
Abstract
The present study introduces a systematic approach using analytical quality by design (AQbD) methodology for the development of a qualified liquid chromatographic analytical method, which is a challenge in herbal medicinal products due to the intrinsic complex components of botanical sources. The ultra-high-performance liquid chromatography-photodiode array-mass spectrometry (UHPLC-PDA-MS) technique for 11 flavonoids in Genkwa Flos was utilized through the entire analytical processes, from the risk assessment study to the factor screening test, and finally in method optimization employing central composite design (CCD). In this approach, column temperature and mobile solvent slope were found to be critical method parameters (CMPs) and each of the eleven flavonoid peaks’ resolution values were used as critical method attributes (CMAs) through data mining conversion formulas. An optimum chromatographic method in the design space was calculated by mathematical and response surface methodology (RSM). The established chromatographic condition is as follows: acetonitrile and 0.1% formic acid gradient elution (0–13 min, 10–45%; 13–13.5 min, 45–100%; 13.5–14 min, 100–10%; 14–15 min, 10% acetonitrile), column temperature 28℃, detection wavelength 335 nm, and flow rate 0.35 mL/min using C18 (50 × 2.1 mm, 1.7 μm) column. A validation study was also performed successfully for apigenin 7-O-glucuronide, apigenin, and genkwanin. A few important validation results were as follows: linearity over 0.999 coefficient of correlation, detection limit of 2.87–22.41, quantitation limit of 8.70–67.92, relative standard deviation of precision less than 0.22%, and accuracy between 100.13 and 102.49% for apigenin, genkwanin, and apigenin 7-O-glucuronide. In conclusion, the present design-based approach provide a systematic platform that can be effectively applied to ensure pharmaceutically qualified analytical data from complex natural products based botanical drug.
Similar content being viewed by others
Introduction
Interest in high-level analytical system for complex pharmaceutical ingredients such as plant extract is increasing in the reality that drug development using natural extracts is increasing worldwide. Botanical drug guidelines of the United States Food and Drug Administration (USFDA) which was revised in 2016, recommends a ‘Totality-of-the-Evidence’ approach that comprehensively utilizes fingerprint analysis, chemical identification, and quantification of active or chemical constituents in the drug substance to characterize the complexity of the botanical sources to ensure consistency in drug quality1,2.
In order to achieve high standard of analytical methods of quality control, quality by design (QbD) approach have been adopted during analytical method development of various pharmaceutical practices3,4,5,6. The QbD is a disciplined approach to understand and control new drug products, based on sound science and quality risk management in diverse pharmaceutical processes7,8. Analytical methods play a significant role in drug product development in the control scheme of constant quality system monitoring of a product lifecycle9. The International Conference on Harmonization (ICH) is preparing to develop a new ICH Quality Guideline (ICH Q14) on Analytical Procedure Development, which will include the QbD concept for analytical methods, termed Analytical Quality by Design (AQbD)10. The AQbD approach begins with determining the analytical target profile (ATP), which is the prospective target of the analytical method development process and relates performance elements based on the intended target criteria11. The selection of critical method attributes (CMAs) is also performed, which directly represent a strong link to the intended criteria such as selectivity, precision, or accuracy in the desired analytical quality. Secondly, parameters that may affect analytical results are identified through a risk assessment approach10. Those highly selected risk factors are known as critical method parameters (CMPs) which should be tested with design of experiment (DoE) methodology and statistical screening. Thirdly, the polynomial relationships between CMAs and CMPs were studied in order to understand the in-depth cause-effect aspects that were statistically designed to identify the influential input variables affecting the representative output variables12. Meanwhile, the DoE is usually conducted twice by screening factors and then response surface methodology for optimizing the analytical method. The purpose of the screening study is to find the high-risk factors through fewer experiments, which is usually performed with designed two-level models such as full factorial design (FFD), fractional factorial design (FrFD), and Plackett-Burman design (PBD)8,12. In addition, an optimization process is conducted to ensure that proper quality is attained in the analytical method by considering the selected high-risk factors during design. The results are interpreted through response surface methodology (RSM) which is a potent statistical technique in mathematical modeling to interpret the designed-responses. Optimized strategic design responses include Box–Behnken design (BBD), central composite design (CCD), Taguchi design (TD), Mixture design, and Doehlert design8,12. Finally, the most appropriate designed point or method operable design region (MODR) is calculated from the RSM and confirmed by the method validation processes13.
