differential gene expression analysis. In total, 548 subjects (476 [86.9%] male) including 514 COPD patients were evaluated. We identified one genome-wide significant SNP (P < 5.0 × 10–8), rs117084279, near PIBF1. We identified an additional 57 SNPs (P < 5.0 × 10–6) associated with emphysema in all subjects, and 106 SNPs (P < 5.0 × 10–6) in COPD patients. Of these candidate SNPs, 2 (rs12459249, rs11667314) near CYP2A6 were expression quantitative trait loci in lung tissue and a SNP (rs11214944) near NNMT was an expression quantitative trait locus in whole blood. Of note, rs11214944 was in linkage disequilibrium with variants in enhancer histone marks in lung tissue. Several genes near additional SNPs were identified in our previous EWAS study with nominal level of significance. We identified a novel SNP associated with quantitative emphysema on CT. Including the novel SNP, several candidate SNPs in our study may provide clues to the genetic etiology of emphysema in Asian populations. Further research and validation of the loci will help determine the genetic factors for the development of emphysema.
Similar content being viewed by others


Introduction
Emphysema and chronic bronchitis are important features of chronic obstructive pulmonary disease (COPD), and recent studies show that imaging features are associated with adverse clinical outcomes of COPD1. Computed tomography (CT) evidence of emphysema is associated with worse prognosis even among subjects without airflow obstruction2,3. However, there is a lack of specific emphysema prevention and treatment techniques in part due to a limited understanding of pathobiological mechanisms.
Smoking is a major risk factor for emphysema, but emphysema is identified in non-smokers and the severity of emphysema among smokers varies greatly4,5,6. Emphysema has a genetic component with significant heritability and a previous study estimated its heritability to be approximately 25%7,8. Previous reports have described genetic determinants of emphysema and airway phenotypes in smokers with or without COPD9, emphysema in the general population10, distinct local histogram emphysema pattern11, and emphysema distribution12.
However, most emphysema genome-wide association studies (GWAS) were performed either exclusively or predominantly in European ancestry individuals. The Korean Obstructive Lung Disease (KOLD) and COPD in dusty areas (CODA) cohorts were constructed in South Korea collecting CT imaging and blood samples that enabled assessments of genetic associations with CT features. We investigated genetic determinants of emphysema severity in Korean cohorts and sought to relate these to gene expression and DNA methylation.
Methods
Study sample
Blood samples of 1056 subjects from the KOLD and CODA cohorts were genotyped. KOLD is a prospective cohort which recruited COPD patients from 16 university hospitals in South Korea. CODA is a prospective cohort conducted on subjects with airflow limitation and healthy volunteers living in dusty areas near cement plants in the Kangwon and Chungbuk provinces of South Korea. Details of the cohorts were described in previous papers13,14. Written informed consent was given by each participant. This study received ethical approval from the Kangwon National University Hospital IRB (KNUH 2012-06-007) and the Asan Medical Center IRB (Approval No. 2005-0345). This study was conformed to the tenets of the Declaration of Helsinki.
Computed tomographic measurements
In the KOLD cohort, CT measurements were obtained using 16-channel multidetector row CT scanner (SOMATOM Sensation; Siemens Medical Systems, Erlangen, Germany). In the CODA cohort, CT measurements were obtained using dual source CT scanner (SOMATOM Definition, Siemens Healthcare, Forchheim, Germany). In both studies, all subjects were scanned at full inspiration in a supine position. Emphysema was calculated as percent of lung area below or equal to the − 950 HU threshold and log-transformed.
Genotyping and quality control
We genotyped all subjects on Axiom KoreanChip 1.0 platform15. We removed low quality SNPs with low variant call rate, excessive heterozygosity and singletons, gender discrepancy, Hardy–Weinberg Equilibrium p < 0.001 and minor allele frequency < 0.01 using Affymetrix Power Tools and PLINK. After quality control, 586,966 SNPs with minor allele frequency of 1% or more were remained. (Fig. 1) Genotype imputation was performed at the Michigan Imputation Server using the HRC r1.1 reference panel. After imputation, SNPs with low imputation quality (R2 < 0.8) and minor allele frequency < 0.01 were removed. We combined original genotype and imputation data for analysis, and finally, 5,992,248 SNPs were analyzed for GWAS. The SNPs locations were based on National Center for Biotechnology Information (NCBI) Build 37.

Study workflow.
