
Abstract
Several TBC1D24 variants are causally involved in the development of profound, prelingual hearing loss (HL) and different epilepsy syndromes inherited in an autosomal recessive manner. Only two TBC1D24 pathogenic variants have been linked with postlingual progressive autosomal dominant HL (ADHL). To determine the role of TBC1D24 in the development of ADHL and to characterize the TBC1D24-related ADHL, clinical exome sequencing or targeted multigene (n = 237) panel were performed for probands (n = 102) from multigenerational ADHL families. In four families, TBC1D24-related HL was found based on the identification of three novel, likely pathogenic (c.553G>A, p.Asp185Asn; c.1460A>T, p. His487Leu or c.1461C>G, p.His487Gln) and one known (c.533C>T, p.Ser178Leu) TBC1D24 variant. Functional consequences of these variants were characterized by analyzing the proposed homology models of the human TBC1D24 protein. Variants not only in the TBC (p.Ser178Leu, p.Asp185Asn) but also in the TLDc domain (p.His487Gln, p.His487Leu) are involved in ADHL development, the latter two mutations probably affecting interactions between the domains. Clinically, progressive HL involving mainly mid and high frequencies was observed in the patients (n = 29). The progression of HL was calculated by constructing age-related typical audiograms. TBC1D24-related ADHL originates from the cochlear component of the auditory system, becomes apparent usually in the second decade of life and accounts for approximately 4% of ADHL cases. Given the high genetic heterogeneity of ADHL, TBC1D24 emerges as an important contributor to this type of HL.
Similar content being viewed by others


Introduction
Increasing use of high throughput DNA sequencing methods has significantly improved the detection rate of genetic alterations causative of hearing loss (HL). It has resulted in discovering novel HL variants and genes and assigning new inheritance patterns to known HL genes1,2. Several HL genes are causally involved in the development of both autosomal recessive and dominant forms of hereditary HL. Another level of complexity is provided by the diversity of clinical presentation. Some HL genes may lead to isolated HL but also syndromes that do not necessarily include HL as one of their phenotypic features3. This phenomenon is well-exemplified by the TBC1D24 gene.
Recessive variants detected in TBC1D24 may cause a spectrum of phenotypes, beginning with a mild form of familial infantile myoclonic epilepsy (FIME; OMIM #605021) and encompassing early-infantile epileptic encephalopathy 16 (EIEE16; OMIM #615338) and progressive myoclonic epilepsy (PME4) that represent a combination of epilepsy with other variable features and ending with DOORS syndrome (deafness, onychodystrophy, osteodystrophy, mental retardation and seizures; OMIM #220500), a syndromic form of HL5. Alterations in TBC1D24 were also found in patients with isolated HL inherited in autosomal recessive (DFNB86; OMIM #614617) or autosomal dominant manner (DFNA65; OMIM #616044). While the involvement of TBC1D24 in the development of a recessive form of HL is documented, with ten pathogenic variants identified so far6,7,8,9,10, only two TBC1D24 pathogenic variants have been reported in the context of autosomal dominant HL (ADHL)1,11,12.
More than 40 different TBC proteins (TBC domain-containing proteins) are present in humans. TBC1D24 (and its homolog from D. melanogaster—Skywalker) contains a unique combination of TBC and TLDc domains. The TBC domain is considered to serve as GTPase-activating protein (GAP) that promotes GTP conversion to GDP in Rab proteins, which inactivates them. The Rab proteins are a crucial component of vesicular trafficking and are involved in vesicle formation, cargo transport along the cytoskeleton, and membrane fusion13. In 2006, a dual-finger mechanism of the TBC-Rab activity was proposed by Pan et al.14. According to this mechanism, both the TBC domain and Rab supply residues crucial for GTP hydrolysis.
Interestingly, a different mechanism was postulated for the TBC domain of Skywalker, as the region that supposedly interacts with Rab, is substantially different from that observed for other TBC proteins15. This alternative hypothesis explains the TBC1D24 impact on vesicular traffic by the ability of its TBC domain to interact directly with the membrane. This hypothesis originates from the observation of the positively charged region unique for the TBC1D24 orthologs, constituting residues that are conserved among this family and are not observed in other TBC proteins. The binding is presumed to occur through direct interactions with phosphoinositides in the lipid bilayer as the TBC domain from Skywalker does not bind to membranes devoid of these lipids. The estimated Kd (dissociation constant) for IP3 binding is around 0.019 mM, similar to other phosphoinositide-binding proteins. Moreover, changes of the positively charged interface (e.g., Arg79Cys, Arg281Cys) significantly weaken the interactions with IP3 (Kd rises to 0.32 and 0.18 mM, respectively) and produce several phenotypes that can be linked to impaired vesicle transport, such as seizures. The role of TLDc, the second TBC1D24 domain is more obscure. However, oxidative stress sensing or resistance has been demonstrated in cell cultures16.
