
Abstract
Various proteins play a decisive role in the pathology of different neurodegenerative diseases. Nonetheless, most of these proteins can only be detected during a neuropathological assessment, although some non-specific biomarkers are routinely tested for in the cerebrospinal fluid (CSF) as a part of the differential diagnosis of dementia. In antemortem CSF samples from 117 patients with different types of neuropathologically confirmed neurodegenerative disease with dementia, we assessed total-tau (t-tau), phosphorylated-tau (181P) (p-tau), amyloid-beta (1–42) (Aβ42), TAR DNA binding protein (TDP)-43, progranulin (PGRN), and neurofilament light (NfL) chain levels, and positivity of protein 14-3-3. We found t-tau levels and the t-tau/p-tau ratios were significantly higher in prion diseases compared to the other neurodegenerative diseases. Statistically significant differences in the t-tau/Aβ42 ratio predominantly corresponded to t-tau levels in prion diseases and Aβ42 levels in AD. TDP-43 levels were significantly lower in prion diseases. Additionally, the TDP-43/Aβ42 ratio was better able to distinguish Alzheimer’s disease from other neurodegenerative diseases compared to using Aβ42 alone. In frontotemporal lobar degeneration, PRGN levels were significantly higher in comparison to other neurodegenerative diseases. There is an increasing need for biomarkers suitable for diagnostic workups for neurodegenerative diseases. It appears that adding TDP-43 and PGRN to the testing panel for neurodegenerative diseases could improve the resolution of differential diagnoses.
Similar content being viewed by others


Introduction
Neurodegenerative diseases are characterized by an accumulation of specific proteins1,2. This accumulation leads to the formation of intracellular inclusions and/or extracellular deposits that usually consist of misfolded, proteolytically cleaved, or covalently modified forms of native proteins1,2,3,4. The formation of the misfolded proteins can be the result of a stochastic process or a gene mutation involved in the pathology of the disease. Genetic alterations can also increase brain levels of these proteins, which can then lead to an abnormal accumulation. Protein aggregates can be found in two different forms: (1) smaller, soluble, and oligomeric or (2) less soluble or insoluble, and fibrillar1,2.
Prion diseases are a rare group of fatal neurodegenerative disorders characterized by a progressive loss of neurons, spongiform changes in the neuropil of the deep cortical layers, and the cerebellar cortex or subcortical grey matter and deposition of the abnormal prion protein5,6,7. The most common prion disease is sporadic Creutzfeldt-Jakob disease (CJD)7. Common clinical features include rapidly progressive dementia, ataxia, spasticity, rigidity, and myoclonus7,8.
In Alzheimer disease (AD), the pathological changes are caused by neuritic plaques formed by accumulated amyloid-beta and intraneuronal neurofibrillary tangles; which are composed of hyperphosphorylated tau and result in neuronal cell loss and visible brain atrophy, predominantly in the hippocampus, entorhinal cortex, and the association areas of the neocortex9,10. Progressive impairment of episodic memory and other cognitive functions are the characteristic features of AD, and the diagnosis is supported by biomarkers, in particular, increased total tau (t-tau) and phosphorylated 181 P tau (p-tau) levels with low amyloid-beta 1–42 (Aβ42) levels in the cerebrospinal fluid (CSF), and positive amyloid PET9,11,12.
Dementia with Lewy bodies (DLB) is characterized by an accumulation of abnormal alpha-synuclein in Lewy bodies and Lewy neurites in the brainstem, limbic system, and cortical areas. Clinical features of DLB include fluctuating cognition with pronounced variations in attention and alertness, recurrent visual hallucinations, and spontaneous motor features characteristic of parkinsonism13,14. In patients with DLB, AD-related pathology often co-occurs15.
The behavioral variant of frontotemporal dementia (bvFTD), another progressive dementia within the spectrum of frontotemporal lobar degenerations (FTLD) involving behavioral, language, and motor manifestations, is related to the aggregation of two crucial pathologically misfolded proteins—tau and TDP-43. Clinically, bvFTD is characterized by early behavioral and cognitive manifestations caused by the degeneration of cortical areas of the frontal and temporal lobes, sometimes with the involvement of subcortical brain regions16,17,18,19.
TAR DNA binding protein (TDP)-43 is in the heterogeneous nuclear ribonucleoprotein family. It is mainly located in the nucleus, albeit a small amount can also be found in the cytosol since the protein is known to shuttle between the nucleus and the cytosol18,20,21. In pathological conditions, TDP-43 is mostly present in the cytosol and aggregates into hyperphosphorylated, ubiquitinated, and truncated forms consisting of C-terminal fragments of various lengths18,22,23,24. Proteolytic cleavage can be mediated by progranulin (PGRN), a 593 amino acid cysteine-rich glycoprotein encoded by the GRN gene. Mutations in this gene are detected in approximately 5–20% of familial bvFTD cases and 1–5% of sporadic bvFTD cases; they usually result in null mutations that cause haploinsufficiency. Mutations in the GRN gene can lead to a reduction in PGRN levels and cytosolic accumulation of TDP-43; it can also cause neurodegeneration18,25,26,27,28,29.
Neurofilaments are the main structural components of the axonal and dendritic cytoskeleton and are composed of light (NfL), medium, and heavy neurofilament chains30,31. Being the smallest of the three components, NfL can be easily released into the CSF and, in a variety of neurological disorders, is proportional to the degree of axonal damage32,33.
The aim of our study was to examine levels of selected protein CSF biomarkers in neuropathologically confirmed cases of prion diseases, AD, frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP), frontotemporal lobar degeneration with tau inclusions (FTLD-tau), and DLB and to determine which of the aforementioned CSF biomarkers, or their ratios, would be helpful in the differential diagnosis of dementia, in cases with overlapping clinical symptoms.
