
Abstract
IL-17A and IL-17F are both involved in the pathogenesis of neutrophilic inflammation observed in COPD and severe asthma. To explore this, mice deficient in both Il17a and Il17f and wild type (WT) mice were exposed to cigarette smoke or environmental air for 5 to 28 days and changes in inflammatory cells in bronchoalveolar lavage (BAL) fluid were determined. We also measured the mRNA expression of keratinocyte derived chemokine (Kc), macrophage inflammatory protein-2 (Mip2), granulocyte–macrophage colony stimulating factor (Gmcsf) and matrix metalloproteinase-9 (Mmp9 ) in lung tissue after 8 days, and lung morphometric changes after 24 weeks of exposure to cigarette smoke compared to air-exposed control animals. Macrophage counts in BAL fluid initially peaked at day 8 and again on day 28, while neutrophil counts peaked between day 8 and 12 in WT mice. Mice dual deficient with Il17a and 1l17f showed similar kinetics with macrophages and neutrophils, but cell numbers at day 8 and mRNA expression of Kc, Gmcsf and Mmp9 were significantly reduced. Furthermore, airspaces in WT mice became larger after cigarette smoke exposure for 24 weeks, whereas this was not seen dual Il17a and 1l17f deficient mice. Combined Il17a and Il17f deficiency resulted in significant attenuation of neutrophilic inflammatory response and protection against structural lung changes after long term cigarette smoke exposure compared with WT mice. Dual IL-17A/F signalling plays an important role in pro-inflammatory responses associated with histological changes induced by cigarette smoke exposure.
Similar content being viewed by others


Introduction
The IL-17 family of cytokines has six members, of which IL-17A and IL-17F are highly homologous and form a homodimer or a heterodimer and bind to a complex receptor of IL-17RA and IL-17RC, so that they share similar biological effects1,2,3 and appear to work in an overlapping way3,4. Several studies suggest that IL-17A and/or IL-17F are associated with airway inflammatory diseases, such as severe asthma and COPD5,6. Alveolar macrophages from COPD patients and healthy subjects express both IL-17A and IL-17F7 and cells positively immunostained for IL-17A are increased in the airway submucosa in patients with severe asthma and COPD8. Sputum IL-17A as well as CXCL8 levels are also reported to be higher in more severe COPD patients, and are inversely related to airway obstruction9. In patients with COPD, sputum levels of IL-17A are increased during exacerbations, and come down again after recovery, whereas sputum IL-17F levels show lesser increases10. Likewise, IL-17A and IL-17F are increased in asthma patients, with the levels higher in more severe disease and associated with increased sputum neutrophils11. These data suggest that IL-17A and IL-17F are both involved in the pathogenesis of neutrophilic inflammation observed in COPD patients and some with severe asthma.
The role of IL-17 in airway inflammations in COPD has been investigated using cigarette smoke exposure in mice. Treatment with a neutralizing antibody against IL-17A partially restored neutrophil recruitment to the lung induced by cigarette smoke exposure for 10 weeks12. Cigarette smoke exposure for 4 weeks resulted in the recruitment of neutrophils to the lung, and the enhanced expression of IL-17A, but not IL-17F. Neutrophil recruitment to the lung after cigarette smoke exposure was reduced in IL-17A gene-deficient mice13,14, although protection against the increase in alveolar size was not observed2,14. In another COPD mouse model induced by intratracheal elastase, there was an increase in lung Il17a expression and neutrophil recruitment to the lung, as well as alveolar airspace enlargement. Both neutrophil recruitment to the lung and airspace enlargement in murine model of elastase-induced emphysema were again only partially attenuated in Il17a deficient mice15. Infectious sinusitis is reported to occur exclusively in mice deficient both in Il17a and Il17f genes, while mice deficient in either Il17a or Il17f did not show the same phenotype4. In addition, another study reported that IL-17F expression was increased in Il17a -deficient mice, while IL-17A treatment suppresses Il17f expression, all suggesting that these related cytokines may overlap their functions and why double il17a/f knock-out mice are more informative16. Therefore, to explore in both acute neutrophilic inflammation and its chronic consequences, the present study investigated Il17a/f double-knockout mice exposed to cigarette smoke for over 28 days to study the inflammatory response and then for 24 weeks to study structural changes in peripheral lung .