While quality control systems based on the AQbD approach are applied widely in the field of pharmaceuticals, few application studies have been conducted on botanical extracts14,15,16. Since botanical extracts have complex and diverse phytochemicals as active ingredients, the selection of optimal analytical conditions is not simple. Also, it is quite challenging to screen the analytical parameters (i.e. buffer pH, organic solvent type, gradient slope, column temperature, etc.) that must be optimized by DoE technique.
In this paper, a systematic design-based approach to optimize a liquid chromatographic analytical method for major constituents of Genkwa Flos was investigated to suggest an analytical platform for how to consider CMAs and identify CMPs in an integrated case study with a botanical source. Compared to the routine One-Factor-At-a-Time (OFAT) approach, which tried one variable and collected one response, the current total quality approach utilizes scientific designing models and statistic expertise to finally obtain less experiment time, robust, precise, and easily validated analytical method9.
The flower buds of Daphne genkwa (Genkwa Flos, Thymelaeaceae) have been widely used as traditional oriental medicine in East Asia, China and Korea, and continue to draw great attention for their diverse pharmacological efficacy17,18,19. Previous phytochemical studies on D. genkwa revealed diverse chemical components including diterpenoids, flavonoids, lignans, and coumarins20,21,22. In recent years, genkwa flavonoids, as the main active constituents of Genkwa Flos, have been reported to exhibit remarkable pharmacological activities such as anti-inflammatory23, immunoregulation24, anti-tumor activity in colorectal cancer25, and anti-rheumatoid arthritis activity26. In order to exploit Genkwa Flos as a main ingredient of botanical drug, it is necessary to develop a robust and reliable analytical method for quality control, which is able to identify and quantify multiple components in botanical extracts in order to assure the consistency of pharmacological efficacy of herbal drug products.
CMPs were determined by risk assessment and factor screening experimental data in sequence. CMAs were established by equations that can be expressed as a single number by collecting the resolution of multiple peaks. After developing the optimized method by central composition design (CCD), the method validation was carried out in order to evaluate the soundness of the methods.
Results and discussion
Characterization of flavonoids using UHPLC-PDA-MS analysis
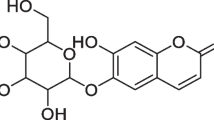
UHPLC-PDA-MS system was utilized for the identification of flavonoids in Genkwa Flos. High-resolution mass data from Time-of-Flight (TOF) analyzer combined with UV–Visible absorption spectral pattern enabled to identify known flavonoids from Genkwa Flos extracts by direct comparison with those of previous researches22,23 and/or reference standard solutions. A total of eleven identified flavonoids were listed in Table 1 providing their retention time, λmax, quasi-molecular ion, observed mass, mass difference, and molecular formula. Those were also tagged as peak 1 to peak 11 in the UHPLC chromatogram obtained at 335 nm (Fig. 1A) which are apigenin 5-O-glucoside, apigenin 7-O-glucoside, yuanhuanin, apigenin 7-O-glucuronide, genkwanin 5-O-primeveroside, genkwanin 5-O-glucoside, genkwanin 4′-O-rutinoside, tiliroside, apigenin, 3′-hydroxygenkwanin, and genkwanin as eluted in order.

Representative UHPLC chromatogram of Genkwa Flos extract tagged with characteristic 11 flavonoid peaks (A) and their chemical structures (B). Kinetex-C18 50× 2.1 mm, 1.7 μm column; mobile phase-A: 0.1% formic acid in water, mobile phase-B: acetonitrile; 335 nm detection; column temperature 28 ℃; 0.35 mL/min; gradient Time (min):%B, 0:10, 13:45, 13.5:100, 14:10, 15:10 used for the chromatogram.
Analytical target profile (ATP) and critical method attributes (CMAs)
The first step in AQbD-based method development is to define the ATP for stepwise and scientific procedures7. An analytical procedure which is able to quantitatively determine the specified eleven flavonoids in Genkwa Flos is a target of this study. Various elements of ATP such as analytical technique and instrument requirement were summarized as the intended target criteria (Supplementary Table S1). After ATP set-up, the potential CMAs were considered based on preliminary studies and review of the literature8,9. The general key CMA is the resolution (Rs) of critical peaks4,15,27, which may be a critical attribute to avoid peak overlap for selective identification in liquid chromatography. Finally, the CMAs, corresponding to ATP, were established as countable peak number (Yn) and resolution (Y1–11 and Ysum) after substantial consideration based on the modeling of experimental studies.