Total tau, p-tau, and Aβ42 are routinely used in the clinical diagnosis of AD (NIA-AA diagnostic criteria for AD11), and protein 14–3-3 is part of the current clinical diagnostic criteria for CJD (revised WHO criteria for CJD)34. In other dementia subtypes (bvFTD, DLB, and others), CSF analysis is not routinely done in clinical practice. When combined with clinical and neuropsychological profiling and imaging, CSF biomarkers may facilitate clinical differentiation of AD from CJD or bvFTD35,36,37,38. However, many published CSF studies in patients with dementia do not include exhaustive neuropathological verification.
The use of CSF biomarkers in clinical settings for the diagnosis of neurodegenerative dementia is of increasing interest. However, some studies have reported variability in sensitivity and specificity of biomarkers as well as variability in laboratory results, and few neuropathology correlations from larger studies are available. Moreover, the routine use of CSF is available only for AD and CJD. Our hypothesis was that—based on neuropathological data from a larger cohort—the combination of available but not yet validated biomarkers could be helpful in discriminating between different neurodegenerations mimicking CJD and in the differential diagnosis of dementia in cases with overlapping clinical symptoms.
Material and methods
Materials
Our study was designed as a retrospective study; the unifying feature was the single center of neuropathology, the National Reference Laboratory for Diagnosis of Human Prion Disease. In our center, we performed standardized brain autopsies and proceeded to a unified analysis of CSF samples from patients followed in different neurology departments over the entire Czech Republic. Patients had a clinical diagnosis of possible or probable CJD as well as differential diagnoses that included other dementia disorders (Table S1). The reliability of the clinical diagnosis and both the quality and comprehensiveness of the reported data, however, largely depended on the referring site (institutional bias). In some cases, the clinical diagnosis was largely appropriate if compared to neuropathology results, while in other cases, both the accuracy and quality of reported data were suspect.
To obtain a homogenous cohort, our study included only cases with a precisely confirmed neuropathological disorder; cases with a predominant comorbid neurodegenerative disease were excluded. Clinical data associated with the biochemical analysis were eligible for retrospective analysis once the autopsy had been completed.
Our cohort included 117 CSF samples from neuropathologically confirmed cases of prion diseases (i.e., sporadic and familial CJD and Gerstmann–Sträussler–Scheinker syndrome (GSS)) (n = 37), and non-prion diseases: AD (n = 39), FTLD-TDP (n = 15), FTLD-tau (predominantly progressive supranuclear palsy (PSP) cases) (n = 15), and DLB (n = 11).
The patients or their relatives agreed with the storage of CSF samples and brain tissue for research purposes and signed informed consent. The study protocol was approved by the Central Ethics Committee of Thomayer University Hospital and the Institute for Clinical and Experimental Medicine (Prague, Czech Republic). All research was performed in accordance with relevant guidelines and regulations. The privacy of patients was fully respected during statistical analysis.
CSF analysis
All patients underwent a single lumbar puncture and collection. All samples were analyzed in the National Reference Laboratory for Diagnosis of Human Prion Disease. CSF samples were centrifuged at 5000 RPM for 5 min and stored in polypropylene tubes at − 80 °C in aliquots to avoid thawing and refreezing. The storage interval was variable; however, at − 80 °C, storage time does not impact sample quality39,40. CSF levels of t-tau, p-tau, and Aβ42 were measured during routine diagnostic testing using commercially available enzyme-linked immunoassay (ELISA) kits (INNOTEST hTAU Ag, cat. #80323/81572, INNOTEST PHOSPHO-TAU(181P), cat. #80317/81574, INNOTEST β-AMYLOID(1–42), cat. #80324/81576, all Innogenetics/FUJIREBIO) according to the manufacturer’s protocol. Our laboratory has extensive experience determining CSF biomarkers and successfully participates in the Alzheimer’s Association’s external quality control program.
The presence of 14-3-3 beta protein was determined using a standardized western blot protocol (adapted from41) followed by all laboratories for the diagnosis of CJD and followed EURO-CJD standards, with stringent control quality. We performed a standardized qualitative western blot analysis for 14-3-3 in doublets. A weak positive test was interpreted to mean that one sample load was positive, the other one negative (the positive control was always positive).
TDP-43, PGRN, and NfL were evaluated using commercially available ELISA kits (Human TAR DNA-binding protein 43, TARDBP/TDP-43 ELISA Kit, cat. #MBS705899, MyBioSource; Progranulin (human) ELISA Kit, cat. #AG-45A-0018YEK-KI01, AdipoGen; NF-light ELISA, cat. #10-7001 CE, UmanDiagnostics) according to the manufacturer’s protocol. For the TDP-43, PGRN, and NfL assay, internal validations were performed. The intra-assay coefficients of variation (CV) were 8.1%, 5.2%, and 4.8%, respectively, and the inter-assay CVs were 17.1%, 14.9%, and 11.2%, respectively.
Neuropathology
All patients underwent an autopsy with a subsequent brain neuropathological investigation using a standardized protocol, i.e., after 3–4 weeks of fixation in 10% neutral buffered formalin. Paraffin-embedded tissue sections (4 µm) were taken from different regions and diagnosed using standardized recommendations42. A definite diagnosis of prion disease was confirmed through neuropathological examination and western blot detection of the proteinase K resistant form of the prion protein. A diagnosis of AD was based on neurofibrillary tangles and amyloid-beta deposits in a specific region of the brain (using the “ABC” scoring system)43. Characteristic neuropathological findings were seen in FTLD-tau, namely PSP cases, such as oligodendroglial and astroglial cytoplasmic inclusions and neuropil threads, and were scored according to Williams and Kovacs44,45. Specific TDP-43 neuronal inclusions and dystrophic neurites were present in specific brain regions in cases with FTLD-TDP, which fulfilled the diagnostic criteria for Harmonized classification46. A diagnosis of DLB was performed following the criteria of McKeith and Braak using antibodies against alpha-synuclein47.