Results
Time course of inflammatory response to inhaled cigarette smoke
BAL fluid macrophage counts in WT mice peaked at day 8 of cigarette exposure, and then declined to day 12 prior to another gradual increase up to day 28. Il17a and Il17f dual deficient mice showed similar kinetics although macrophage counts were significantly lower at day 8 and day 28 (p < 0.05, Fig. 1A). On the other hand, neutrophil counts peaked between day 8 and 12 in both groups of animals, followed by a decline towards the day 28. Neutrophil counts in Il17a and Il17f dual deficient mice were significantly lower both at day 8 and day 28 (p = 0.025 and p < 0.001 respectively) (Fig. 1B). Lymphocyte counts peaked at day 12 in the WT mice and the Il17a and Il17f dual -deficient mice showed a trend of increase in lymphocyte counts, but this was not statistically significant (Fig. 1C).

Time course of inflammatory cell counts in interleukin-17A/F (IL-17A/F) deficient mice compared to wild-type mice, following exposure to cigarette smoke over 28 days. Macrophages (A), neutrophils (B) and lymphocyte (C) in bronchoalveolar lavage (BAL) fluid are shown. Values are means ± SD of minimum of 6 animals per group (*p < 0.05, **p < 0.01, ***p < 0.001 compared with the group of day 0; †p < 0.05, ††p < 0.01, †††p < 0.001 compared between wild-type and Il-17A/F deficient mice on the same conditions of exposure).
Inflammatory response to cigrette smoke in IL-17-deficient mice at day 8
The mRNA levels of MIP-2 on day 8 were significantly increased in Il17a and Il17f dual deficient mice as well as in the WT mice, with no significant difference (Fig. 2A). Kc and Gmcsf expression was induced by cigarette smoke exposure in both genotypes, but the levels were significantly reduced in Il17a and Il17f dual deficient mice compared to control animals (Fig. 2B,C). The time course of the cellular response, as well as enhancement of mediator expression, after smoke exposure is consistent with previous reports17,18,19,20.

Inflammatory response induced by cigarette smoke exposure in Il17a and Il17f dual deficient mice compared to wild-type mice. Lung tissue mRNA expression of Mip2 (A), Kc (B), and Gmcsf (C). Closed bars represent IL-17A/F deficient mice; open bars represent wild-type mice. Values are means ± SD of 4 to 8 animals per group (*p < 0.05, compared with air control group in the same genotype; †p < 0.05 compared between Il17a and Il17f dual deficient and wild-type mice).
Airspace enlargement and neutrophilic infiltrate
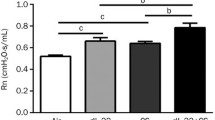
Histological analysis of the lungs revealed a neutrophilic infiltrate and airspace enlargement after cigarette exposure. In WT mice, the mean linear intercept (MLI) (Fig. 3A) and the alveolar space area (Fig. 3B) were significantly increased after 24 weeks of cigarette smoke-exposure, while alveoli per unit area decreased (shown in Figs. 3C, 4A,B). In contrast, there was no alveolar changes in Il17a and Il17f dual deficient animals (Figs. 3A–C, 4C,D).

Airspace enlargement after cigrette smoke exposure for 24 weeks were compared between IL-17A/F deficient mice and wild type mice by determining mean linear intercepts (MLI) (A), area of average airspace area (B), and the number of alveoli in an unit area (C) on histologic samples of haematoxylin–eosin (HE) staining (low magnification). Closed bars represent Il17a and Il17f dual deficient mice; open bars represent wild-type mice. Values are means ± SD (n = 4 to 8 per group, †p < 0.05 compared with wild-type animals on environmental exposure).

