
Abstract
Determining the cause of focal segmental glomerulosclerosis (FSGS) has crucial implications for evaluating the risk of posttransplant recurrence. The degree of foot process effacement (FPE) on electron micrographs (EM) of native kidney biopsies can reportedly differentiate primary FSGS from secondary FSGS. However, no systematic evaluation of FPE in genetic FSGS has been performed. In this study, percentage of FPE and foot process width (FPW) in native kidney biopsies were analyzed in eight genetic FSGS patients and nine primary FSGS patients. All genetic FSGS patients showed segmental FPE up to 38% and FPW below 2000 nm, while all primary FSGS patients showed diffuse FPE above 88% and FPW above 3000 nm. We reviewed the literature which described the degree of FPE in genetic FSGS patients and identified 38 patients with a description of the degree of FPE. The degree of FPE in patients with mutations in the genes encoding proteins associated with slit diaphragm and cytoskeletal proteins was varied, while almost all patients with mutations in other FSGS genes showed segmental FPE. In conclusion, the present study suggests that the degree of FPE in native kidney biopsies may be useful for differentiating some genetic FSGS patients from primary FSGS patients.
Similar content being viewed by others


Introduction
Focal segmental glomerulosclerosis (FSGS) is one of the most frequent causes of end-stage kidney disease in children, and recurrence after kidney transplantation is a major challenge because of its association with poor graft survival1. FSGS is described as a renal histologic lesion with diverse causes and pathogenicity. Subclasses of FSGS include primary, genetic, and secondary forms, the latter of which comprises maladaptive, viral, and drug-induced FSGS2,3,4,5. Primary FSGS is caused by circulating factors and has a high risk of posttransplant recurrence, while other forms have very low risk of recurrence1. Therefore, identifying the cause of FSGS in each patient has crucial implications for the treatment strategy for kidney transplantation in these patients.
Advancements in next-generation sequencing techniques have allowed for rapid and efficient genetic variant detection. It has been proposed that genetic testing should be performed in all patients with child-onset steroid-resistant nephrotic syndrome6. However, genetic testing may not be feasible in some situations, especially when insurance coverage is not available for the test7. In addition, a negative test result does not exclude genetic disease, as novel mutations in undiscovered genes may be missed3. Therefore, thorough clinicopathologic evaluations remain an indispensable measure to identify the cause of FSGS.
Deegens et al. analyzed the differences in foot process width (FPW) between patients with primary FSGS versus those with secondary FSGS and found the effacement to be most severe in those with primary FSGS. Foot process was relatively preserved in secondary FSGS, with little overlap between the two subclasses8. Sethi et al. described that FSGS patients with nephrotic syndrome showed diffuse foot process effacement (FPE) in electron microscopy (EM) images, whereas those without nephrotic syndrome showed segmental FPE. The authors concluded that EM findings in native kidney biopsies are useful for differentiating primary FSGS from secondary FSGS9.
However, to date, no systematic evaluation of FPE in genetic FSGS has been performed. In this study, we analyzed the degree of FPE by EM analysis of native kidney biopsies in a case series with genetic FSGS and also reviewed the literature describing the degree of FPE in genetic FSGS. Additionally, we examined the degree of FPE in patients with a definitive diagnosis of primary FSGS who had a posttransplant recurrence. Finally, we examined whether the degree of FPE seen in EM images can differentiate genetic FSGS from primary FSGS.
Results
Baseline demographics and clinical data
There were no significant differences observed between primary FSGS and genetic FSGS with respect to age at disease onset, sex, time from onset to end-stage kidney disease, urinary protein excretion at kidney biopsy, and the Columbia classification (Table 1). The proportion of patients with edema was significantly higher in patients with primary FSGS than in those with genetic FSGS. Notably, five of eight patients with genetic FSGS met the criteria of nephrotic syndrome at kidney biopsy, and two of the five who met the criteria showed systemic edema during the clinical course. No patients with maladaptive FSGS presented with nephrotic syndrome or systemic edema.