Statistics
The basic statistical characteristics, i.e., median values with interquartile range, were calculated for quantitative variables, while frequencies were used to describe discrete variables. Statistical analyses were performed using GraphPad Prism 5 (La Jolla, CA, USA). The statistical analysis used the Kruskal–Wallis test for comparison of all groups together, and Dunn’s multiple comparisons tests, and consequently the Mann–Whitney nonparametric test for comparisons between two groups (for non-normally distributed data), all at a significance level of p < 0.05. The Spearman’s rank-order correlation was used to state correlations between the 14-3-3 protein and the concentrations of analytes in the CSF and between all combinations of analytes concentrations from all five neurodegenerative diseases (i.e., all five groups together). From the ROC curve analysis, 95% confidence intervals (CI) were obtained, and cut-off values were determined by maximizing the Youden index.
Results
All results are summarized in tables and graphs. Table 1 is a summary of demographic data and biomarker concentrations.
Isolated biomarker levels in neurodegenerative disorders
TDP-43 levels were significantly lower in prion diseases compared to the other groups (Fig. 1a). On the other hand, NfL (Fig. 1b) and t-tau (Fig. 1d) levels were significantly higher in prion diseases compared to the other groups. In addition, t-tau levels were significantly lower in FTLD-tau compared to AD and FTLD-TDP (Fig. 1d); p-tau differed only slightly between AD and FTLD-tau (Fig. 1e). Aβ42 levels were significantly lower in AD (Fig. 1f). Various significances of PGRN levels were found: PGRN was lower in AD and DLB compared to FTLD-TDP and FTLD-tau, and there was only a weak difference between prion disease and FTLD-tau (Fig. 1c).

Scatter dot plots represent values of (a) TDP-43, (b) NfL, (c) PGRN, (d) t-tau, (e) p-tau, and (f) Aβ42 individually for prion diseases, AD, FTLD-TDP, FTLD-tau, and DLB. Bars represent medians with the interquartile range. Statistically significant p-values represent the exact two-tailed p-values of the Mann–Whitney test (p < 0.05). TDP-43, TAR DNA binding protein-43; PGRN, progranulin; NfL, neurofilament light chain; t-tau, total-tau; p-tau, phosphorylated-tau (181P); Aβ42, amyloid-beta (1–42); AD, Alzheimer disease; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; FTLD-tau, frontotemporal lobar degeneration with tau inclusions; DLB, dementia with Lewy bodies.
Combination of biomarkers in neurodegenerative disorders
We then calculated paired ratios for every possible combination (Table 2). The TDP-43/Aβ42 ratio (Fig. 2a) was better at differentiating AD from the other groups (Fig. 3a) and prion disease from FTLD-TDP (Fig. 3b) than Aβ42 alone (Fig. 1f). The TDP-43/PGRN ratio was significantly higher in AD compared to FTLD-tau (p = 0.0024). The TDP-43/NfL ratio was significantly higher in AD than in FTLD-TDP and FTLD-tau (p = 0.0059 and 0.0195, respectively).

Scatter dot plots represent values of (a) TDP-43/Aβ42 ratio, (b) PGRN/p-tau ratio, (c) NfL/p-tau ratio, (d) t-tau/p-tau ratio, (e) t-tau/Aβ42 ratio, and (f) p-tau/Aβ42 ratio individually for prion diseases, AD, FTLD-TDP, FTLD-tau, and DLB. Bars represent medians with the interquartile range. Statistically significant p-values represent the exact two-tailed p-values of the Mann–Whitney test (p < 0.05). TDP-43, TAR DNA binding protein-43; NfL– neurofilament light chain; t-tau, total-tau; p-tau, phosphorylated-tau (181P); Aβ42, amyloid-beta (1–42); AD, Alzheimer disease; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; FTLD-tau, frontotemporal lobar degeneration with tau inclusions; DLB, dementia with Lewy bodies.

ROC diagrams of the TDP-43/Aβ42 ratio (solid line) and Aβ42 values (dotted line) in (a) AD vs. non-AD disease cases: for the TDP-43/Aβ42 ratio AUC 0.8695, CI 0.806 to 0.933, cut-off 6.737, p < 0.0001, for Aβ42 AUC 0.7840, CI 0.692 to 0.876, cut-off 425.5 pg/ml, p < 0.0001, (b) prion diseases vs. FTLD-TDP: for the TDP-43/Aβ42 ratio AUC 0.8072, CI 0.671 to 0.943, cut-off 9.537, p = 0.0006, for Aβ42 AUC 0.6036, CI 0.430 to 0.777, cut-off 867.0 pg/ml, p non-significant. TDP-43, TAR DNA binding protein-43; Aβ42, amyloid-beta (1–42); AD, Alzheimer disease; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; AUC, area under the curve; CI, 95% confidence interval.
The PGRN/Aβ42 ratio was slightly better at discriminating prion disease from FTLD-TDP (p = 0.0263) and AD from DLB (p = 0.0007) than Aβ42 alone (Fig. 1f). The PGRN/p-tau ratio (Fig. 2b) was significantly lower in AD than in FTLD-TDP and FTLD-tau (p = 0.0017 and 0.0007, respectively) and significantly higher in FTLD-tau than in prion disease and DLB (p = 0.0237 and p = 0.0170, respectively). The PGRN/p-tau ratio was better at differentiating AD from FTLD-tau (Fig. 4a) and FTLD-TDP and FTLD-tau from prion disease and DLB (Fig. 4b) than p-tau alone (Fig. 1e). The PGRN/t-tau ratio was significantly lower in prion diseases compared to the other groups (p < 0.0001) and significantly higher in FTLD-tau compared to AD (p = 0.0005) and DLB (p = 0.0224) and could differentiate FTLD-tau from AD and DLB better than t-tau (Fig. 1d). The PGRN/NfL ratio was significantly lower in prion diseases than in the FTLD-TDP group (p = 0.0026).