Representative histology with haematoxylin and eosin staining (low magnification) after exposure for 24 weeks, showing the comparison in airspace enlargement between wild-type mice on air (control, A), wild-type mice on cigarette smoke exposure (B), dual deficient mice on air (C), dual deficient mice on cigarette smoke exposure (D). Representative histology with haematoxylin and eosin staining (high magnification) as well as immuno-histochemistry (high magnification) against MMP-9 were shown in wild type mice on air (E,I) and those on cigarette smoke exposure (F,J) in comparison with dual deficient mice on air (G,K), and those after cigarette smoke exposure (H,L), respectively. It was shown that neutrophils were strongly positive in (J). Scale bar represents 100 micro-meter. Arrows indicate neutrophils.
MMP-9 positive cells
Neutrophils were recruited to the alveoli in wild-type mice exposed to cigarette smoke for 8 days (Fig. 4E,F), but not in Il17a and Il17f dual deficient animals (Fig. 4G,H). MMP-9 positive cells identified by immunostaining were increased in the alveolar walls of WT mice exposed to cigarette smoke (Fig. 4J vs I; K vs L), but this was significantly blunted (p < 0.05) in Il17a and Il17f dual deficient animals (Fig. 5A). This was confirmed by measuring Mmp9 mRNA levels, demonstrating that cigarette smoke exposure induced Mmp9 expression in both groups but that with lower expression in Il17a and Il17f dual deficient mice (Fig. 5B).

After 8 days of exposure, neutrophil recruitment was determined by the cell counts with MMP-9 immunostaining (A) and by the increased MMP-9 mRNA (B). Values are means ± SD of 4 to 8 animals per group (*p < 0.05 compared with the control animals on environmental exposure, and †p < 0.05 compared between Il17a and Il17f dual deficient mice and wild-type mice).
Discussion
The present study has demonstrated that Il17a and Il17f dual deficiency had a marked suppressive effect on macrophage and neutrophil recruitment to the lung after cigarette smoke exposure. Furthermore, Il17a and Il17f dual deficient mice were protected against enlargement of airspaces (emphysema) due to long-term cigarette exposure.
Previous studies showed that IL-17A in mice was a major mediator in short-term (2–10 weeks) cigarette smoke-induced airway inflammation, and in acute emphysema models using elastase12,13,14,15, whereas another study showed that neither IL-17A nor MMP-9 increased in expression after the elastase instillation in mice21. In addition, Il17a deficiency alone partially prevented the inflammatory responses12,13,15, and airspace enlargement was not protected (14% increase in mean chord length)2,14.
In contrast to these models of acute exposure12,13,14,15,21 and those focussing on the role of IL-17A2,14, the present model employed chronic (24 weeks) and low-level pro-inflammatory (to cigarette smoke) conditions, which can lead to emphysematous changes in wild-type mice. These lung inflammatory and structural changes were prevented by the dual Il17a and Il17f deficiency after 24 weeks, with significant attenuation of neutrophil recruitment by over 70% after 28 days of inhalation, compared to WT control animals. On the one hand, the disparities in the lung structural changes after long-term cigaretet smoke exposure, between mice deficient with Il17a in a literature2,14 and those dually deficient of Il17a and Il17f in the present study, are likely to be explained by overlapping functions between IL-17A and IL-17F3,4,22. On the other hand, IL-17A is associated with frequent acute exacerbations10,23,24, whereas IL-17F has been linked to lung cancer25, and asthma26,27 suggesting that IL-17A and F may have multiple, but indipendent roles in airway pathogenesis.
Regarding the overlapping function of IL-17A and F, previous studies showed that both IL-17A and IL-17F share the activation of p38 MAP kinase signalling, leading to the migration of both B cells and neutrophils, as well as production of chemokines, such as CCL-19 and CXCL-1328,29. Furthermore, IL-17 synergises with various inflammatory pathways mediated by NF-κB, cFOS, STAT-1 and Ras-Raf6. IL-17 enhances GM-CSF and CXCL-8 release in human cells in vitro30,31,32,33,34 and in mice in vivo35. Likewise, KC expression, as well as the subsequent morphometric changes, were upregulated and extended in our mouse model in the presence of IL-17A/F, together with increasing trend of other cytokines, such as TNF-α at day 817,18, whereas these responses are markedly abrogated in the absence of IL-17A and F, especially after chronic exposure.