For this study we carefully selected 102 HL families with pedigrees showing an autosomal dominant mode of inheritance. We found that in four of them, HL developed due to a TBC1D24 pathogenic variant. For three of the families, the identified variants were novel, not previously associated with hearing impairment. Stimulated by the relatively high frequency of TBC1D24 variants in the ADHL cohort, we focused on molecular aspects of detected TBC1D24 variants and clinical features of the TBC1D24-related HL. In this paper, we provide extensive characteristics of this type of HL.
Materials and methods
Ethics approval
All tested subjects gave informed consent for participation in the study following the tenets of the Declaration of Helsinki. The study was approved by the ethics committee at the Institute of Physiology and Pathology of Hearing (KB.IFPS.25/2017).
Study subjects
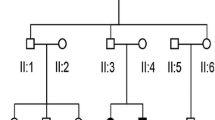
A total of 102 multigenerational families with HL occurring in at least three generations were selected for the study. In all recruited families, HL was transmitted from at least one male family member to offspring, which strongly indicated an autosomal dominant mode of HL inheritance. From this group, four families with TBC1D24 pathogenic variants (n = 29 HL patients and n = 22 unaffected individuals) were selected for further evaluation (Fig. 1A–D).

Four families with TBC1D24 pathogenic variants. Black symbols indicate individuals affected by HL; open symbols indicate unaffected individuals; diagonal line denotes the deceased individuals. YOB year of birth. In Family 1 (A) TBC1D24 c.533C>T (p.Ser178Leu), in Family 2 (B) TBC1D24 c.553G>A (p.Asp185Asn), in Family 3 (C) TBC1D24 c.1461C>G (p.His487Gln) and in Family 4 (D) TBC1D24 c.1460A>T (p.His487Leu) were identified as causative for ADHL.
Audiometry and hearing threshold data analysis
Pure-tone audiometry (PTA) was performed in individuals from the tested families. Hearing thresholds were measured with the AC40 clinical audiometer (Interacoustics, Middelfart, Denmark) for frequencies 125–8000 Hz with 10/5 dB descending-ascending threshold estimation procedure17. The degree of HL was described as mild (21–40 dB), moderate (41–70 dB), severe (71–90 dB) or profound (> 90 dB). All available previous PTA results were also analyzed. Symmetry of hearing thresholds was validated, and mean binaural air conduction thresholds (dB hearing level, dBHL) were calculated. Patient III.10 from Family 2 has been excluded from further comparisons because his audiograms revealed steeply-sloping high-frequency HL different from all his family members. His HL most likely developed due to the acoustic trauma experienced in adolescence; none of his children had HL (Supplementary Fig. S1). Binaural hearing threshold data from families with TBC1D24 p.Ser178Leu and p.Asn307His pathogenic variants reported in previous studies were also collected1,11,12.
Based on the obtained PTA data (n = 81 binaural hearing thresholds) from individuals with different TBC1D24 pathogenic variants, the age-related typical audiograms (ARTA) were constructed as described previously18. A cross-sectional linear regression analysis of threshold on age was performed for every hearing frequency, and then characteristic hearing thresholds were predicted for fixed ages (10–80 years). The HL progression was determined and expressed as the annual threshold deterioration (ATD; dB per year). The progression was considered significant if the regression coefficient (slope) was significantly different from 0 at p < 0.05.
The ARTA were also constructed separately for TBC1D24 p.Ser178Leu (Family 1 together with the previously published data for this variant1,11), as well as for p.Asp185Asn (Family 2) and p.His487Leu (Family 4). There were no longitudinal PTA data for separate ARTA calculations for the remaining TBC1D24 p.His487Gln variant (Family 3). To evaluate differences between characteristic HL patterns observed in patients with different TBC1D24 variants, threshold features arrays were calculated19. The data were plotted and compared using the chi-square goodness of fit test with significance level p < 0.05.