Genetic mutations
Pathogenic mutations identified in patients with genetic FSGS (patient numbers 1–8) are shown in Supplementary Table 1. The affected genes were NUP107 in three patients10, WT1 in two patients, and LAMB2, INF2, and NUP9311 in one patient each. No patients with primary FSGS had pathogenic mutations in the 64 genes analyzed in the present study.
The degree of FPE in each group
Percentage of FPE in primary, genetic and maladaptive FSGS patients is shown in Fig. 1. Percentage of FPE in genetic FSGS patients ranged from 0 to 38%, while that in primary FSGS patients ranged from 88 to 100%. Therefore, all patients with genetic FSGS showed segmental FPE and all patients with primary FSGS showed diffuse FPE (Fig. 1). Percentage of FPE was significantly higher in primary FSGS patients than in genetic FSGS patients (p = 0.0003). Percentage of FPE in maladaptive FSGS patients ranged from 0 to 38%.

The degree of FPE (%FPE), shown as the percentage of capillary wall surface that was covered by podocyte foot processes uninterrupted by filtration slits. All patients with genetic FSGS (eight patients) showed segmental FPE ranging from 0 to 38%, while all patients with primary FSGS (nine patients) showed diffuse FPE ranging from 88 to 100%. Percentage of FPE was significantly higher in primary FSGS patients than in genetic FSGS patients (p = 0.0003). Percentage of FPE of maladaptive FSGS (three patients) ranged from 0 to 38%.
FPW in primary, genetic, and maladaptive FSGS patients is shown in Fig. 2. FPW of all genetic FSGS patients was below 2000 nm, while that of all primary FSGS patients was above 3000 nm. FPW of all maladaptive FSGS patients was below 1500 nm. FPW was significantly larger in primary FSGS than in genetic FSGS (p = 0.0006) (Fig. 2). Representative electron micrographs in a patient with primary FSGS and a patient with genetic FSGS are shown in Fig. 3A,B, respectively.

Foot process width of patients with primary, genetic and maladaptive FSGS patients. Median FPW was 4504 nm (range, 3534–5722 nm), 1719 nm (range, 647–1960 nm), and 1203 nm (range, 1047–1402 nm) in primary, genetic, and maladaptive FSGS patients, respectively. FPW was significantly larger in primary FSGS patients than in genetic FSGS patients (p = 0.0006).

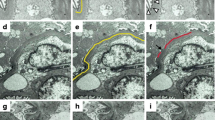
Representative electron micrographs of a patient with (A) primary FSGS and one with (B) genetic FSGS. (A) The patient (No. 13) with primary FSGS showed 100% FPE. All capillary loops were fully covered by FPE. (B) The patient (No. 1) with genetic FSGS (NUP107 mutation) showed segmental (0%) FPE with no capillary loops fully covered by FPE. The thin white arrows indicate preserved foot processes, and the thick white arrows point to effaced foot processes. Lower panels show images with a higher magnification. Original magnification: 3000× in (A) and (B). The scale bar denotes 10 μm.
The relationships between the amount of proteinuria and the degree of FPE (Supplementary Fig. 1)
Because less patients with genetic FSGS patients showed nephrotic syndrome compared to primary FSGS patients (Table 1), we examined the relationships between the amount of proteinuria and the degree of FPE. The amount of proteinuria correlated with neither percentage of FPE (r = 0.44; p = NS) nor FPW (r = 0.39; p = NS).