ROC diagrams of the PGRN/p-tau ratio (solid line) and p-tau values (dotted line) in (a) AD vs. FTLD-tau: for the PGRN/p-tau ratio AUC 0.8000, CI 0.668 to 0.933, cut-off 92.1, p = 0.0007, for p-tau AUC 0.6786, CI 0.517 to 0.841, cut-off 36.0 pg/ml, p = 0.0436, (b) FTLD-tau vs. DLB: for the PGRN/p-tau ratio AUC 0.7818, CI 0.606 to 0.958, cut-off 137.6, p = 0.0158, for p-tau AUC 0.5788, CI 0.352 to 0.805, cut-off 50.5 pg/ml, p non-significant. PGRN, progranulin; p-tau, phosphorylated-tau (181P); AD, Alzheimer disease; FTLD-tau, frontotemporal lobar degeneration with tau inclusions; DLB, dementia with Lewy bodies; AUC, area under the curve; CI, 95% confidence interval.
The NfL/Aβ42 ratio was significantly higher in FTLD-TDP compared to DLB (p = 0.0401). For the NfL/p-tau ratio, there was a slightly significant difference between AD and FTLD-TDP/FTLD-tau (Fig. 2c). The NfL/t-tau ratio was significantly higher in FTLD-tau compared to DLB (p = 0.0486).
The t-tau/p-tau ratio was significantly higher in prion diseases compared to the other groups and was significantly higher in FTLD-TDP compared to FTLD-tau and DLB (Fig. 2d). The t-tau/Aβ42 ratio was significantly higher in prion diseases compared to the other groups, significantly higher in AD compared to FTLD-tau and DLB, and in FTLD-TDP compared to FTLD-tau (Fig. 2e). Finally, the p-tau/Aβ42 ratio was significantly higher in AD compared to the other groups (Fig. 2f).
Correlation of biomarkers with the 14–3-3 protein
We also correlated the concentrations of all biomarkers with each other and with the 14-3-3 protein. We found a negative correlation between TDP-43 levels and NfL and t-tau levels and a positive correlation between NfL and t-tau levels (Table 3). There was also a positive correlation between 14-3-3 positivity and NfL and t-tau levels but a negative correlation between 14-3-3 positivity and TDP-43 levels (Table 3).
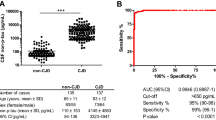
Since 14–3-3 positivity is an important biomarker for CJD, we compared 14–3-3 positivity and t-tau levels in prion diseases and non-prion diseases using ROC curves (Fig. 5). The AUC values were 0.844 and 0.926 (both p < 0.0001). For 14-3-3 positivity in prion diseases, the sensitivity was 75.7%, and the specificity was 86.3%. For t-tau, the cut-off was assessed to be 1200 pg/ml; both values were higher with a sensitivity of 89.2% and a specificity of 93.8%. When we applied both variables together, the AUC was 0.922 (p < 0.0001), which gave the highest sensitivity (94.6%) but the lowest specificity (82.5%). 14-3-3 positivity did not correlate with the duration of prion diseases (p = 0.5260).

ROC diagrams of protein 14–3-3 positivity, CI 0.753 to 0.934 (solid line), t-tau values higher than 1200 pg/ml, CI 0.868 to 0.984 (dashed line), and the combination of protein 14–3-3 positivity and t-tau values higher than 1200 pg/ml, CI 0.861 to 0.983 (dotted line) in prion vs. non- prion disease cases. t-tau, total tau; AUC, area under the curve; CI, 95% confidence interval.
Age and gender
We correlated biomarker levels with age and gender to see if there was a trend in biomarker levels despite the ongoing neurodegeneration. In our cohort, the correlation of biomarkers with age and gender did not show any significant differences.
Discussion
The differential diagnosis of rapidly progressive dementia is a challenging issue since the rapidity of disease evolution is not the only criterion for a prion origin. Other biomarkers for probable CJD (updated WHO criteria34) may be absent (e.g., caudate hyperintensities on MRI or periodic patterns on EEG), only present in advanced stages of the disease, or simply unavailable (RT-QuIC); in these situations, CSF analysis can help differentiate CJD from other neurodegenerative disorders (in particular comorbid neurodegeneration).
We focused on CSF biomarkers, and the strength of our study was the availability of neuropathology for all patients. The main findings of our study are:
-
First, t-tau levels and the t-tau/p-tau ratio are significantly higher in prion diseases compared to the other neurodegenerative diseases.
-
Second, differences in the t-tau/Aβ42 ratio are statistically significant and correspond predominantly to t-tau levels in prion disease and Aβ42 levels in AD.
-
Third, TDP-43 levels are significantly lower in prion diseases.
-
Fourth, the TDP-43/Aβ42 ratio is better at distinguishing AD from other neurodegenerative diseases than Aβ42 alone.
Both t-tau levels and the t-tau/p-tau ratio were significantly higher in prion diseases compared to the other groups. This corresponds with previous studies38,48, where they assessed a low p-tau/t-tau ratio to be an appropriate marker for sCJD. This finding is related to the fact that t-tau reflects the massive neuronal cell loss associated with prion diseases; p-tau (as a marker of hyperphosphorylated tau associated with neurofibrillary pathology) is thus normal in prion diseases but increased in AD and to a lesser extent in other tauopathies49,50,51,52. In our study, we did not find any significant differences in p-tau levels between diseases when all groups were compared together. Nonetheless, medians of all groups were higher than the median of healthy controls estimated in our lab53. In a study by Riemenschneider et al.38, they found significantly higher CSF p-tau levels in AD compared to CJD (p < 0.001) and FTD (p = 0.001). We found only slightly higher p-tau levels in AD in comparison to FTLD-tau (p < 0.0445). This might have been caused by relatively small groups in both studies and, in particular, by the lack of neuropathological confirmation for most of their cases.