The recruited neutrophils after cigarette smoke exposure were associated with increased MMP-9 levels in BAL fluid in Il10 deficient mice17, suggesting that cigarette smoke exposure induced pulmonary neutrophil recruitment via enhancing the expression of chemoattractants and that recruited neutrophils resulted in the elevation of Mmp9 expression in the lung tissue36, which in turn destroyed elastin fibres in the extracellular matrix of the alveoli, resulting in enlarged alveolar airspaces. According to the present study, Il17a and Il17f dual deficiency inhibited these processes by blocking neutrophil chemoattractants and thus neutrophil recruitment. This may explain the negative result of a clinical trial of antibody against IL-17RA in severe asthma, since it had no inclusion criteria for neutrophilic inflammation37. Both studies using mice deficient with il17ra14,38, and a clinical study showing the no efficacy of blocking IL-17A to COPD patients39 strongly indicate that inhibition of both IL-17A and IL-17F, or alternative inhibition of their receptor, IL-17RA, may be needed for effective suppression of neutrophilic airway inflammation.
Limitations
The present study has some limitations. First, since mouse models of COPD do not completely mimic the human disease, the pro-inflammatory effect of Il17a and Il17f may not occur in human COPD. However, several studies have demonstrated that the overlapping mechanisms between IL-17A and IL-17F are shared both in humans and mice2,4,22. Second, the lung morphometric measurements in the air-exposed mice with dual deletion of Il17a and Il17f appeared to range between those of air-exposed and cigarette smoke exposed wild type mice, suggesting some additional mechanism is involved. Third, protein levels and mRNA expression may dissociate, and the measuring protein level of these chemoattractants may indicate possible post-transcriptional effects of IL-17 deficiency on KC and MIP-2. However, a previously published report showed that cigarette smoke exposure in mice increases protein levels of KC and MIP-2 in BAL fluid in parallel to the upregulation of mRNA levels40. In addition, even low-level but chronic gene expression may play a crucial role in the development of chronic inflammatory disorders, in which the mediator levels in BAL fluid may be unmeasurable because of dilution, so that mRNA levels are more reliable measurements. Forth, the present study did not include any physiologic assessments, and is restricted to the histologic analysis of the lungs to show alveolar wall destruction, but previous studies have shown that mice exposed to cigarette smoke using the same model show exercise-induced impairment in oxygenation19. In addition, elevation in IL-17A levels and decrease in IL-10 levels are described in COPD patients9,41, and IL-17/IL-10 ratio was associated with disease severity, as measured by % predicted FEV1 values in COPD patients41, suggesting that IL-17 deficiency is likely to be associated with impairment in physiologic measures. Fifth, comparison between the effect of dual deficiency of il17a and il17f and single deficiency of il17a in a literature2,14 showed that the former was exclusively associated with restored airspace enlargement after cigarette smoke exposure for 24 weeks. However, this difference may possiblly be accounted for by il17f deficiency, which will be addressed by the comparison between mice dually deficient with il17a and il17f and those simply with il17f deficiency. Finally, other MMPs, such as MMP-2 and MMP-12, have also been shown to be involved in the pathogenesis of COPD42,43. However, the present study focused exclusively on the association between IL-17 and MMP-9, because they are separately described as major players in COPD development19,41, and their association is well described in cellular experiments36.
Concluding remarks
In conclusion, Il17a and Il17f dual deficient mice showed reduced neutrophilic inflammation in responses to short-term cigarette exposure and the complete protection from airspace enlargement induced by long-term exposure, which was not found in il17a deficient mice. This suggests that both IL-17A and F play an interactive role in the development of emphysema, so that these cytokines or their common receptors need a total block to provide the maximum benefit in COPD and severe neutrophilic asthma.