Literature review of articles and case reports describing the degree of FPE in genetic FSGS patients
A total of 1768 articles were identified using the predefined search strategy. By screening the study titles and abstracts, 1111 were considered not eligible as they did not address EM findings of patients with pathogenic mutations in the genes analyzed in this study. Subsequently, 640 of the remaining 657 studies were excluded after full review for the following reasons: 88 articles described patients with congenital or infantile nephrotic syndrome; 552 articles did not provide description of the FPE. Together with eight articles found by manual search, a total of 25 articles consisting of one review article, two case series, and 22 case reports describing a total of 38 cases were included12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36. Mutated genes identified in these 38 patients included CD2AP, KIRREL1, TRPC6, ACTN4, INF2, CRB2, PLCE1, WT1, NUP93, LAMB2, ITGA3, and COL4A3. Patients with NPHS1 mutations were excluded because the disease onset was in infancy in all patients. Our study included three patients who were described in the previous reports10,11 and five patients who were not described previously. The degree of FPE in a total of 46 patients from the literature and the present study is summarized in Table 2. Patients with pathogenic mutations in the genes that encode proteins associated with slit diaphragm, such as NPHS212,13,14, CD2AP15, KIRREL116, and TRPC617,18,19 showed diffuse FPE, except for one case with NPHS2 mutations13. Patients with mutations in the genes that encode cytoskeletal proteins, such as ACTN420,21,22,23 and INF224,25,26, showed varied degrees of FPE, with some patients showing segmental FPE and others showing diffuse FPE. All patients with mutations in the genes that encode other functioning proteins associated with podocytes and glomerular basement membrane (GBM) showed segmental FPE, except for one case with a WT1 mutation31.
Discussion
This study is the first to examine the degree of FPE in a case series of genetic FSGS patients and compare them with those in children with a definitive diagnosis of primary FSGS who had posttransplant recurrence. Children with maladaptive FSGS were also analyzed and showed segmental FPE (Fig. 1), which was consistent with a previous report9. Furthermore, FPW in all maladaptive FSGS patients was lower than 1500 nm, which was also consistent with the description by Deegens et al.8. All patients with genetic FSGS included in this study showed segmental FPE (%FPE < 40%), while all patients with primary FSGS showed diffuse FPE (%FPE > 80%) (Fig. 1). Additionally, FPW of all genetic FSGS patients was below 2000 nm, while that of all primary FSGS patients was above 3000 nm (Fig. 2). Therefore, our results suggest that the degree of FPE seen in EM images may be helpful to discriminate between some genetic FSGS patients and primary FSGS patients.
Several studies showed that the degree of FPE correlated with the amount of proteinuria37,38. Sethi et al. reported that FSGS patients presenting with nephrotic syndrome and diffuse FPE in EM images are likely to have primary FSGS9. In the present study, the amount of proteinuria correlated with neither percentage of FPE nor FPW (Supplementary Fig. 1). Additionally, urine protein excretion was not significantly different between patients with primary FSGS and those with genetic FSGS (Table 1). Notably, some patients with genetic FSGS presented with nephrotic syndrome and/or systemic edema, suggesting that these clinical manifestations are less helpful to discriminate between primary FSGS and genetic FSGS. Therefore, our study suggested that the degree of FPE seen in EM images may contribute to identifying primary FSGS and some cases of genetic FSGS, regardless of the presence or absence of nephrotic syndrome.
Our literature review identified 38 patients with genetic FSGS whose EM images were analyzed for the degree of FPE. As shown in Table 2, previous case reports and our results suggest that patients with mutations in the genes encoding slit diaphragm-associated proteins showed diffuse FPE, whereas those with mutations in the genes that encodes cytoskeletal scaffold and membrane proteins showed varied degrees of FPE. Almost all patients with mutations in the genes that encodes other proteins associated with podocytes and the GBM showed segmental FPE. The functions and localization of affected podocyte genes may impact the degree of FPE in genetic FSGS patients. For NPHS2, truncating or homozygous R138Q mutations resulted in earlier onset of disease before six years of age, while it was significantly later in patients with any other NPHS2 mutation, indicating a genotype–phenotype correlation39. Additionally, two siblings have been reported to have different clinical features with the degree of FPE: one showed diffuse FPE, while the other showed segmental, despite having the same genotype of NPHS2 mutations13. Similar findings were reported in siblings who had ACTN4 mutations21. These studies highlight a complex relationship between genotype, environmental factors, and epigenetic phenomena that is responsible for significant variability in the phenotype of a gene mutation. Combined with the results obtained from our patients, segmental FPE seen in EM images is strongly suggestive of genetic FSGS rather than primary FSGS.