The observed increase in the t-tau/p-tau ratio in FTLD-TDP in comparison with FTLD-tau and DLB is in line with previous observations54,55,56. However, in our cohort, the weak significance would appear to suggest that it has limited utility for the differential diagnosis of the FTLD subgroups in clinical practice, as previously described19. Additionally, only a weak significance was found in p-tau levels between AD and FTLD-tau. In contrast to the study by Irwin et al., our results indicate that p-tau alone is not an appropriate biomarker for the differential diagnosis of the FTLD subgroups57.
Unsurprisingly, reduced Aβ42 levels and reduced Aβ42/p-tau ratios were seen in AD compared to all other groups, which agrees with previous works focused on CSF biomarkers in AD patients52,58. Similar to observations by Bian et al., we found a significantly lower t-tau/Aβ42 ratio in FTLD-tau samples in comparison with AD and FTLD-TDP59. In our study, the differences in the t-tau/Aβ42 ratio were statistically significant among all groups, corresponding primarily to t-tau levels in prion diseases and Aβ42 levels in AD. In addition, a significantly lower t-tau/Aβ42 ratio was found in FTLD-tau in comparison with AD and the FTLD-TDP group. Our results suggest that the t-tau/Aβ42 ratio might serve as a useful tool for distinguishing various neurodegenerative diseases.
Higher NfL levels reflect ongoing rapidly progressive neurodegeneration in brain tissue and strongly correlate with white and grey matter atrophy10,60,61,62. This might be the reason we found, in line with recent observations, the highest NfL levels in prion diseases63. In our study, the median NfL level was 2.7-fold higher in FTLD-TDP than in AD and 1.5-fold higher in FTLD-tau than in AD, which also confirms previously published reports10,19,64. However, none of these relationships were statistically significant due to four AD cases with extremely elevated CSF NfL levels. In our study, NfL levels were not significantly higher in FTLD-TDP compared to FTLD-tau, which was in contrast with another study62. Additionally, Holmberg et al. found extremely elevated levels in PSP patients60. We included PSP patients in the FTLD-tau group. However, even when PSP samples were evaluated separately, NfL levels were not dramatically increased (except in patients with prion diseases, see above).
We detected significantly lower TDP-43 levels in prion disease samples in comparison with other disease groups. To our knowledge, there are no similar studies measuring TDP-43 in prion diseases using ELISA, and only one study concerning TDP-43 in prion diseases. The study reports a lack of TDP-43 aggregates in histopathological samples from human brains with prion disease65. In AD, FTLD-TDP, FTLD-tau, and DLB, the concentration of TDP-43 overlapped among all four groups in concordance with the TDP-43 pathology found in AD66,67 and DLB67,68. Surprisingly, there were no differences in TDP-43 levels between FTLD-TDP and FTLD-tau69,70.
Additionally, we found that the TDP-43/Aβ42 ratio was better able to distinguish AD from other neurodegenerative diseases than Aβ42 alone.
Our next findings were that PGRN levels were higher in FTLD-tau compared to prion diseases, AD, and DLB, and PGRN levels were higher in FTLD-TDP compared to AD and DLB. Lower PGRN levels in prion disease samples could be attributed to fulminant neuronal cell loss during the pathology of prion disease. Previously, PGRN levels were associated predominantly with frontal dysfunction in bvFTD71. This finding correlates with our results, i.e., higher PGRN levels were found in FTLD in general compared with other neurodegenerative diseases. PGRN alone or in a ratio with t-tau, p-tau, or Aβ42 could be a useful biomarker for resolving different neurodegenerative disorders.
Detection of 14-3-3 in the CSF is still a diagnostic criterion of probable CJD72. Collins et al.73 found that there was no association between the time of sampling and 14-3-3 protein positivity. This corresponds with our results since we found no correlation between the disease duration and 14-3-3 protein positivity. Nonetheless, this biomarker is not only present in the CSF of CJD patients since it shows rapid ongoing neuronal destruction in a variety of progressive neurological disorders74. Since t-tau levels are dramatically increased in patients with prion diseases51, we compared the ROC curves of 14-3-3, t-tau and their combinations in prion vs. non-prion diseases. Our results indicate that t-tau levels or t-tau levels combined with 14-3-3 positivity work better for detecting ongoing prion disease than 14-3-3 positivity alone, which we previously described75. We confirmed these results by comparing a large number of CSF samples with autopsy-confirmed neurodegeneration (examined in our department over the last 20 years (data not yet published)). For 14-3-3 positivity, the sensitivity was 63.1%, and the specificity was 81.1%. For elevated t-tau levels (> 1,200 pg/ml), both values were higher, with a sensitivity of 87.5% and a specificity of 91.5%.
Unlike the study by Bahl et al.48, where they found 95% sensitivity at 75% specificity for 14-3-3 protein positivity, our results show lower sensitivity and higher specificity. On the other hand, they stated that the combination of elevated t-tau levels with 14-3-3 protein positivity could be more helpful in the differential diagnosis of sCJD, having specificity and sensitivity similar to our results. Similar findings were published by Sanchez-Juan et al.76. In another study, Hamlin et al.77 found that t-tau levels could better predict sCJD than 14-3-3 protein positivity. They also found that the combination of t-tau high levels with the 14-3-3 protein positivity is not superior to the single use of t-tau, which was confirmed by our study. It seems, total tau can be a helpful tool in the differential diagnosis of sCJD and thus should be measured in addition to 14-3-3, especially when RT-QuIC is unavailable due to technical reasons and/or cost.
Our study was designed as a retrospective one, and we examined autopsy and CSF samples from patients referred as possible or probable CJD and other neurodegenerative dementias in the differential diagnosis (Supplementary Table S1). One could argue that our CSF findings would have enabled us to shift the initial clinical diagnosis of sporadic CJD in some patients to another neurodegeneration, but in fact, this shift has not occurred. Retrospectively, it seems obvious that some patients should not have been considered as sporadic CJD any longer, but we cannot modify the clinicians’ decision-making a posteriori. This standpoint, however, was one of the motivations we had to realize this study to learn from clinical-neuropathological correlations and to improve our interpretation of biomarker values in neurodegenerative diseases. This approach shows both the strengths and weaknesses of our study. Our data were confirmed by a definite neuropathological diagnosis contrary to many previously published studies; however, from a clinical point of view, protein biomarker levels do not track the real clinical evolution of patients.