Methods
Animals
C57BL/6J (B6) male mice (wild type) were purchased from CLEA Japan Inc (Tokyo, Japan) and mice deficient in both Il17a and Il17f of B6 genetic background were kind gift from Professor Y Iwakura (Tokyo University of Science). Mice dual deficient in Il17a and Il17f were obtained from pairs of dual deficient mice. The parental pairs were genotyped and confirmed to be dual deficient, using the primer sets for genomic genes for IL-17A and F (shown in Table S1)4,44. Animals were acclimatized for a week before the study. Male mice were exclusively used in this study, because female mice have three-day long estrous cycle of MIP-2 levels and neutrophilic migration to vagina45,46, which were avoided in our laboratory17,18,19,20. All experimental animals were housed in a specific pathogen-free unit at the animal facility with sterile bedding, food, and water. The number of the mice was decided according to the previously published study from other researchers, who used 4 to 10 mice in a group13. All the methods performed were reviewed and approved by the Experimental Animal Ethics Committee at Kyorin University (No.32, 2009), in accordance with the guideline by the Ministry of Education, Culture, Sports, Science and Technology, Japan47, and furthermore, with ARRIVE guidelines48.
Exposure of mice to cigarette smoke
Two experiments were conducted, one investigating the time-course of inflammatory cellular response and the other measuring structural changes after cigarette smoke exposure in Il17a/f deficient mice compared to their wild-type counterparts, using the method previously described with some modifications17,18,19,20. In the former experiments, WT mice, aged 6–10 weeks old were exposed to cigarette smoke daily for 3, 5, 8, 12 and 28 days in a consecutive way by placing mice in a nose-only cigarette smoke inhalation experiment system for small animals (INH06- CIGR02A; MIPS, Osaka, Japan). Mice were exposed to compressed air with 6% cigarette smoke for 1 h/day, which were generated from commercially available, unfiltered Peace cigarettes (20 cigarettes/day, with 28 mg tar and 2.3 mg nicotine per cigarette; Japan Tobacco Inc., Tokyo, Japan).
In the long-term exposure studies, Il17a/f-deficient mice (n = 10 per group), aged 6–10 week old, and age-matched wild-type mice (n = 5–10 per group) were exposed to cigarette smoke daily or to continuous environmental air, starting at day 0, for 0, 5, 8, 12 and 28 days to make cellularity assessment at day 0,5,8, 12 and 28, respectively, for 8 days to measure mRNA levels, as well as histologic analyses, and for 24 week to make morphometric analyses on airspace enlargement (Figure S1).
Exposed and control mice were sacrificed 24 h after the last exposure, and bronchoalveolar lavage (BAL) was performed and lung tissue was sampled immediately upon sacrifice. BAL fluid was collected as described previously17,18,19,20. Briefly, mice were anesthetized and sacrificed with an overdose of pentobarbital (100 mg/kg i.p.; Schering-Plough, Kenilworth, NJ) 24 h after the last exposure to cigarette smoke. One mL aliquot of Hanks’ balanced salt solution (HBSS, Thermo Fisher Scientific, MA, USA) was instilled via the tracheal cannula and BAL fluid was recovered by gentle manual aspiration and repeated for 3 times. In each animal, approximately 95% of the BAL fluid was recovered. BAL fluid was centrifuged at 300×g for 5 min, and cell pellets were resuspended in 0.25 ml of HBSS. Total cells were counted in a Buerker chamber. Following staining with Diff Quick solution (International Reagents, Kobe, Japan), differential cell number (macrophages, neutrophils, lymphocytes) were counted on cytocentrifuged preparations (Auto smear CF-120; Sakura Finetek, Tokyo, Japan).
Histology and immunohistochemistry
Histological and immunohistochemical analyses were conducted, according to a previously published study17. Briefly, anti-mouse MMP-9 polyclonal antibody (Abcam, Cambridge, UK) was used for immunohistochemical analysis at a dilution of 1:1000. A heat-induced epitope retrieval technique was used for immunohistochemical analysis. Three micron thick sections were obtained from the paraffin blocks. Immunohistochemical staining was performed using an automated immunostainer (Ventana Inc, Tuscot, AZ). All sections used for immunohistochemistry were counterstained with hematoxylin. Tissue sections processed without the addition of primary antibody were used for the negative control. Four slides were evaluated from each mouse; 10 independent high-power fields were randomly photographed, and the number of positively stained cells was counted and evaluated by a blinded observer.