This study is limited by a small sample size obtained from a single medical center as well as the diversity of genetic FSGS. The mutated genes identified in this study were different from the genes previously reported from Western countries. These studies described that the most frequently affected genes were NPHS2 and WT1 in patients with FSGS or steroid-resistant nephrotic syndrome at age of onset ≥ one year40,41. It has been reported that mutations in the NPHS2 genes are rarely identified in Japanese children with FSGS42,43. Additionally, our study did not examine adult FSGS patients. Further studies in a larger number of patients with mutations in different genes are needed to fully investigate the degree of FPE in genetic FSGS patients.
In conclusion, our study suggests that the degree of FPE in native kidney biopsies may be useful for differentiating some genetic FSGS cases from primary FSGS cases, which will help with the evaluation of the risk of recurrence before kidney transplantation.
Patients and methods
Study population (Fig. 4)

Study population in the present study. FSGS, focal segmental glomerulosclerosis; KT, kidney transplantation; EM, electron microscopy.
In this study, patients with congenital or infantile nephrotic syndrome were excluded, because they greatly differ in clinical manifestations and genetic background from FSGS patients with later onset40,41,44. A total of 64 patients with FSGS who underwent kidney transplantation at our institution between January 1, 1989 and December 31, 2018 were identified. No organs were procured from prisoners. All transplantations were performed at Tokyo Women’s Medical University. Thirty-seven kidney transplant recipients who were not analyzed by EM in their native kidney biopsies and three patients without sufficient clinical data to determine the presence of nephrotic syndrome were also excluded from this study. Of the remaining 24 patients, 9 showed posttransplant recurrence, while 15 did not. Clinical characteristics of the patients who showed posttransplant recurrence (patient numbers 9 to 17), and thus were diagnosed as having primary FSGS, are shown in Supplementary Table 2.
All 24 patients underwent genetic testing. We performed whole-exome sequencing using peripheral blood mononuclear cells with a focus on 64 genes currently known to be associated with FSGS (Supplementary Table 3). Of the 15 patients without posttransplant recurrence of FSGS, eight had pathogenic mutations in the genes associated with FSGS. Clinical characteristics of these eight patients with genetic FSGS (patient numbers 1 to 8) are shown in Supplementary Table 4. The remaining seven patients did not have any pathogenic mutations in the genes associated with FSGS. Because they did not experience posttransplant recurrence and may have as yet undiscovered genetic mutations associated with FSGS, a definitive diagnosis of primary FSGS could not be made, and thus were excluded from this study. Consequently, nine patients with a definitive diagnosis of primary FSGS and eight patients with genetic FSGS were included in this study. Three patients with maladaptive FSGS, diagnosed based on their clinical manifestations and native kidney biopsies between 1989 and 2018, were also included in this study to determine whether they show segmental FPE as previously reported9. The causes of maladaptive FSGS in these patients were bilateral hypoplastic kidneys, cyanotic congenital heart disease, and obesity-related nephropathy in one patient each. All three patients with maladaptive FSGS neither progressed to end-stage kidney disease nor underwent kidney transplantation. This study was approved by the ethical committees of Tokyo Women’s Medical University (approval number #4866-R3). All procedures performed in studies were in accordance with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individuals participating in this study.
Definitions
Nephrotic syndrome has been defined as the presence of a urinary protein to creatinine ratio above 2.0 g/g45 and a serum total protein level ≤ 6.0 g/dl46. The serum total protein level, instead of the serum albumin level, was used to define nephrotic syndrome, because some patients in this study lacked records of serum albumin levels at native kidney biopsies. End-stage kidney disease was diagnosed when a patient required chronic dialysis or kidney transplantation. A diagnosis of posttransplant recurrence of FSGS was based on the presence of at least one of the following criteria: (1) clinical recurrence of the nephrotic syndrome; (2) graft biopsy showing diffuse FPE by EM; (3) histological identification of FSGS by light microscopy in the absence of transplant glomerulopathy or any other apparent cause of proteinuria47.