Biomarkers have become very useful in the diagnostic workup for neurodegenerative diseases, and their role will certainly increase in the near future due to increasing evidence of comorbid neuropathologies occurring in the same patient, which would certainly have an impact on the clinical presentations.
Conclusion
Our results suggest that adding TDP-43 or PGRN to the testing panel of CSF biomarkers could enhance the differential diagnosis in neurodegenerative dementias and that total tau in association with protein 14-3-3 could be a useful biomarker for sCJD when RT-QuIC is unavailable. However, further investigation on a broader spectrum of verified neuropathologies is needed before new biomarkers can enter routine clinical practice.
References
Golde, T. E. & Miller, V. M. Proteinopathy-induced neuronal senescence: a hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res. Ther. 1, 5. https://doi.org/10.1186/alzrt5 (2009).
Golde, T. E., Borchelt, D. R., Giasson, B. I. & Lewis, J. Thinking laterally about neurodegenerative proteinopathies. J. Clin. Invest. 123, 1847–1855. https://doi.org/10.1172/JCI66029 (2013).
Forman, M. S., Trojanowski, J. Q. & Lee, V.M.-Y. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 10, 1055–1106. https://doi.org/10.1038/nm1113 (2004).
Ross, C. A. & Poirier, M. A. Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl), S10–S17. https://doi.org/10.1038/nm1066 (2004).
Budka, H. et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol. 5, 459–466. https://doi.org/10.1111/j.1750-3639.1995.tb00625.x (1995).
Kovacs, G. G. & Budka, H. Molecular pathology of human prion diseases. Int. J. Mol. Sci. 10, 976–999. https://doi.org/10.3390/ijms10030976 (2009).
Kovacs, G. G. et al. Tau pathology in Creutzfeldt-Jakob disease revisited. Brain Pathol. 27, 332–344. https://doi.org/10.1111/bpa.12411 (2016).
Mackenzie, G. & Will, R. Creutzfeldt-Jakob disease: recent developments. F1000Res 6, 2053. https://doi.org/10.12688/f1000research.12681.1 (2017).
Clark, C. M. et al. Cerebrospinal fluid tau and β-amyloid how well do these biomarkers reflect autopsy-confirmed dementia diagnoses?. Arch. Neurol. 60, 1696–1702. https://doi.org/10.1001/archneur.60.12.1696 (2003).
Scherling, C. S. et al. CSF neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann. Neurol. 75, 116–126. https://doi.org/10.1002/ana.24052 (2014).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. https://doi.org/10.1016/j.jalz.2011.03.005 (2011).
McKhann, G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. https://doi.org/10.1212/wnl.34.7.939 (1984).
Donaghy, P. C. & McKeith, I. G. The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis. Alzheimers Res. Ther. 6, 46. https://doi.org/10.1186/alzrt274 (2014).
Garcia-Esparcia, P. et al. Dementia with Lewy bodies: molecular pathology in the frontal cortex in typical and rapidly progressive forms. Front. Neurol. 8, 89. https://doi.org/10.3389/fneur.2017.00089 (2017).
Hall, S. et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch. Neurol. 69, 1445–1452. https://doi.org/10.1001/archneurol.2012.1654 (2012).
Snowden, J. S., Neary, D. & Mann, D. M. A. Frontotemporal dementia. Br. J. Psychiatry. 180, 140–143. https://doi.org/10.1192/bjp.180.2.140 (2002).
Baker, M. et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. https://doi.org/10.1038/nature05016 (2006).
Goedert, M., Ghetti, B. & Spillantini, M. G. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006254. https://doi.org/10.1101/cshperspect.a006254 (2012).
Goossens, J. et al. Diagnostic value of cerebrospinal fluid tau, neurofilament, and progranulin in definite frontotemporal lobar degeneration. Alzheimers Res. Ther. 10, 31. https://doi.org/10.1186/s13195-018-0364-0 (2018).
King, O. D., Gitler, A. D. & Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80. https://doi.org/10.1016/j.brainres.2012.01.016 (2012).
Davis, S. A. et al. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 678, 8–15. https://doi.org/10.1016/j.neulet.2018.04.053 (2018).
Zhang, Y.-J. et al. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 27, 10530–10534. https://doi.org/10.1523/JNEUROSCI.3421-07.2007 (2007).
Davidson, Y. et al. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 113, 521–533. https://doi.org/10.1007/s00401-006-0189-y (2007).
Igaz, L. M. et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem. 284, 8516–8524. https://doi.org/10.1074/jbc.M809462200 (2009).
Van Damme, P. et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 181, 37–41. https://doi.org/10.1083/jcb.200712039 (2008).
Ahmed, Z. et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 177, 311–324. https://doi.org/10.2353/ajpath.2010.090915 (2010).
Rademakers, R., Neumann, M. & Mackenzie, I. R. A. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434. https://doi.org/10.1038/nrneurol.2012.117 (2012).
Meeter, L. H. H. et al. Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement. Geriatr. Cogn. Disord. Extra. 6, 330–340. https://doi.org/10.1159/000447738 (2016).
Chang, M. C. et al. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 214, 2611–2628. https://doi.org/10.1084/jem.20160999 (2017).
Bacioglu, M. et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 91, 56–66. https://doi.org/10.1016/j.neuron.2016.05.018 (2016).
Olsson, B. et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol. 76, 318–325. https://doi.org/10.1001/jamaneurol.2018.3746 (2019).