Quantitative reverse transcriptional polymerase chain reaction (qRT-PCR)
Quantitative reverse transcriptional polymerase chain reaction (qRT-PCR) were conducted, according to a previously published study17,18. Briefly, following recovery of BAL fluid samples, whole lungs were rapidly excised en bloc and frozen in liquid nitrogen. Gene expression levels in samples of lung tissue were determined using qRT-PCR. Total RNAwas extracted from whole lung samples using an RNeasy kit (Qiagen, Hamburg, Germany), cDNA was prepared using the Im Prom II reverse transcription system (Promega, Madison, WI, USA) for reverse transcription, and all procedures were conducted according to the manufacturer’s instructions. Murine Kc, Gmcsf, Mmp9, and Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) were measured using real-time PCR, which was performed with reaction mixtures containing SYBRPremix Ex Taq (Takara, Tokyo, Japan) on a 7500 real-time PCRsystem (Applied Biosystems, Foster City, CA). Target cDNAs were amplified using the primers listed in Table 1. The relative amount of each target gene transcript was determined by dividing the calculated value of each target gene by the calculated value of Gapdh, as an internal control for each sample.
Image analysis and lung morphometric analysis
Airspace enlargement was quantified using Image J software (National Institute of Health, https://imagej.nih.gov/ij/), according to the method previously published with some modification49,50,51. Briefly, a line grid was used to count the number of lines that ended on or intercepted alveolar tissue and automatically analyzed by the software, and mean linear intercepts (MLI) were calculated from the data49,50,51 Alveolar walls were also detected automatically, and the total area were calculated, in addition to the count of alveoli. If each air space becomes larger, MLI and air space area are both increased, while alveolar counts are lower. For the assess of airspace enlargement, three indices were employed: mean linear intercept (MLI), area of each alveolar airspace, and the number of alveoli per unit area; theoretically, if airspace is enlarged, MLI increases, the area of each alveolar airspace becomes larger, and the number of alveoli per unit area decreases.
Statistical analysis
Statistical analyses of the data were made, using the PRISM version 4, (GraphPad Software, San Diego, CA, USA) and Statistical Package for Social Science (SPSS) version 17.0 for Windows (SPSS Inc., Chicago, IL, USA). Values are expressed as mean ± standard error (SE). Kruskal–Wallis tests, in conjunction with post-hoc Dunn’s tests, were performed for comparisons between four groups, if the former test revealed statistically significant. Mann–Whitney U-tests were performed in order to make an estimation of the comparison between two groups. Two-sided P-values < 0.05 were considered to be statistically significant.
Statement of ethics
The experiments and protocols were approved by the Experimental Animal Ethics Committee at Kyorin University.
Abbreviations
- BAL:
-
Bronchoalveolar lavage
- COPD:
-
Chronic obstructive pulmonary disease
- JAK:
-
Janus activated kinase
- KC:
-
Keratinocyte-derived chemokine
- STAT:
-
Signal transduction activated transcription factor
References
Iwakura, Y., Ishigame, H., Saijo, S. & Nakae, S. Functional specialization of interleukin-17 family members. Immunity 34, 149–162 (2011).
Ritzmann, F. & Beisswenger, C. Preclinical studies and the function of IL-17 cytokines in COPD. Ann. Anat. 237, 151729 (2021).
Beringer, A., Noack, M. & Miossec, P. IL-17 in chronic inflammation: From discovery to targeting. Trends Mol. Med. 22, 230–241 (2016).
Ishigame, H. et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30, 108–119 (2009).
Li, X., Bechara, R., Zhao, J., McGeachy, M. J. & Gaffen, S. L. IL-17 receptor-based signaling and implications for disease. Nat Immunol. 20, 1594–1602 (2019).
Mc Geachy, M. J., Cua, D. J. & Gaffen, S. L. The IL-17 family of cytokines in health and disease. Immunity 50, 892–906 (2019).
Eustace, A. et al. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest 139, 1089–1100 (2011).
Doe, C. et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest 138, 1140–1147 (2010).
Maneechotesuwan, K., Kasetsinsombat, K., Wongkajornsilp, A. & Barnes, P. J. Decreased indoleamine 2,3-dioxygenase activity and IL-10/IL-17A ratio in patients with COPD. Thorax 68, 330–337 (2013).