Kidney pathology evaluation
Pathological findings of native kidney biopsies, including EM images, were analyzed in all patients. Light microscopy evaluation of kidney biopsies included staining with hematoxylin and eosin, periodic acid-Schiff, Masson’s trichrome, and periodic acid-methenamine-silver stain. Toluidine blue stained semi-thin sections were examined, and non-segmentally sclerosed glomeruli were identified for EM studies. Each biopsy was classified according to the Columbia classification48.
Degree of FPE in EM images
We examined the degree of FPE using two methods, which was described by Sethi et al. and Deegens et al.8,9. Percentage of FPE was defined as the percentage of capillary wall surface that was covered by podocyte foot processes uninterrupted by filtration slits9. In brief, eight capillary loops within one glomerulus that was neither globally sclerosed nor collapsed were analyzed by EM at a magnification of 1000× to 3000× for each patient. If foot processes were preserved or only partially effaced in one loop, this loop was not judged as diffuse effacement. Percentage of FPE was defined as the percentage of the eight loops that showed complete effacement: 100%, all loops showed complete effacement; 88%, one of eight loops did not show complete effacement; 75%, two of eight loops did not show complete effacement. No more than eight capillary loops on electron micrographs were eligible for analysis because of the retrospective nature of this study, although the previous study used 10 loops in each patient9.
Average FPW was calculated by dividing the total number of foot processes by the total length of the GBM8. Eight capillary loops within one glomerulus that was neither globally sclerosed nor collapsed were analyzed by EM at a magnification of 1000× to 3000× for each patient. ImageJ software (National Institutes of Health, USA) was used to measure the length of the GBM for each loop. Also, for each loop the number of foot processes was manually counted.
Immunosuppression regimen through kidney transplantation
Five patients (four of nine patients with primary FSGS and one of eight patients with genetic FSGS), who underwent kidney transplantation between April 1983 and January 2001, were treated with immunosuppressive regimens consisting of calcineurin inhibitor (cyclosporine or tacrolimus), azathioprine or mizoribine, and methylprednisolone49. Antilymphocyte globulin or deoxyspergualin was used as an induction agent. In the remaining 12 patients who underwent kidney transplantation between May 2002 and December 2018, the immunosuppression regimens consisted of induction with an anti-CD25 antibody (basiliximab), followed by maintenance treatment with corticosteroid, calcineurin inhibitor and mycophenolate mofetil47.
Prophylactic maneuver for recurrence of FSGS
In four of nine patients with primary FSGS, two to four sessions of plasmapheresis were performed prior to living-donor kidney transplantation. A single dose of rituximab (375 mg/m2) was also administered in one patient before living-donor kidney transplantation in 201250. Patients with genetic FSGS did not receive the prophylactic maneuver.
Whole-exome analysis
Whole-exome analysis was performed using a previously described method11,42. In brief, genomic DNA was extracted from peripheral blood. Exon capture was performed with a commercial kit (SureSelect Human All Exon Kit v5; Agilent Technologies, Santa Clara, CA, USA). Exon libraries were sequenced (HiSeq 2000 platform; Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Paired 100-base pair reads were aligned to the reference human genome (University of California Santa Cruz hg19) using the Burrows–Wheeler Aligner (Version 0.7.3a)51. Single-nucleotide variants and indels were identified as previously described52. We focused on the variants of 64 genes associated with FSGS and steroid-resistant nephrotic syndrome (Supplementary Table 3). Mitochondrial genome was not interrogated. Next, variant filtering on the basis of population frequency was performed to include only minor allele frequencies of < 1% of healthy control population databases53,54. Variants that were protein-truncating, highly conserved across species, and predicted to be deleterious based on at least two of three programs’ prediction scores from the web-based prediction programs PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (Sorting Intolerant From Tolerant) (http://sift.bii.a-star.edu.sg/), and MutationTaster (http://www.mutationtaster.org) were kept for analysis.