Preliminary studies
To carry out design-based method development studies, several preliminary tests were performed in different columns (i.e., length, particle size, manufacturer), using various solvents (i.e., acetonitrile, methanol), and acidified water (i.e., non-acidified, 0.1% acetic acid, 0.1% formic acid). Also, the detection wavelength for analyte was tested to acquire the greatest specific detection. The purpose of these attempts is to reduce variables by fixing those three parameters, but guarantee the best peak symmetry with the least working time. The achievement results were organized in Supplementary Table S2, and final decision to C18 (50 × 2.1 mm, 1.7 μm) column, acetonitrile and 0.1% formic acid water solvent system, and 335 nm detection wavelength, respectively.
Risk assessment studies
Quality risk management (QRM) allows us to control the entire process and recognize high-risk parameters that will affect the final quality of the analytical method28. We endeavored to establish QRM through risk assessment studies including experimental instruments and analytical parameters as shown in Fig. 2, an Ishikawa fishbone cause-effect diagram. From the cause-effect diagram, potential factors in performing liquid chromatography could be identified and a subsequent step, the organized failure effect in each of the potential factors were calculated with a risk priority number (RPN) to sort out the high risk factors29. Following the guidance of ICH Q1130, RPN numbers were calculated with the equation ‘Severity \(\times\) Probability \(\times\) Detectability’ to allocate risk in each failure mode. The risk assessment and control strategy are summarized in Table 2. Those parameters, column temperature (X1), flow rate (X2), injection volume (X3), and gradient slope, indicate highly influential factors, which are calculated greater than 10 RPN. Practically, when designing the models, the gradient slope was converted into run time (X4), because the initial and final percentages of acetonitrile solvent were fixed at 10 to 45 (Table 3). Thus, these four parameters were thereby selected for the further factor screening studies. The parameters counted less than 10 RPN were controlled as the constant.

Ishikawa Fishbone in Six Sigma of the UHPLC-PDA performance.
Factor screening studies
A (42) full factorial design (FFD), 4-factors and 2-levels, was performed for finding relatively fewer significant parameters from a list of higher risk potentially affecting the chosen CMAs, peak numbers (Yn). Since Yn generally reflects the integral quality of chromatographic separation, we chosen it for the FFD which is roughly executed at just 2-levels (Low and High). The selected high risk factors during risk assessment studies were identified as column temperature (X1), flow rate (X2), injection volume (X3), and run time (X4). The main effect(s) were estimated by selecting the first-order polynomial models, which were drawn out per Eq. (1):
In the equation, Yn is the studied CMAs, which is number of countable flavonoid peaks, when examined in each of 19 runs as depicted in Table 3. Those experimental runs were constructed randomly. A Pareto chart and Main effect plots (Fig. 3) show the significant influence of column temperature (X1) and run time (X4) on the studied CMAs, as these parameter frequencies were found to cross the corresponding α-value. As observed in Fig. 3B, the countable peak numbers (Yn) showed a negative correlation to column temperature (X1), but a positive effect by run time (X4). According to the statistical results (Table 4), the fitted model was very suitable to the experimental data by p-value under 0.05 with lack-of-fit larger than 0.05. Thus, factors such as column temperature (X1) and run time (X4) were selected as the CMPs for further optimization studies, and the other minor effective factors were kept as constant values. The flow rate (X2) was adjusted to 0.35 mL/min, while the injection volume (X3) was fixed at 1.0 μL.

Pareto chart (A) and main effect plots (B) obtained during factor screening of critical method attributes (CMAs), Yn peak numbers.
Response surface analysis
The subsequent chromatographic method optimization was executed by selecting the second-order quadratic polynomial model, where a central composite design (CCD) model designed with level 1.41421α were conducted with fourteen experimental runs (Table 5). The analyzed CMPs were column temperature (X1) and run time (X4) and studied at five different equidistant levels, i.e. low axial (− 1.41421), low factorial (− 1), central (0), high factorial (+ 1), and high axial (+ 1.41421). Meanwhile, the potential CMAs were newly chosen as Y1–11, which are the resolution (Rs) of each of the identified eleven flavonoid peaks listed in Table 1. Since botanical extracts have numerous phytochemicals, the resolution of each eleven peaks were defined between the closest eluted peaks. In detail, when calculate Y8 for the peak number 8 shown in Fig. 1A, the closest peak is just behind one eluted at 7.326 min. Besides, the first peak resolution (Y1) and second peak resolution (Y2) were of equal value, because the peaks are not totally separated or completely resolved by the UHPLC system and the closest eluting potential interference was each other. Furthermore, in several experimental runs (Table 5), the Y1 and Y2 were Rs = 0, indicating that those two peaks completely overlapped or co-eluted. The USP resolution equation using the baseline peak width drawn by lines tangent to the peak at 50% height was conducted for absolutely divided peaks, but USP Resolution (HH) using the peak width at half-height multiplied by a constant was utilized when calculated for overlapping peaks31.