Genome-wide association analysis
We performed linear regression on natural log transformed emphysema index adjusted for age, sex, smoking status (never smoker, ex-smoker and current smoker), pack-years of smoking, and study center using PLINK (version 1.19)16. We also performed analyses only in COPD patients. We defined genome-wide significance as P < 5 × 10–8 and defined ‘candidate’ markers of interest at P < 5 × 10–6. Local association plots were generated around 600 kb in either direction of lead SNPs using LocusZoom in Asian genome (hg10/1000 Genome NOV 2014 ASN)17. Recombination rates were obtained using HapMap Phase II data18.
Epigenetic marks and DNase I hypersensitivity regions in top SNPs
Potential functional information of SNPs which exhibit high linkage disequilibrium (LD) (r2 > 0.8, 1000 G Phase1, Asian population as reference) with the top associated SNPs was obtained using the HaploReg database (version 4.1) and the single-tissue expression quantitative trait loci (eQTL) data in the genotype-tissue expression (GTEx) consortium (lung and whole blood)19,20. The P-value threshold for significant eQTL was set at 5 × 10–4 in GTEx database.
Epigenome-wide association study (EWAS) related to emphysema
We performed EWAS of emphysema in blood DNA from 100 CODA subjects using the Infinium HumanMethylation450 platform. EWAS methods have been previously described in a study of emphysema index21.
Comparison with previously published results
We performed look-ups of top SNPs in previously reported quantitative emphysema GWAS results which combined four study populations (COPDGene, ECLIPSE, National Emphysema Treatment Trial/Normative Aging Study, and GenKOLS)9. We also investigated whether the nearest genes of top SNPs were reported in previous differential expressed gene (DEG) analysis related to emphysema22.
Results
Baseline characteristics of study population
Baseline characteristics of study subjects are shown in Table 1. The mean age of all subjects was 71 years and 83.9% of all subjects were ever-smokers. Among a total of 548 subjects, 514 COPD patients who were defined as post-bronchodilator FEV1/FVC less than 0.7 and 34 subjects with normal spirometry had quantitative CT emphysema data (Fig. 1).
GWAS in all subjects and COPD patients only
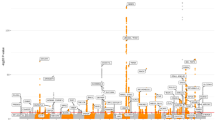
The Qq plot did not show systematic inflation in GWAS test statistics (lambda value 0.99 and 0.99 in all subjects and COPD patients, respectively (Supplementary Fig. S1). rs117084279 (MAF, 0.021), near the PIBF1 gene was genome-wide significant in all subjects (Table 2, Fig. 2). Fifty seven SNPs in 19 loci identified in all subjects, and 106 SNPs in 16 loci in COPD patients, reached a pre-defined suggestive significance level (P < 5.0 × 10–6) forming the candidate SNPs (Table 2). Among the candidate SNPs, 24 SNPs overlapped in both groups. The top 20 SNPs of each group are shown in Tables 2 and 3.

Regional plot for top SNPs in all subjects. Regional plot for ± 600 kb from top SNPs on PIBF1 (a), KLF12 (b), KCNJ3 (c) and rs11214944 (d) in total patient main model.
eQTL results
We sought to determine whether candidate SNPs are located in regions that have effect on regulation of gene expression in lung and whole blood using the GTEx database. Three candidate SNPs in all subjects and 8 candidate SNPs in COPD patients were identified as eQTL in lung or whole blood in GTEx (Tables 4 and 5).
Comparing enhancer regions with GWAS results
To further explore the functional role of candidate SNPs, we examined our candidate SNPs and SNPs in LD with our candidate SNPs using the HaploReg database. We found 67 lead variants in candidate SNPs of all subjects and 69 lead variants in candidate SNPs of COPD patients were in LD (r2 > 0.8) with SNPs in promoter or enhancer histone marks or DNase I in lung tissue (Supplementary Table S1-A,B).
Look-ups in DEG and EWAS data
Comparing with DEGs identified in a preceding emphysema RNA-seq analysis, MACROD2 upregulation in emphysema overlapped with a region identified in the GWAS in all subjects. Similarly, DOCK1, LARGE and ERAL1 also overlapped loci noted genes that is associated with SNPs identified in COPD patients (Table 6). In addition, several annotated genes of our candidate SNPs were also identified in our EWAS study related to emphysema with nominal level of significance (Supplementary Table S2). We plotted results of GWAS and functional studies together in Fig. 3.

Integrating results of GWAS and functional studies.
Look-ups in COPDGene data
In COPDGene emphysema results, we did not find any replicated SNP with our results (Supplementary Table S3).