Degree of FPE in genetic FSGS literature review
We performed a comprehensive literature search of the PubMed database (up to June 2020) to identify review articles, original articles and case reports that described the degree of FPE in FSGS patients with identified mutated genes that were analyzed in the present study (Supplementary Table 3). Articles and reports that described patients with congenital or infantile nephrotic syndrome were excluded. We developed a search strategy that used a combination of text words and Medical Subject Headings, which included the following: “genetic,” “genetic testing,” “genes,” “focal segmental glomerulosclerosis,” and each name of 64 genes listed in Supplementary Table 3. The search was limited to human studies published in English. We further reviewed the reference lists of the selected studies for additional publications.
Statistical analysis
Statistical analysis was performed for the comparisons between primary FSGS and genetic FSGS patients. Data were expressed as medians with 25th and 75th percentiles and were compared using the Mann–Whitney U test. Categorical data were analyzed using chi-squared test or Fisher’s exact test as appropriate. Spearman’s rank correlation coefficients were calculated to assess the relationship between the amount of proteinuria and the degree of FPE. For all statistical tests, a p value < 0.05 was considered statistically significant.
Data availability
The dataset generated and analyzed in the current study are available from the corresponding author upon reasonable request.
References
Cosio, F. G. & Cattran, D. C. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. 91, 304–314 (2017).
D’Agati, V. D., Kaskelj, F. J. & Falk, R. J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 365, 2398–2411 (2011).
De Vriese, A. S., Sethi, S., Nath, K. A., Glassock, R. J. & Fervenza, F. C. Differentiating primary, genetic, and secondary FSGS in adults: A clinicopathologic approach. J. Am. Soc. Nephrol. 29, 759–774 (2018).
Sethi, S., Glassock, R. J. & Fervenza, F. C. Focal segmental glomerulosclerosis: Towards a better understanding for the practicing nephrologist. Nephrol. Dial. Transplant. 30, 375–384 (2015).
Zand, L., Glassock, R. J., De Vriese, A. S., Sechi, S. & Fervenza, F. C. What are we missing in the clinical trials of focal segmental glomerulosclerosis?. Nephrol. Dial. Transplant. 32, i14–i21 (2017).
Preston, R., Stuart, H. M. & Lennon, R. Genetic testing in steroid-resistant nephrotic syndrome: Why, who, when and how?. Pediatr. Nephrol. 34, 195–210 (2019).
Gbadegesin, R. A., Winn, M. P. & Smoyer, W. E. Genetic testing in nephrotic syndrome—Challenges and opportunities. Nat. Rev. Nephrol. 9, 179–184 (2013).
Deegens, J. K. et al. Podocyte foot process effacement as a diagnostic tool in focal segmental glomerulosclerosis. Kidney Int. 74, 1568–1576 (2008).
Sethi, S., Zand, L., Nasr, S. H., Glassock, R. J. & Fervenza, F. C. Focal and segmental glomerulosclerosis: Clinical and kidney biopsy correlations. Clin. Kidney J. 7, 531–537 (2014).
Miyake, N. et al. Biallelic mutations in nuclear pore complex subunit NUP107 case early-childhood-onset steroid-resistant nephrotic syndrome. Am. J. Hum. Genet. 97, 555–566 (2015).
Hashimoto, H. et al. In vivo expression of NUP93 and its alteration by NUP93 mutations causing focal segmental glomerulosclerosis. Kidney Int. Rep. 31, 1312–1322 (2019).
Ardiles, L. G., Carrasco, A. E., Carpio, J. D. & Mezzano, S. A. Late onset of familial nephrotic syndrome associated with a compound heterozygous mutation of the podocin-encoding gene. Nephrology 10, 553–556 (2005).
Lepori, N., Zand, L., Sethi, S., Fernandez-Juarez, G. & Fervenza, F. C. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin. Kidney J. 11, 179–190 (2018).