In order to evaluate efficiently the total quality of separation in chromatographic fingerprints derived from each experimental run, one hypothetic score was introduced as total summation of Ys values of each peaks. In the design space, the Y1 to Y11 peaks were integrated as one value of Ysum by Eq. (3), which represents the estimated response for the experimental correlation with the two selected CMPs. Also, in order to prevent the value of a few peaks from dominating the overall result, it was necessary to determine the maximum value of each variable. A resolution over 1.5 usually indicates great separation, and when it is greater than 2, the peak is considered to be completely separated32. Hence, before integrating, the resolution values greater than 2 were set to 2 as shown in Eq. (2):
where Yi represents ith peak resolution after normalizing by Eq. (2), and the minimum to maximum response followed by Eq. (3) is 0 to 22, respectively. The randomly experimented fourteen runs to the selected CMAs are tabulated in Table 5 with the studied CMPs levels and designed experimental schedule. To clarify the CCD results, Minitab software ver. 18 was utilized for deriving ANOVA analysis and statistical optimization. Equation (4) is obtained by substituting the experimental data into a mathematical mode encompassing both main effects and interactions reflecting the second-order quadratic polynomial model.
ANOVA analysis was performed to statistically verify the model, which illustrates a statistically highly significant model (p < 0.05) and reasonable values of R2 (95.09% for determination and 90.89% for adjusted). The results are given in Table 4, it is also apparent that two CMPs in the first-order (X1, X4) and second-order (X1·X1, X4·X4) terms were significant, whereas the interaction correlation (X1·X4) was not significant. Those statistical results are also confirmed by observing the Pareto chart, Main effect plots, and Interaction plot shown in Fig. 4.

Pareto chart (A), main effect plots (B), and interaction plots (C) obtained during center composite design (CCD) studies of critical method attributes (CMAs), Ysum; summarizes the eleven resolutions.
Selection of optimum chromatographic solution
To obtain the optimized chromatographic method, the CCD design space was further studied in response surface analysis by using Statistica software ver. 13.3.0, carried out for the specific CMAs, Ysum. The 3D response surface (Fig. 5A) and 2D contour plot (Fig. 5B) revealed individual and plausible interaction(s) in factors and responses. Both column temperature (X1) and run time (X4) have a similarly curved plot, which is gradually increasing and decreasing at around the central level (0). Specifically, the central level of column temperature (X1) was 35 ℃ and run time (X4) was 14 min, respectively. As observed from Eq. (4), those patterns also may be inferred to be parabolic curves, which mean the response with a maximum value can be calculated by mathematical computing works. Finally, the optimum UHPLC-PDA performance solution with a maximum response Ysum of 18.80 was adjusted mathematically to the column temperature of 28.2861 ℃ and run time of 13.1784 min as portrayed in diagrams in Fig. 6. The verification step was studied to appraise model suitability and the repeatability results were near the predicted value of Ysum with a very acceptable %RSD and %RE (Table 6).

3D response surface plot (A) and 2D contour plot (B) depicting the interaction of two critical method parameters (CMPs) on the Ysum.

Optimization diagrams calculated mathematically.
Analytical method validation studies
The purpose of validating an analytical method is to demonstrate that the proposed method is suited for its intended use by satisfying the expectations of ATP. At first, method validation of UHPLC fingerprint was performed to determine the precision and stability. The same test solution (30 mg/mL) of the Genkwa Flos, which was injected six times in one day for precision test. Next the same test solution was analyzed 0 and 24 h after the preparation of test solution for stability test. The results were summarized in Supplementary Table S3 as calculated %RSD values of relative retention time (RRT) and relative peak area (RPA) of each peak which were calculated relative to the selected marker peak, apigenin 7-O-glucuronide (peak 4). All %RSD values of RRT and RPA of eleven peaks were under 1%, indicating the commendable precision and stability of the fingerprint method.