Fialová, L., Bartoš, A. & Švarcová, J. Neurofilament light chains in serum and cerebrospinal fluid and status of blood-CSF barrier in the selected neurological diseases. Cesk. Slov. Neurol. N. 81, 185–192. https://doi.org/10.14735/amcsnn2018185 (2018).
Gaetani, L. et al. Neurofilament light chain as a biomarker in neurological disorders. J Neurol. Neurosurg. Psychiatry. 90, 870–881. https://doi.org/10.1136/jnnp-2018-320106 (2019).
CDC. CDC’s Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD). (2018). Available at: https://www.cdc.gov/prions/cjd/diagnostic-criteria.html. Accessed 29 Oct 2020.
Hu, W. T. et al. Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology 75, 2079–2086. https://doi.org/10.1212/WNL.0b013e318200d78d (2010).
Hales, C. M. & Hu, W. T. From frontotemporal lobar degeneration pathology to frontotemporal lobar degeneration biomarkers. Int. Rev. Psychiatry. 25, 210–220. https://doi.org/10.3109/09540261.2013.776522 (2013).
Kapaki, E., Kilidireas, K., Paraskevas, G. P., Michalopoulou, M. & Patsouris, E. Highly increased CSF tau protein and decreased beta-amyloid (1–42) in sporadic CJD: a discrimination from Alzheimer’s disease?. Neurol. Neurosurg. Psychiatry. 71, 401–403. https://doi.org/10.1136/jnnp.71.3.401 (2001).
Riemenschneider, M. et al. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol. Psychiatry 8, 343–347. https://doi.org/10.1038/sj.mp.4001220 (2003).
Teunissen, C. E. et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 73, 1914–1922. https://doi.org/10.1212/WNL.0b013e3181c47cc2 (2009).
Willemse, E. A., Koel-Simmelink, M. J., Durieux-Lu, S., van der Flier, W. M. & Teunissen, C. E. Standard biobanking conditions prevent evaporation of body fluid samples. Clin. Chim. Acta. 442, 141–145. https://doi.org/10.1016/j.cca.2015.01.036 (2015).
Green, A. J. et al. Use of 14-3-3 and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt–Jakob disease. J. Neurol. Neurosurg. Psychiatry 70, 744–748. https://doi.org/10.1136/jnnp.70.6.744 (2001).
Kovacs, G. G. & Budka, H. Current concepts of neuropathological diagnostics in practice: neurodegenerative diseases. Clin. Neuropathol. 29, 271–288. https://doi.org/10.5414/npp29271 (2010).
Montine, T. J. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. https://doi.org/10.1007/s00401-011-0910-3 (2012).
Williams, D. R. et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 130, 1566–1576. https://doi.org/10.1093/brain/awm104 (2007).
Kovacs, G. G. et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 140, 99–119. https://doi.org/10.1007/s00401-020-02158-2 (2020).
Mackenzie, I. R. A. et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. https://doi.org/10.1007/s00401-011-0845-8 (2011).
McKeith, I. G. et al. Diagnosis and management of dementia with Lewy bodies. Neurology 89, 88–100. https://doi.org/10.1212/WNL.0000000000004058 (2017).
Bahl, J. M. et al. The diagnostic efficiency of biomarkers in sporadic Creutzfeldt-Jakob disease compared to Alzheimer’s disease. Neurobiol. Aging. 30, 1834–1841. https://doi.org/10.1016/j.neurobiolaging.2008.01.013 (2009).
Blennow, K., Vanmechelen, E. & Hampel, H. CSF total tau, Aβ42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol. Neurobiol. 24, 87–97. https://doi.org/10.1385/MN:24:1-3:087 (2001).
Sjögren, M. et al. Both total and phosphorylated tau are increased in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 70, 624–630. https://doi.org/10.1136/jnnp.70.5.624 (2001).
Skillbäck, T. et al. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease. JAMA Neurol. 71, 476–483. https://doi.org/10.1001/jamaneurol.2013.6455 (2014).
Struyfs, H. et al. Cerebrospinal fluid P-tau181P: biomarker for improved differential dementia diagnosis. Front. Neurol. 6, 138. https://doi.org/10.3389/fneur.2015.00138 (2015).
Bartoš, A., Smětáková, M., Říčný, J., Nosková, L. & Fialová, L. Determination of tau proteins and β-amyloid 42 in cerebrospinal fluid by ELISA methods and preliminary normative values. Cesk. Slov. Neurol. N. 82, 533–540. https://doi.org/10.14735/amcsnn2019533 (2019).
Hu, W. T. et al. Reduced CSF p-Tau181 to Tau ratio is a biomarker for FTLD-TDP. Neurology 81, 1945–1952. https://doi.org/10.1212/01.wnl.0000436625.63650.27 (2013).
Borroni, B. et al. CSF p-Tau181/Tau ratio as biomarker for TDP pathology in Frontotemporal Dementia. Amyotroph. Lateral Scler. Frontotemporal. Degener. 16, 86–91. https://doi.org/10.3109/21678421.2014.971812 (2015).
Pijnenburg, Y. A. L., Verwey, N. A., van der Flier, W. M., Scheltens, P. & Teunissen, C. E. Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimers Dement. (Amst) 1, 505–512. https://doi.org/10.1016/j.dadm.2015.11.001 (2015).
Irwin, D. J. et al. Ante mortem CSF tau levels correlate with post mortem tau pathology in FTLD. Ann. Neurol. 82, 247–258. https://doi.org/10.1002/ana.24996 (2017).
Shaw, L. M. et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413. https://doi.org/10.1002/ana.21610 (2009).
Bian, H. et al. CSF biomarkers in frontotemporal lobar degeneration with known pathology. Neurology 70, 1827–1835. https://doi.org/10.1212/01.wnl.0000311445.21321.fc (2008).
Holmberg, B., Rosengren, L., Karlsson, J. E. & Johnels, B. Increased cerebrospinal fluid levels of neurofilament protein in progressive supranuclear palsy and multiple-system atrophy compared with Parkinson’s disease. Mov. Disord. 13, 70–77. https://doi.org/10.1002/mds.870130116 (1998).