Benetti, E. et al. mRNA sequencing of a novel NPHS2 intronic mutation in a child with focal and segmental glomerulosclerosis. Saudi J. Kidney Dis. Transpl. 25, 854–857 (2014).
Tsvetkov, D. et al. A CD2AP mutation associated with focal segmental glomerulosclerosis in young adulthood. Clin. Med. Insites. Case. Rep. 14, 15–19 (2016).
Solanki, A. K. et al. Mutations in KIRREL1, a slit diaphragm component, cause steroid-resistant nephrotic syndrome. Kidney Int. 96, 883–889 (2019).
Liakopoulos, V. et al. Familial collapsing focal segmental glomerulosclerosis. Clin. Nephrol. 75, 362–368 (2011).
Hofstra, J. M. et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 28, 1830–1838 (2013).
Oo, S. Z. M. W. H., Freese, M. E., Holanda, D. G. & Thomas, C. P. Spontaneous remission of genetic, apparent primary, FSGS presenting with nephrotic syndrome challenges traditional notions of primary FSGS. J. Nephrol. https://doi.org/10.1007/s40620-020-00837-7 (2020).
Kaplan, J. M. et al. Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 (2000).
Pollak, M. R., Alexander, M. P. & Henderson, J. M. A case of familial kidney disease. Clin. J. Am. Soc. Nephrol. 2, 1367–1374 (2007).
Henderson, J. M., Alexander, M. P. & Pollak, M. R. Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. J. Am. Soc. Nephrol. 20, 961–968 (2009).
Kakajiwala, A. K., Meyers, K. E., Bhatti, T. & Kaplan, B. S. Rapid progression to end-stage renal disease in a child with a sporadic ACTN4 mutation. Clin. Nephrol. Case Stud. 23, 14–18 (2015).
Lee, H. K. et al. Variable renal phenotype in a family with an INF2 mutations. Pediatr. Nephrol. 26, 73–76 (2011).
Brown, E. J. et al. Mutations in the formin protein INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 42, 72–76 (2010).
Sanchez-Ares, M. et al. A novel mutation, outside of the candidate region for diagnosis, in the inverted formin 2 gene can cause focal segmental glomerulosclerosis. Kidney Int. 83, 153–159 (2012).
Watanabe, S. et al. Long-term clinicopathologic observation in a case of steroid-resistant nephrotic syndrome caused by a novel Crumbs homolog 2 mutation. Nephrology 23, 697–702 (2018).
Udagawa, T. et al. Altered expression of Crb2 in podocytes expands a variation of CRB2 mutations in steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 32, 801–809 (2017).
Fan, J. et al. A case report of CRB2 mutation identified in a Chinese boy with focal segmental glomerulosclerosis. Medicine (Baltimore) https://doi.org/10.1097/MD.0000000000012362 (2018).
Li, J. et al. WT1 mutation and podocyte molecular eexpression in a Chinese Frasier syndrome patient. Pediatr. Nephrol. 22, 2133–2136 (2007).
Benetti, E. et al. A novel WT1 gene mutation in a three-generation family with progressive isolated focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 5, 698–702 (2010).
Denamur, E. et al. Mother-to-child transmitted WT1 splice-site mutation is responsible for distinct glomerular diseases. J. Am. Soc. Nephrol. 10, 2219–2223 (1999).
Baun, D. A. et al. Mutations in nuclear pore genes NUP93, NUP205, and XPO5 cause steroid resistant nephrotic syndrome. Nat. Genet. 48, 457–465 (2016).
Mohney, B. G. et al. A novel mutation of LAMB2 in a multi-generational Mennonite family reveals a new phenotypic variant of Pierson syndrome. Ophthalmology 118, 1137–1144 (2011).
Nicolaou, N. et al. Gain of glycosylation in integrin α3 causes lung disease and nephrotic syndrome. J. Clin. Invest. 122, 4375–4387 (2012).
Malone, A. F. et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 86, 1253–1259 (2014).
Powell, H. R. Relationship between proteinuria and epithelial cell changes in minimal lesion glomerulopathy. Nephron 16, 310–317 (1976).