Next, we studied the quantitative method validation using three standard compounds of apigenin 7-O-glucuronide, apigenin, and genkwanin, which were identified as major components by chromatography (Fig. 1). Since the assigned eleven flavonoids were all 2-phenylchromen-4-one backbone flavones, those three peaks with the highest % area in the Fig. 1 were selected as representatives for verification of the optimized analytical method. Standard calibration curves of three compounds for linearity were derived in the range of 0.9765–500.00 μg/mL or 31.25–2000.00 μg/mL with the high values of the coefficient of correlation (0.999), respectively (Table 7). The linear calibration plots with corresponding residual plots are depicted in Supplementary Fig. S1, where none of the points were observed as outliers in the studied range of each concentration. Detection limit (DL) and Quantitation limit (QL) were also drawn out from the linearity test, indicating a sensitive method for quantification of those flavonoids. Precision, a measure of repeatability, was evaluated by intra-day and inter-day variability. As shown in Table 7, the %RSD value of content in the intra-day and also inter-day variability tests were found to be with a reasonable value as under 0.22, respectively. Accuracy of the method was confirmed by spiked and triplicate injections of known standard concentrations into the sample solution. Percentage recovery for the three compounds’ test concentrations studied ranged from 100.13% to 102.49% (Table 7), with their %RSD values less than 0.85.
Discussion
System suitability has been checked with the systematically optimized chromatographic method and found to be well within ICH criteria11 except resolution, as represented in Fig. 1. Among the eleven flavonoid peaks, resolution of peaks 1, 2, 3, 6, and 9 were under 1.5, which is the remaining challenge for a detailed trial of the isocratic and gradient mixed solvent system or to consider other factors. Meanwhile, an accurate and precise chromatographic method also depends on the %RSD values for injection repeatability precision, tailing factor9, plate count13, and capacity factor distribution11, so those criteria also must be considered as CMAs. However, the only criteria of resolution was selected for CMAs because %RSD and tailing factor were estimated to great precision and symmetry over the entire experiment. Also, when performed CCD studies of those parameters, plate count (> 2000), and capacity factor (> 1), were evaluated as proper in the overall 14 runs of experimental design work as tabulated in Supplementary Table S4.
To apply the AQbD approach, a thorough study on the characteristic of the analyte must be accomplished. The risk assessment studies were conducted carefully to achieve the optimized analytical method that is able to quantify diverse flavonoids from all of the other detected interferences with a substantial acceptable resolution, selectivity, and good efficiency. Thus, optimizing the selected CMPs as column temperature (X1) and run time (X4) the resolution of eleven identified flavonoid peaks were well resolved as mentioned and represented in Fig. 1.
Conclusion
The present study adopted a novel AQbD approach to develop a sensitive, robust, and accurate UHPLC-PDA-MS method for the identification and quantification of flavonoids in Genkwa Flos extract. In this approach, a methodical data collection process was conducted to identify the CMPs and CMAs through serial experiments of preliminary tests, risk assessment, full factorial design, and central composite design (CCD). Moreover, a new attempt to express target multiple peak resolutions as a single value was proposed by integrating all analytical peak data, and it provides a direction of how to handle CMAs in developing an analytical method of botanical extracts containing diverse components. The quantitative models depicted by a 3D surface plot with a 2D contour plot between two potential parameters, column temperature (X1) and run time (X4), were successfully constructed to facilitate finding the most suitable conditions for the chromatographic analysis. In conclusion, an AQbD-based quantitative multi-component analytical method is successfully developed and can serve as a template for other herbal medicinal product cases.
Material and methods
Standards and reagents
Apigenin (CAS no. 520-36-5, > 98.6%), apigenin 7-O-glucuronide (CAS no. 29741-09-1, > 98.8%), and genkwanin (CAS no. 437-64-9, > 98.0%) were purchased from Chem Faces, Wuhan, China. All of the other reagents were supplied by Duksan Pure Chemicals Co., Ltd., Ilsan, South Korea. For the analytical studies, HPLC-grade water, methanol, and acetonitrile were purchased from Fisher Scientific, Waltham, MA, USA; high purity nitrogen gas was provided by Shinyang Oxygen Co., Ltd., Seoul, South Korea.