Discussion
In this study, we identified one genome-wide significant SNP in a novel candidate gene (PIBF1) and candidate SNPs (P < 5.0 × 10–6) for quantitative emphysema on CT in all subjects and COPD patients in Korean cohorts. We further found candidate SNPs (at a more liberal threshold of significance) were often eQTL in lung tissue, in linkage disequilibrium with variants in promotor or enhancer histone markers, or DNase I hypersensitivity sites in lung tissue or whole blood. Moreover, several SNPs were located near genes reported in preceding emphysema RNA-seq analysis or preceding EWAS studies.
In all subjects, we identified 2 candidate SNPs which were also identified as eQTL in lung tissue, near CYP2A6. CYP2A6 has been associated with COPD and emphysema, and also smoking habits23,24. Genetic polymorphisms of CYP2A6 result in altered activity of the CYP2A6 protein, affecting nicotine metabolism and smoking behavior9,25,26. This effect was also evaluated in the Asian population27,28. However, a relationship between this region and COPD and emphysema has not yet been described. Smoking is an important risk factor for the development of emphysema; therefore, it is possible that this variant contributes to the development of emphysema through smoking. Further research is needed to elucidate the causal relationship.
One of our candidate SNPs (rs11214944) near NNMT was identified as an eQTL and in LD with SNPs lying in enhancer histone marks in lung tissue. NNMT has also been identified as a gene differentially expressed according to severity of COPD29,30. Moreover, NNMT has been identified as differentially expressed in moderate emphysema compared to mild emphysema by more than sixfold31. Intriguingly, in a previous study, NNMT was one of differentially expressed genes in IL-6 signaling related to airway inflammation and remodeling32. Although the study was focused on airway inflammation, not emphysema, considering that IL-6 and its signaling play a main function in emphysema pathogenesis, we could expect association of NNMT and emphysema through IL-633.
Although the MAF was very low, we identified one SNP, rs117084279, near PIBF1, that was genome-wide significant. Interestingly, PIBF1 has also been identified as one component of centriolar satellite proteins and has an essential role in primary cilia formation and ciliary protein recruitment34,35. By whole genome siRNA-based functional genomics screen, mutation in PIBF1 has been known to induce hereditary ciliopathy disease36. It has been well known that cigarette smoking causes structural and functional abnormality of cilia in bronchial epithelial cell37,38,39,40. Also, cigarette smoke is suggested to be responsible for genes associated with altered ciliary growth41. The relationship between ciliary function in airway epithelial cell and emphysema has not been well elucidated but our novel SNP could provide a potential link between them.
Likewise, KLF12and KCNJ3 are genes near our candidate SNPs, which were also found to be associated with our EWAS results. Of them, KCNJ3 was identified to associated with lung function and airway obstruction in previous studies, though there is no data on emphysema24,42. There is insufficient data to clarify that KLF12 is related to emphysema in lung. However, previous studies suggested that KLF12 gene, also known as the AP-2rep gene, functions as a transcriptional repressor of the AP-2α gene through a set of overlapping cis-regulatory promoter elements and a reciprocal regulation of both genes43,44. AP-2α is known to be involved in ras oncogene-mediated transformation and myc-mediated programmed apoptotic cell death45,46. Of interest, one study indicated that AP-2α protein was increased in lung of cigarette smoke exposure induced COPD rat model and this was also associated with increased cell apoptosis47. There are several mechanisms that likely contribute to the pathogenesis of emphysema48,49,50,51,52. One of them is apoptosis. Both animal COPD and human lung model suggest that apoptosis might be involved in the development of emphysema53,54,55. However, it has been unclear whether there is a direct relationship between AP-2α and apoptosis of cells in lung, results of our study yield novel insight of development and progression of emphysema and further experimental study is warranted.
In addition, we identified SNPs at a suggestive level of significance near MACROD2 which is DEG identified in preceding emphysema RNA-seq analysis. MACROD2 was associated with COPD and lung function in previous studies, but there is lack of data on the association with emphysema directly56,57,58. Although the reference study identified DEG according to presence of emphysema instead of quantitative value, integration with our GWAS result helps to identify meaningful genes among numerous genes22.
Although we did not find replicated SNPs in lookup results with COPDGene results, differential expression of genes and DNA methylation through high linkage disequilibrium with the genes could be suggested as a potential functional mechanism.