Sjögren, M. et al. Neurofilament protein in cerebrospinal fluid: a marker of white matter changes. J. Neurosci. Res. 66, 510–516. https://doi.org/10.1002/jnr.1242 (2001).
Landqvist Waldö, M. et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol. 13, 54. https://doi.org/10.1186/1471-2377-13-54 (2013).
Abu-Rumeileh, S. et al. Comparison between plasma and cerebrospinal fluid biomarkers for the early diagnosis and association with survival in prion disease. J. Neurol. Neurosurg. Psychiatry 91, 1181–1188. https://doi.org/10.1136/jnnp-2020-323826 (2020).
Sjögren, M. et al. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology 54, 1960–1964. https://doi.org/10.1212/wnl.54.10.1960 (2000).
Isaacs, A. M. et al. Lack of TAR-DNA binding protein-43 (TDP-43) pathology in human prion diseases. Neuropathol. Appl. Neurobiol. 34, 446–456. https://doi.org/10.1111/j.1365-2990.2008.00963.x (2008).
Amador-Ortiz, C. et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 61, 435–445. https://doi.org/10.1002/ana.21154 (2007).
Higashi, S. et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 1184, 284–294. https://doi.org/10.1016/j.brainres.2007.09.048 (2007).
Nakashima-Yasuda, H. et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229. https://doi.org/10.1007/s00401-007-0261-2 (2007).
Feneberg, E., Gray, E., Ansorge, O., Talbot, K. & Turner, M. R. Towards a TDP-43-based biomarker for ALS and FTLD. Mol. Neurobiol. 55, 7789–7801. https://doi.org/10.1007/s12035-018-0947-6 (2018).
Majumder, V., Gregory, J. M., Barria, M. A., Green, A. & Pal, S. TDP-43 as a potential biomarker for amyotrophic lateral sclerosis: a systematic review and meta-analysis. BMC Neurol. 18, 90. https://doi.org/10.1186/s12883-018-1091-7 (2018).
Körtvélyessy, P. et al. Progranulin and amyloid-β levels: relationship to neuropsychology in frontotemporal and Alzheimer’s disease. J. Alzheimers Dis. 46, 375–380. https://doi.org/10.3233/JAD-150069 (2015).
Zerr, I. et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 132, 2659–2668. https://doi.org/10.1093/brain/awp191 (2009).
Collins, S. J. et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 129, 2278–2287. https://doi.org/10.1093/brain/awl159 (2006).
Cuadrado-Corrales, N. et al. Impact of the clinical context on the 14-3-3 test for the diagnosis of sporadic CJD. BMC Neurol. 6, 25. https://doi.org/10.1186/1471-2377-6-25 (2006).
Rohan, Z., Smetakova, M., Kukal, J., Rusina, R. & Matej, R. Proteinase-activated receptor 2 and disease biomarkers in cerebrospinal fluid in cases with autopsy-confirmed prion diseases and other neurodegenerative diseases. BMC Neurol. 15, 50. https://doi.org/10.1186/s12883-015-0300-x (2015).
Sanchez-Juan, P. et al. CSF tests in the differential diagnosis of Creutzfeldt–Jakob disease. Neurology 67, 637–643. https://doi.org/10.1212/01.wnl.0000230159.67128.00 (2006).
Hamlin, C. et al. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt–Jakob disease. Neurology 79, 547–552. https://doi.org/10.1212/WNL.0b013e318263565f (2012).
Peng, L., Bi, C., Xia, D., Mao, L. & Qian, H. Increased cerebrospinal fluid neurofilament light chain in central nervous system inflammatory demyelinating disease. Mult. Scler. Relat. Disord. 30, 123–128. https://doi.org/10.1016/j.msard.2019.02.009 (2019).
Mukaetova-Ladinska, E. B., Monteith, R. & Perry, E. K. Cerebrospinal fluid biomarkers for dementia with Lewy bodies. Int. J. Alzheimers Dis. 2010, 536538. https://doi.org/10.4061/2010/536538 (2010).
Oeckl, P., Steinacker, P., Feneberg, E. & Otto, M. Neurochemical biomarkers in the diagnosis of frontotemporal lobar degeneration: an update. J. Neurochem. 138, 184–192. https://doi.org/10.1111/jnc.13669 (2016).
Mehta, P. D. et al. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch. Neurol. 57, 100–105. https://doi.org/10.1001/archneur.57.1.100 (2000).
Karikari, T. K. et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet 19, 422–433. https://doi.org/10.1016/S1474-4422(20)30071-5 (2020).
Jack, C. R. Jr. et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562. https://doi.org/10.1016/j.jalz.2018.02.018 (2018).
Acknowledgements
The authors wish to thank Thomas Secrest for the revision of the English version of this article.
Funding
This study was supported by the Ministry of Health, Czech Republic (Conceptual development of research organization VFN64165, General University Hospital in Prague and Thomayer University Hospital in Prague, TUH, 00064190), by the Grants Agency of the Ministry of Health (NV19-04-00090 and NV18-04-00179), and by Charles University (Projects Progress Q27/LF1, Progress Q35/LF3, and GAUK 142120).
Author information
Authors and Affiliations
Contributions
RM, MB, and ZS were involved in planning the project. MB and ZS performed the measurements. RM supervised the project. MB analyzed the experimental data and drafted the manuscript with support from RR. All authors have reviewed and approve the contents of the manuscript and have agreed to submit the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bruzova, M., Rusina, R., Stejskalova, Z. et al. Autopsy-diagnosed neurodegenerative dementia cases support the use of cerebrospinal fluid protein biomarkers in the diagnostic work-up. Sci Rep 11, 10837 (2021). https://doi.org/10.1038/s41598-021-90366-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90366-5
- Springer Nature Limited