Plant material and preparation of extracts
The flower bud of Daphne genkwa, which is a MFDS (Ministry of Food and Drug Safety of Republic of Korea) certified herbal medicine, was purchased from the Kyung-dong drugstore in Seoul, South Korea. The botanical origin was identified by Prof. Young Pyo Jang who is the head of Medicinal Herb Garden of College of Pharmacy, Kyung Hee University. A Voucher specimen (KHUP-2103) is deposited at the Herbarium of College of Pharmacy, Kyung Hee University, South Korea. Acquiring all plant samples and manufacturing extracts were carried out in compliance with the IUCN Policy Statement on Research Involving Species at Risk of Extinction (https://portals.iucn.org/library/efiles/documents/PP-003-En.pdf) and the Convention on International Trade in Endangered Species of Wild Fauna and Flora https://cites.org. The sample was ground and then powdered with 850 μm mesh sieves. Using 56% acetone in water as the extraction solvent, all flavonoid components were extracted by a shaking extraction procedure. The detailed list of extraction parameters are as follows: agitation speed of 150 rpm, shaking time of 12 h, and extraction temperature of 65 ℃. The concentration of the sample solution was fixed in all experimental sections as 30 mg/mL.
Instrumentation and UHPLC-PDA-ESI/MS conditions
A Waters AQCUITYTM H-class UPLC system (Waters Corp., Milford, MA, USA) was used for the UHPLC analysis. The system composed of a photo diode array (PDA) detector, quaternary solvent and sample manager, cooling auto sampler, and column oven. The operating software was Empower-3 software (Waters Corp.). A Kinetex-C18 column (2.1 mm × 50 mm i.d., particle size 1.7 μm, Phenomenex, Torrance, CA, USA) was used for all the chromatographic analysis. The sample was maintained at 25 ℃ and the UV/Visible detector wavelength was fixed at 335 nm in all experiments. The mobile phase was composed of acetonitrile and acidified water with 0.1% formic acid. The column oven, flow rate, injection volume, and solvent gradient system were screened by experimental design.
To identify and assign flavonoids, the mass spectrometric studies were carried out on an AccuTOF® single-reflection TOF mass spectrometer (JEOL, Tokyo, Japan) equipped with an ESI probe. Some important parameters of mass spectrometry were as follows: positive ion mode, mass range m/z 100—1500, needle voltage—2000 V, orifice-1 voltage—80 V, ring lens voltage—10 V, orifice-2 voltage—5 V. Nebulizing and desolvation gas was nitrogen. The desolvation temperature was 250 °C and the orifice-1 temperature was set to 80 °C. Mass Center System (version 1.3.7b, JEOL, Tokyo, Japan) was operating software and mass calibration was conducted using the YOKUDELNA kit (JEOL, Tokyo, Japan).
Statistical analysis
In current study, two design of experiments, full factorial design (FFD) and central composite design (CCD), were constructed and also statistical analyzed using Minitab software ver. 18 (Minitab Inc., State College, PA, USA). The statistically significant coefficients (p < 0.05) per analysis of variance (ANOVA) were used in framing the polynomial equation followed by the evaluation of the fit of the two models. Parameters evaluated for appropriate fitting of the models including coefficient of correlation (R2), lack of fit, F-value, and P-value are listed, respectively. Among them, the result of CCD was also studied in response surface analysis utilizing Statistica software ver. 13.3.0 (TIBCO Software Inc., Palo Alto, CA, USA).
Chromatographic method validation analysis
After defining the design model, the analytical operating point was validated per the International Conference on Harmonization (ICH) guideline Q2 (R1) and the parameters are described below33. Among the eleven identified flavonoids, three major eluates were chosen for study in this validation process, which are apigenin 7-O-glucuronide, apigenin, and genkwanin.
Linearity and range
To confirm linearity, working standards of apigenin 7-O-glucuronide in the range of 31.25–2000.00 μg/mL, apigenin and genkwanin in the range of 0.9765–500.00 μg/mL were prepared by a serial dilution process and then analyzed. From regression analysis, three regression lines along with the regression equation and least squares were derived by each of the standard compounds, respectively.
Detection limit and quantitation limit
Following the guideline Q2 (R1), there are several approaches for calculating Detection limit (DL) and Quantitation limit (QL), we chose the method “Based on the Standard Deviation of the Response (s) and the Slope (α)33” for this study. In Eqs. (5) and (6), the slope (α) was derived from each slope of the three analytical curves. The standard deviation of the response (s) was determined based on the residual standard deviation of each regression line.
Precision
Repeatability and Intermediate Precision were performed with a known concentration of the analyte (30 mg/mL) to investigate precision. On the same day, two samples at 100% of the test concentration were studied by six determinations each for the repeatability test. One sample was prepared for chromatographic analysis by six determinations on the next day testing for Intermediate Precision. All results were assessed as the percentage relative error by converted reference contents.