Inability to detect replicated SNPs in previous study is one of our limitations. This might be owing to small sample size, another limitation of our study, which can increase the false positive rates and decrease the statistical power. Even though a genome-wide significant SNP being identified, in view of both the relatively low MAF of the SNP and the small sample size of our study, further replication studies with larger population sizes are needed. Ethnic differences also could contribute to the inability of replication, and further Asian studies on emphysema are needed. Third, our study was not able to perform a meta-analysis with GWAS on emphysema or COPD in Asian population due to the lack of data. Comparing the results of meta-analysis with GWASs in different ethnicity would facilitate to elucidate the ethnic specificity. Fourth, the functional and biological impacts of the SNPs on emphysema are not identified in our study. Functional and integrated analysis may lead to a better understanding of pathophysiology of emphysema in Asian population. Also, we could not find out causal effects on emphysema of SNPs identified in our study. In further studies or meta-analysis including our study is needed to explore causal effects of the SNPs using Mendelian Randomization59,60,61,62,63. Finally, we had an interest in exploring whether the genetic cause could be a determining factor in emphysema regardless of smoking. Therefore, we focused on the results of the population that includes more never-smokers. However, owing to a small number of non-COPD subjects included in the total population, characteristics between the total and COPD populations may not be significantly different.
Conclusions
In a genome-wide association study of emphysema in Korean COPD, we identified a new genome-wide significant association and several associations at suggestive significance. Ours is the first GWAS related to quantitative emphysema in Korean population. Further analysis of including replication in other independent cohorts and functional studies would yield insights into the development of emphysema. In particular, this work may be a starting point to investigate the aspects of the pathobiology of emphysema that are shared or unique across differing ancestries.
References
Bhatt, S. P. et al. Imaging advances in chronic obstructive pulmonary disease: Insights from COPDGene. Am. J. Respir. Crit. Care Med. 199, 286–301 (2019).
Oelsner, E. C. et al. Association between emphysema-like lung on cardiac computed tomography and mortality in persons without airflow obstruction: A cohort studyemphysema-like lung on CT and all-cause mortality. Ann. Intern. Med. 161, 863–873 (2014).
Oh, Y. M. et al. Emphysematous phenotype is an independent predictor for frequent exacerbation of COPD. Int. J. Tuberc. Lung Dis. 18, 1407–1414 (2014).
Gietema, H. A. et al. Distribution of emphysema in heavy smokers: impact on pulmonary function. Respir. Med. 104, 76–82 (2010).
Agusti, A. et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir. Res. 11, 122 (2010).
Auerbach, O., Hammond, E. C., Garfinkel, L. & Benante, C. Relation of smoking and age to emphysema. Whole-lung section study. N. Engl. J. Med. 286, 853–857 (1972).
Zhou, J. J. et al. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am. J. Respir. Crit. Care Med. 188, 941–947 (2013).
Patel, B. D. et al. Airway wall thickening and emphysema show independent familial aggregation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 178, 500–505 (2008).
Cho, M. H. et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am. J. Respir. Crit. Care Med. 192, 559–569 (2015).
Manichaikul, A. et al. Genome-wide study of percent emphysema on computed tomography in the general population. The Multi-Ethnic Study of Atherosclerosis Lung/SNP Health Association Resource Study. Am. J. Respir. Crit. Care Med. 189, 408–418 (2014).
Castaldi, P. J. et al. Genome-wide association identifies regulatory Loci associated with distinct local histogram emphysema patterns. Am. J. Respir. Crit. Care Med. 190, 399–409 (2014).
Boueiz, A. et al. Genome-wide association study of the genetic determinants of emphysema distribution. Am. J. Respir. Crit. Care Med. 195, 757–771 (2017).
Oh, Y.-M. et al. Blood eosinophil count as a prognostic biomarker in COPD. Int. J. Chronic Obstr. Pulm. Dis. 13, 3589–3596 (2018).
Lee, H. et al. Inflammatory biomarkers and radiologic measurements in never-smokers with COPD: A cross-sectional study from the CODA cohort. Chronic. Respir. Dis. 15, 138–145 (2018).
Moon, S. et al. The Korea Biobank Array: Design and identification of coding variants associated with blood biochemical traits. Sci. Rep. 9, 1382 (2019).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Pruim, R. J. et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Cox, D. G. et al. GWAS in the SIGNAL/PHARE clinical cohort restricts the association between the FGFR2 locus and estrogen receptor status to HER2-negative breast cancer patients. Oncotarget 7, 77358–77364 (2016).
Ward, L. D. & Kellis, M. HaploReg v4: Systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877-881 (2016).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science (New York, NY) 348, 648–660 (2015).