Roos, A. B. et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 192, 428–437 (2015).
Sorbello, V. et al. Nasal IL-17F is related to bronchial IL-17F/neutrophilia and exacerbations in stable atopic severe asthma. Allergy 70, 236–240 (2015).
Shen, N., Wang, J., Zhao, M., Pei, F. & He, B. Anti-interleukin-17 antibodies attenuate airway inflammation in tobacco-smoke-exposed mice. Inhal. Toxicol. 23, 212–218 (2011).
Chang, Y. et al. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 306, L132–L143 (2014).
Voss, M. et al. Il-17A contributes to maintenance of pulmonary homeostasis in a murine model of cigarette smoke-induced emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L188–L195 (2015).
Kurimoto, E. et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir. Res. 14, 5 (2013).
von Vietinghoff, S. & Ley, K. IL-17A controls IL-17F production and maintains blood neutrophil counts in mice. J. Immunol. 183, 865–873 (2009).
Higaki, M. et al. Interleukin-10 modulates pulmonary neutrophilic inflammation induced by cigarette smoke exposure. Exp. Lung Res. 41, 525–534 (2015).
Mikura, S. et al. Erythromycin prevents the pulmonary inflammation induced by exposure to cigarette smoke. Transl. Res. 158, 30–37 (2011).
Nakamaru, Y. et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 23, 2809–2810 (2009).
Nakamura, M. et al. Clarithromycin ameliorates pulmonary inflammation induced by short term cigarette smoke exposure in mice. Pulm. Pharmacol. Ther. 35, 60–66 (2015).
Limjunyawong, N., Craig, J. M., Lagassé, H. A., Scott, A. L. & Mitzner, W. Experimental progressive emphysema in BALB/cJ mice as a model for chronic alveolar destruction in humans. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L662-676 (2015).
Cypowyj, S., Picard, C., Maródi, L., Casanova, J. L. & Puel, A. Immunity to infection in IL-17-deficient mice and humans. Eur. J. Immunol. 42, 2246–2254 (2012).
Wei, B. & Sheng, L. C. Changes in Th1/Th2-producing cytokines during acute exacerbation chronic obstructive pulmonary disease. J. Int. Med. Res. 46, 3890–3902 (2018).
Zou, Y. et al. Serum IL-1β and IL-17 levels in patients with COPD: Associations with clinical parameters. Int. J. Chron. Obstruct. Pulmon. Dis. 12, 1247–1254 (2017).
Li, C. et al. IL-17F expression correlates with clinicopathologic factors and biological markers in non-small cell lung cancer. Pathol. Res. Pract. 215, 152562 (2019).
De Luca, A. et al. The IL-17F/IL-17RC axis promotes respiratory allergy in the proximal airways. Cell Rep. 20, 1667–1680 (2017).
Chen, R. et al. IL-17F, rather than IL-17A, underlies airway inflammation in a steroid-insensitive toluene diisocyanate-induced asthma model. Eur. Respir. J. 53, 1801510 (2019).
Brusselle, G. G., Joos, G. F. & Bracke, K. R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378, 1015–1026 (2011).
Halwani, R. et al. IL-17 enhances chemotaxis of primary human B cells during asthma. PLoS ONE 9, e114604. https://doi.org/10.1371/journal.pone.0114604 (2014).
Dragon, S. et al. IL-17 enhances IL-1beta-mediated CXCL-8 release from human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L1023–L1029 (2007).
Mizunoe, S. et al. Synergism between interleukin (IL)-17 and Toll-like receptor 2 and 4 signals to induce IL-8 expression in cystic fibrosis airway epithelial cells. J. Pharmacol. Sci. 118, 512–520 (2012).
Numasaki, M., Tomioka, Y., Takahashi, H. & Sasaki, H. IL-17 and IL-17F modulate GM-CSF production by lung microvascular endothelial cells stimulated with IL-1beta and/or TNF-alpha. Immunol. Lett. 95, 175–184 (2004).
Laan, M. et al. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-alpha. Eur. Respir. J. 21, 387–393 (2003).