Accuracy
Calculating the percentage recovery of analyzed spiked samples was used for the accuracy test. Three known amount of each standard solutions: 125, 250, and 500 μg/mL of apigenin 7-O-glucuronide; 15.625, 31.25, and 62.5 μg/mL of apigenin; 31.25, 62.5, and 125 μg/mL of genkwanin were spiked with respect to the analyte (30 mg/mL) solution. The recovery studies were carried out three times showing that the percentage recovery and also percentage relative error were calculated to be accurate.
References
Lee, S. L. A Totality-of-evidence approach to ensuring therapeutic consistency of naturally derived complex mixtures. In The Science and Regulations of Naturally Derived Complex Drugs Vol. 32 (eds Sasisekharan, R. et al.) 265–270 (Springer, 2019).
Wu, C. et al. Scientific and regulatory approach to botanical drug development: A US FDA perspective. J. Nat. Prod. 83, 552–562 (2020).
Bandopadhyay, S., Beg, S., Katare, O., Sharma, T. & Singh, B. Integrated analytical quality by design (AQbD) approach for the development and validation of bioanalytical liquid chromatography method for estimation of valsartan. J. Chromatogr. Sci. 58, 606–621 (2020).
Bommi, S., Jayanty, S., Tirumalaraju, S. R. & Bandaru, S. Quality by design approach to develop stability indicating method to quantify related substances and degradation products of sacubitril by high performance liquid chromatography. J. Chromatogr. Sci. 58, 844–858 (2020).
Sharma, T., Khurana, R. K., Jain, A., Katare, O. & Singh, B. Development of a validated liquid chromatographic method for quantification of sorafenib tosylate in the presence of stress-induced degradation products and in biological matrix employing analytical quality by design approach. Biomed. Chromatogr. 32, e4169. https://doi.org/10.1002/bmc.4169 (2018).
Kumar, D. D., Ancheria, R., Shrivastava, S., Soni, S. L. & Sharma, M. Review on pharmaceutical quality by design (QbD). Asian J. Pharm. Res. Dev. 7, 78–82 (2019).
Panda, S., Beg, S., Bera, R. & Rath, J. Implementation of quality by design approach for developing chromatographic methods with enhanced performance: A mini review. J. Anal. Pharm. Res. 2, 1–4 (2016).
Sahu, P. K. et al. An overview of experimental designs in HPLC method development and validation. J. Pharm. Biomed. Anal. 147, 590–611 (2018).
Tome, T., Žigart, N., Časar, Z. & Obreza, A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: Overview and recent advances. Org. Process Res. Dev. 23, 1784–1802 (2019).
International Conference on Harmonization (ICH); Tripartite Guidelines. ICH Q2 (R1) Q14 (2018). https://database.ich.org/sites/default/files/Q2R2-Q14_EWG_Concept_Paper.pdf. Accessed 15 Jan 2021.
Sharma, G., Thakur, K., Raza, K. & Katare, O. Stability kinetics of fusidic acid: Development and validation of stability indicating analytical method by employing analytical quality by design approach in medicinal product(s). J. Chromatogr. B 1120, 113–124 (2019).
Dejaegher, B. & Van Heyden, Y. The use of experimental design in separation science. Acta Chromatogr. 21, 161–201 (2009).
Gupta, K. Analytical quality by design: A mini review. Biomed. J. Sci. Tech. Res. 1, 1–5 (2017).
Dai, S. et al. Robust design space development for HPLC analysis of five chemical components in Panax notoginseng saponins. J. Liq. Chromatogr. Relat. Technol. 39, 504–512 (2016).
Gong, X. et al. Development of an analytical method by defining a design space: A case study of saponin determination for Panax notoginseng extracts. Anal. Methods 8, 2282–2289 (2016).
Zhang, H. et al. Establishing the chromatographic fingerprint of traditional Chinese medicine standard decoction based on quality by design approach: A case study of Licorice. J. Sep. Sci. 42, 1144–1154 (2019).
Li, F. et al. Daphnane-type diterpenes with inhibitory activities against human cancer cell lines from Daphne genkwa. Bioorg. Med. Chem. Lett. 23, 2500–2504 (2013).
Zhou, D. C. et al. Comprehensive evaluation on anti-inflammatory and anti-angiogenic activities in vitro of fourteen flavonoids from Daphne Genkwa based on the combination of efficacy coefficient method and principal component analysis. J. Ethnopharmacol. 268, 113683. https://doi.org/10.1016/j.jep.2020.113683 (2021).