Lee, M. K., Hong, Y., Kim, S.-Y., Kim, W. J. & London, S. J. Epigenome-wide association study of chronic obstructive pulmonary disease and lung function in Koreans. Epigenomics 9, 971–984 (2017).
Jeong, I., Lim, J.-H., Oh, D. K., Kim, W. J. & Oh, Y.-M. Gene expression profile of human lung in a relatively early stage of COPD with emphysema. Int. J. Chronic Obstr. Pulm. Dis. 13, 2643–2655 (2018).
Korytina, G. F. et al. Association of genes involved in nicotine and tobacco smoke toxicant metabolism (CHRNA3/5, CYP2A6, and NQO1) and DNA repair (XRCC1, XRCC3, XPC, and XPA) with chronic obstructive pulmonary disease. Mol. Biol. 48, 823–834 (2014).
Minematsu, N. et al. Association of CYP2A6 deletion polymorphism with smoking habit and development of pulmonary emphysema. Thorax 58, 623–628 (2003).
Benowitz, N. L., Swan, G. E., Jacob, P. 3rd., Lessov-Schlaggar, C. N. & Tyndale, R. F. CYP2A6 genotype and the metabolism and disposition kinetics of nicotine. Clin. Pharmacol. Ther. 80, 457–467 (2006).
Mwenifumbo, J. C. & Tyndale, R. F. Genetic variability in CYP2A6 and the pharmacokinetics of nicotine. Pharmacogenomics 8, 1385–1402 (2007).
Yuan, J.-M. et al. CYP2A6 genetic polymorphisms and biomarkers of tobacco smoke constituents in relation to risk of lung cancer in the Singapore Chinese Health Study. Carcinogenesis 38, 411–418 (2017).
Liu, T. et al. Association between daily cigarette consumption and hypertension moderated by CYP2A6 genotypes in Chinese male current smokers. J. Hum. Hypertens. 27, 24–30 (2013).
Savarimuthu Francis, S. M. et al. Genes and gene ontologies common to airflow obstruction and emphysema in the lungs of patients with COPD. PLoS One 6, e17442–e17442 (2011).
Kim, H. C. et al. Expression and functional significance of nicotinamide N-methyl transferase in skeletal muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 181, 797–805 (2010).
Shaw, J. G. et al. Biomarkers of progression of chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 6, 1532–1547 (2014).
Robinson, M. B. et al. IL-6 trans-signaling increases expression of airways disease genes in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L129–L138 (2015).
Ruwanpura, S. M. et al. Interleukin-6 promotes pulmonary emphysema associated with apoptosis in mice. Am. J. Respir. Cell Mol. Biol. 45, 720–730 (2011).
Kim, K. & Rhee, K. The pericentriolar satellite protein CEP90 is crucial for integrity of the mitotic spindle pole. J. Cell Sci. 124, 338 (2011).
Kim, K., Lee, K. & Rhee, K. CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS One 7, e48196 (2012).
Wheway, G. et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087 (2015).
Verra, F. et al. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am. J. Respir. Crit. Care Med. 151, 630–634 (1995).
Harvey, B. G. et al. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J. Mol. Med. (Berl.) 85, 39–53 (2007).
Leopold, P. L. et al. Smoking is associated with shortened airway cilia. PLoS One 4, e8157 (2009).
Simet, S. M. et al. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. Am. J. Respir. Cell Mol. Biol. 43, 635–640 (2010).
Brekman, A., Walters, M. S., Tilley, A. E. & Crystal, R. G. FOXJ1 prevents cilia growth inhibition by cigarette smoke in human airway epithelium in vitro. Am. J. Respir. Cell Mol. Biol. 51, 688–700 (2014).
Burkart, K. M. et al. A genome-wide association study in hispanics/latinos identifies novel signals for lung function. The hispanic community health study/study of Latinos. Am. J. Respir. Crit. Care Med. 198, 208–219 (2018).
Imhof, A. et al. Transcriptional regulation of the AP-2alpha promoter by BTEB-1 and AP-2rep, a novel wt-1/egr-related zinc finger repressor. Mol. Cell. Biol. 19, 194–204 (1999).
Roth, C., Schuierer, M., Günther, K. & Buettner, R. Genomic structure and DNA binding properties of the human zinc finger transcriptional repressor AP-2rep (KLF12). Genomics 63, 384–390 (2000).