Koop, K. et al. Expression of podocyte associated molecules in acquired human kidney diseases. J. Am. Soc. Nephrol. 14, 2063–2071 (2003).
Hinkes, B. et al. Specific Podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 19, 365–371 (2008).
Sadowski, C. E. et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 26, 1279–1289 (2015).
Bierzynska, A. et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 91, 937–947 (2017).
Ogino, D. et al. Analysis of the genes responsible for steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis in Japanese patients by whole-exome sequencing analysis. J. Hum. Genet. 61, 137–141 (2016).
Maruyama, K. et al. NPHS2 mutations in sporadic steroid-resistant nephrotic syndrome in Japanese children. Pediatr. Nephrol. 18, 412–416 (2003).
Jalanko, H. et al. Congenital nephrotic syndrome. Pediatr. Nephrol. 24, 2121–2128 (2009).
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int. Suppl. 2, 139–274 (2012).
Japanese Society of Nephrology. Guidelines for the treatment of nephrotic syndrome. Nihon. Jinzo. Gakkai. Shi. 53, 78–122 (2011).
Hattori, M. et al. Increase of integrin-linked kinase activity in cultured podocytes upon stimulation with plasma from patients with recurrent FSGS. Am. J. Transplant. 8, 1550–1556 (2008).
D’Agati, V. D., Fogo, A. B., Bruijn, J. A. & Jennette, J. C. Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am. J. Kidney Dis. 43, 368–382 (2004).
Ohta, T. et al. Effect of pre- and postoperative plasmapheresis on posttransplant recurrence of focal segmental glomerulosclerosis in children. Transplantation 71, 628–633 (2001).
Chikamoto, H. et al. Pretransplantation combined therapy with plasmapheresis and rituximab in a second living-related kidney transplant pediatric recipient with a very high risk for focal segmental glomerulosclerosis recurrence. Pediatr. Transplant. 16, E286-290 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Mckenna, A. et al. The Genome Analysis Toolkit; a MapReduce framework for analyzing next generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Abecasis, G. R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Acknowledgements
We are indebted to the participants and families who generously participated in this study. This study was also supported by Medical Research Institute, Tokyo Women’s Medical University. We also thank J. Iacona, Ph.D., from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (KAKENHI) from eht Ministry of Education, Culture, Sports, Science and Technology of Japan (Subject ID: JP18K07029 to Kiyonobu Ishizuka, JP18K07830 to Kenichiro Miura, and JP18K07857 to Motoshi Hattori).
Author information
Authors and Affiliations
Contributions
K.I. and K.M. participated in research design, data collection, data analysis, interpretation, writing of the article and final approval of the version to be published. K.I. and K.M. contributed equally to this article. T.H. participated in genetic testing, data analysis and review of the article. N.K. and T.Y. participated in data collection, genetic testing and data analysis. Y.H. participated in research design and data analysis. Ma.H., S.F. and T.O. participated in data collection. Y.Y. participated in data interpretation. Mo.H. participated in research design and performance, data analysis and interpretation and writing of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ishizuka, K., Miura, K., Hashimoto, T. et al. Degree of foot process effacement in patients with genetic focal segmental glomerulosclerosis: a single-center analysis and review of the literature. Sci Rep 11, 12008 (2021). https://doi.org/10.1038/s41598-021-91520-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91520-9
- Springer Nature Limited
This article is cited by
-
Precise clinicopathologic findings for application of genetic testing in pediatric kidney transplant recipients with focal segmental glomerulosclerosis/steroid-resistant nephrotic syndrome
Pediatric Nephrology (2023)
-
Diagnostik und Therapie der Fokal-Segmentalen Glomerulosklerose – 2023
Wiener klinische Wochenschrift (2023)
-
Clinical, pathological, and genetic characteristics of cases with asymptomatic proteinuria not manifesting nephrotic syndrome at onset: a single-center retrospective study
Clinical and Experimental Nephrology (2022)