Zhang, Y. et al. Simultaneous determination of eight active components in chloroform extracts from raw and vinegar-processed Genkwa flos using HPLC-MS and identification of the hepatotoxic ingredients with an HL-7702 cell. Anal. Methods 6, 7022–7029 (2014).
Xu, W. C., Shen, J. G. & Jiang, J. Q. Phytochemical and biological studies of the plants from the genus Daphne. Chem. Biodivers. 8, 1215–1233 (2011).
Hou, X., Hou, X. & Li, L. Chemical constituents from the flower buds of Daphne genkwa (Thymelaeaceae). Biochem. Syst. Ecol. 91, 104055 (2020).
Wang, Z. P. et al. Investigation of potential toxic components based on the identification of Genkwa Flos chemical constituents and their metabolites by high-performance liquid chromatography coupled with a q exactive high-resolution benchtop quadrupole orbitrap mass spectrometer. J. Sep. Sci. 41, 3328–3338 (2018).
Du, W. J. et al. Relationship between the UPLC-Q-TOF-MS fingerprinted constituents from Daphne genkwa and their anti-inflammatory, anti-oxidant activities. Biomed. Chromatogr. 31, e4012. https://doi.org/10.1002/bmc.4012 (2017).
Gao, X. W., Zheng, W. F. & Peng, Y. C. Effect of serum containing total flavonoids from roots of Daphne genkwa on cell immunity in mice. Chin. Tradit. Herbal Drugs. 37, 721–725 (2006).
Du, W. J. et al. Antitumor activity of total flavonoids from Daphne genkwa in colorectal cancer. Phytother. Res. 30, 323–330 (2016).
Sun, Y. W. et al. Anti-rheumatoid arthritis effects of flavonoids from Daphne genkwa. Int. Immunopharmacol. 83, 106384. https://doi.org/10.1016/j.intimp.2020.106384 (2020).
Tome, T., Časar, Z. & Obreza, A. Development of a unified reversed-phase HPLC method for efficient determination of EP and USP process-related impurities in celecoxib using analytical quality by design principles. Molecules 25, 809 (2020).
Charoo, N. A. & Ali, A. A. Quality risk management in pharmaceutical development. Drug Dev. Ind. Pharm. 39, 947–960 (2013).
Lotllikar, M. Quality risk management (QRM): A review. J. Drug Deliv. Ther. https://doi.org/10.22270/jddt.v3i2.447 (2013).
International Conference on Harmonization (ICH); Tripartite Guidelines. ICH Q11 (2020). https://database.ich.org/sites/default/files/Q11%20Guideline.pdf. Accessed 15 Jan 2021.
Shabir, G. A. Validation of high-performance liquid chromatography methods for pharmaceutical analysis: Understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conference on Harmonization. J. Chromatogr. A 987, 57–66 (2003).
Jeong, L. N., Sajulga, R., Forte, S. G., Stoll, D. R. & Rutan, S. C. Simulation of elution profiles in liquid chromatography I: Gradient elution conditions, and with mismatched injection and mobile phase solvents. J. Chromatogr. A 1457, 41–49 (2016).
International Conference on Harmonization (ICH); Tripartite Guidelines. ICH Q2A (R2) (2006). https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf. Accessed 15 Jan 2021.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant Number: 2018M3A9F3081538).
Author information
Authors and Affiliations
Contributions
M.K.K. contributed conception, design of the study, and performed the experiments; S.C.P. conducted statistical analysis; G.P., E.C. and Y.J. performed the experiments and data; M.K.K. wrote the original draft of the manuscript; Y.P.J. administrated project and acquired funding. All authors contributed to manuscript revision and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, M.K., Park, S.C., Park, G. et al. Analytical quality by design methodology for botanical raw material analysis: a case study of flavonoids in Genkwa Flos. Sci Rep 11, 11936 (2021). https://doi.org/10.1038/s41598-021-91341-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91341-w
- Springer Nature Limited
This article is cited by
-
Analytical quality-by-design (AQbD) approach for comprehensive analysis of bioactive compounds from Citrus peel wastes by UPLC
Analytical and Bioanalytical Chemistry (2023)
-
An Application of Quality by Design and Analytical Greenness Assessment Approach for the Development of Erlotinib Stability Indicating Method
Chromatographia (2022)