Kannan, P. et al. N-ras oncogene causes AP-2 transcriptional self-interference, which leads to transformation. Genes Dev. 8, 1258–1269 (1994).
Moser, M. et al. Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev. 11, 1938–1948 (1997).
Li, J. et al. AP-2α expression and cell apoptosis of the lung tissue of rats with COPD and ECV304 cells stimulated by cigarette smoke extract. Chin. Sci. Bull. 56, 1562 (2011).
Wang, X. et al. Associations between maternal vitamin D status during three trimesters and cord blood 25(OH)D concentrations in newborns: A prospective Shanghai birth cohort study. Eur. J. Nutr. 60, 3473–3483 (2021).
Yu, H. et al. LEPR hypomethylation is significantly associated with gastric cancer in males. Exp. Mol. Pathol. 116, 104493 (2020).
Jin, G., Xu, M., Zou, M. & Duan, S. The processing, gene regulation, biological functions, and clinical relevance of N4-acetylcytidine on RNA: A systematic review. Mol. Ther. Nucleic Acids 20, 13–24 (2020).
Chen, J. et al. Genetic regulatory subnetworks and key regulating genes in rat hippocampus perturbed by prenatal malnutrition: Implications for major brain disorders. Aging (Albany NY) 12, 8434–8458 (2020).
Zheng, S. et al. Immunodeficiency promotes adaptive alterations of host gut microbiome: An observational metagenomic study in mice. Front. Microbiol. 10, 2415 (2019).
Demedts, I. K., Demoor, T., Bracke, K. R., Joos, G. F. & Brusselle, G. G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 7, 53–53 (2006).
Tuder, R. M., Petrache, I., Elias, J. A., Voelkel, N. F. & Henson, P. M. Apoptosis and emphysema. Am. J. Respir. Cell Mol. Biol. 28, 551–554 (2003).
Vij, N., Chandramani-Shivalingappa, P., Van Westphal, C., Hole, R. & Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 314, C73-c87 (2018).
Me, O. et al. A comprehensive evaluation of potential lung function associated genes in the SpiroMeta general population sample. PLoS One 6, e19382–e19382 (2011).
Kaneko, Y. et al. The search for common pathways underlying asthma and COPD. Int. J. Chronic Obstr. Pulm. Dis. 8, 65–78 (2013).
Matsson, H. et al. Targeted high-throughput sequencing of candidate genes for chronic obstructive pulmonary disease. BMC Pulm. Med. 16, 146–146 (2016).
Zhang, F. et al. Causal influences of neuroticism on mental health and cardiovascular disease. Hum. Genet. 240, 1267–1281 (2021).
Zhang, F. et al. Genetic evidence suggests posttraumatic stress disorder as a subtype of major depressive disorder. J. Clin. Investig. 27, 145942 (2021).
Yaghi, A. & Dolovich, M. B. Airway epithelial cell cilia and obstructive lung disease. Cells 5, 40 (2016).
Jiang, L. et al. Sex-specific association of circulating ferritin level and risk of type 2 diabetes: A dose-response meta-analysis of prospective studies. J. Clin. Endocrinol. Metab. 104, 4539–4551 (2019).
Wang, X. et al. Genetic support of a causal relationship between iron status and type 2 diabetes: A Mendelian randomization study. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/clinem/dgab454 (2021).
Funding
This study was supported by the National Research Foundation (NRF-2020R1I1A3073499). MHC was supported by R01HL135142, R01 HL137927, R01 HL089856, R01 HL147148. MHC has received grant funding from GSK and Bayer, and speaking and consulting fees from AstraZeneca and Illumina. Edwin K. Silverman received grant support from GSK and Bayer. MKL was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. Role of the sponsors: There was no sponsor for the study.
Author information
Authors and Affiliations
Contributions
W.J.K. contributed to the study concept and design, collection of data, interpretation, review, and revision of the manuscript; S.S. contributed to the interpretation, literature search, and writing and revision of the manuscript; H.C. and J.L. contributed to the analysis of data and writing manuscript; J.K. contributed to the collection of data and literature search; S.H.B. reviewed CT image; S.S.C., J.P., J.H.L., Y.M.O., M.K.L., B.D.H., M.C., and E.S. contributed to the review and revision of the manuscript. All authors provided final approval for publication submission and critically revised the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sin, S., Choi, HM., Lim, J. et al. A genome-wide association study of quantitative computed tomographic emphysema in Korean populations. Sci Rep 11, 16692 (2021). https://doi.org/10.1038/s41598-021-95887-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95887-7
- Springer Nature Limited