Honda, K. et al. IL-17A synergistically stimulates TNF-α-induced IL-8 production in human airway epithelial cells: A potential role in amplifying airway inflammation. Exp. Lung Res. 42, 205–216 (2016).
Hartupee, J. et al. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J. Immunol. 182, 1660–1666 (2009).
Prause, O., Bozinovski, S., Anderson, G. P. & Lindén, A. Increased matrix metalloproteinase-9 concentration and activity after stimulation with interleukin-17 in mouse airways. Thorax 59, 313–317 (2004).
Busse, W. W. et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 188, 1294–1302 (2013).
Chen, K. et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS ONE 6(5), e20333. https://doi.org/10.1371/journal.pone.0020333 (2011).
Eich, A. et al. A randomized, placebo-controlled phase 2 trial of CNTO 6785 in chronic obstructive pulmonary disease. COPD 14, 476–483 (2017).
Betsuyaku, T. et al. Bronchiolar chemokine expression is different after single versus repeated cigarette smoke exposure. Respir. Res. 9, 7. https://doi.org/10.1186/1465-9921-9-7 (2008).
Maneechotesuwan, K., Wongkajornsilp, A., Adcock, I. M. & Barnes, P. J. Simvastatin suppresses airway IL-17 and upregulates IL-10 in patients with stable COPD. Chest 148, 1164–1176 (2015).
Mahor, D. et al. Elevated serum matrix metalloprotease (MMP-2) as a candidate biomarker for stable COPD. BMC Pulm. Med. 20, 302 (2020).
Haq, I., Lowrey, G. E., Kalsheker, N. & Johnson, S. R. Matrix metalloproteinase-12 (MMP-12) SNP affects MMP activity, lung macrophage infiltration and protects against emphysema in COPD. Thorax 66, 970–976 (2011).
Nakae, S. et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17, 375–387 (2002).
Sasaki, S., Nagata, K. & Kobayashi, Y. Regulation of the estrous cycle by neutrophil infiltration in to the vagina. Biochem. Biophys. Res. Commun. 382, 35–40 (2009).
Sonoda, Y. et al. Physiologic regulation of postovulatory neutrophil migration into vagina in mice by a C-X-C chemokine. J. Immunol. 160, 6159–6165 (1998).
Ministry of Education, Culture, Sports, Science and Technology. Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions (Notice No. 71). https://www.mext.go.jp/b_menu/hakusho/nc/06060904.htm.
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. https://doi.org/10.1371/journal.pbio.3000410 (2020).
Chen, Z. H. et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 107, 18880–18885 (2010).
Dunnill, M. S. Quantitative methods in the study of pulmonary pathology. Thorax 17, 320–328 (1962).
Parameswaran, H., Majumdar, A., Ito, S., Alencar, A. M. & Suki, B. Quantitative characterization of airspace enlargement in emphysema. J. Appl. Physiol. 100, 186–193 (2006).
Funding
The present study was supported by Grant-in-Aid for Scientific Research (No. 26461514), Ministry of Education, Science, Sports and Culture, Japan.
Author information
Authors and Affiliations
Contributions
H.W., M.N., S.I., A.K., T.H., Y.I., F.K., H.K., S.K., K.I., P.J.B., H.T. substantially contributed to the conception or design of the work. H.W., M.N., S.I., A.K., H.K. contributed to the data acquisition, and analysis. H.W., S.I., A.K., T.H., Y.I., F.K., H.K., S.K., K.I., P.J.B., H.T. contributed to interpretation of data for the work. H.W., M.N., S.I., A.K., F.K., H.K., S.K., K.I., P.J.B. substantially contributed to drafting the work or revising it critically for important intellectual content. H.W., M.N., S.I., A.K. T.H., Y.I., F.K., H.K., S.K., K.I., P.J.B., H.T. gave final approval of the version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wada, H., Nakamura, M., Inoue, SI. et al. Dual interleukin-17A/F deficiency protects against acute and chronic response to cigarette smoke exposure in mice. Sci Rep 11, 11508 (2021). https://doi.org/10.1038/s41598-021-90853-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90853-9
- Springer Nature Limited