Additional audiological and neurotological evaluation
For all probands (IV.1 Family 1; IV.12 Family 2; IV.7 Family 3 and IV.6 Family 4) additional audiological and neurotological examinations were performed. Assessment of auditory function comprised impedance audiometry, transient evoked otoacoustic emissions (TEOAE) and auditory brainstem responses (ABRs). Acoustic impedance measurements were performed with the Zodiac 901 instrument (Madsen Electronics, Copenhagen, Denmark). Stapedius reflex was analyzed for the frequencies 500, 1000, 2000 and 4000 Hz in the ipsilateral and contralateral modes20. TEOAE were evoked by standard nonlinear click stimulus with an intensity of 80 ± 5 dB peSPL and recorded using the ILO-292 system (Otodynamics Ltd, Hatfield, United Kingdom).
ABRs were recorded using the Integrity V500 system (Vivosonic Inc., Toronto, Canada). The stimulus was 0.1 ms click with alternating polarity presented with the 90 dB normal hearing level (nHL) intensity at repetition rates of 11/s and 37/s. The amplifier bandwidth was 30–1500 Hz and analysis time 11 ms. The number of sweeps required for an averaged response was 1024.
The objective vestibular function was assessed based on the cervical and ocular evoked myogenic potentials (cVEMP, oVEMP) recorded following stimulation with a 500 Hz tone burst (2 ms rise/fall time, 2 ms plateau) presented through 3 M E-A-RTONE insert earphones at an intensity of 97 dB nHL and a presentation rate of 5.1/s. VEMP measurements were performed using the Interacoustics Eclipse module system (EclipsVemp, Interacoustics, Assens, Denmark). oVEMP responses were considered present if they were stronger than noise, and the N1 and P1 latencies replicated exactly across multiple collections. Responses were required to be larger than 1.5 mV to be considered present. oVEMP interaural amplitude was considered significantly asymmetric and abnormal if there was over 33% side-to-side difference21. cVEMP interaural amplitude was considered significantly asymmetric and abnormal, if there was over 36% side-to-side difference22. To evaluate inner ear morphology and vestibulocochlear nerves anatomy, the temporal bone computed tomography was performed on a 64-slice CT scanner (Siemens CT Definition AS, Germany).
Multigene panels and Sanger sequencing
Genomic DNA was isolated from whole blood samples and buccal swabs of available family members. In probands from Families 1 (IV.1), 3 (IV.7) and 4 (IV.6) a custom multigene panel containing 237 HL genes was performed (SeqCap EZ Choice, Roche, Basel, Switzerland) and the DNA libraries were run on MiSeq using 2 × 75 bp paired-end reads23. In the index patient from Family 2 (IV.12) clinical exome sequencing (TruSightOne, Illumina, Cambridge, UK) was performed according to the manufacturer’s protocol. The sample was run on MiSeq using 2 × 150 bp paired-end reads. Bioinformatics analysis was performed as described previously24. Selected reads and candidate variants were verified with the Integrative Genomics Viewer (IGV)25.
The analysis pipeline included variant population frequencies from different population databases, i.e., the UK10K project (https://www.uk10k.org/), the NHLBI GO Exome Sequencing Project (ESP) (https://esp.gs.washington.edu/drupal/) and the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org) (all accessed in 03/2021). Pathogenicity predictions for non-synonymous variants were performed using REVEL26, CADD27, LRT28, PolyPhen-229, SIFT30 and MutationTaster231 computational algorithms. The potential effect of detected variants on TBC1D24 RNA splicing was assessed using SpliceSite Finder, MaxEntScan, NNSPLICE, GeneSplicer algorithms integrated with Alamut Visual Software v2.15 (Interactive Biosoftware, Rouen, Paris). The pathogenic potential of identified variants was evaluated according to standards and guidelines for interpreting sequence variants32,33.
The presence of the candidate pathogenic variants was confirmed by Sanger sequencing and reported based on the TBC1D24 NM_001199107.1 and NP_001186036.1 reference sequences. The primer pairs 5’-GTTCCCCGACATCTCCTTCTG and 5’-TTCTGCTTCAGGGCTTTCTCATT as well as 5’-GATGAAACGGGTTGTGGCTCT and 5’-CAGACCGTTGACCCTCCATAG were used for amplification of TBC1D24 exon 2 and 7, respectively. PCR products were labeled with BigDye Termination cycle sequencing kit v3.1 (Applied Biosystems, Foster City, CA, USA) and sequenced with a 3500xL Genetic Analyzer (Applied Biosystems). Obtained results were analyzed using Variant Reporter Software v1.1 (Applied Biosystems).
In silico protein analysis
Multiple sequence alignment (MSA) of TBC and TLDc domains of TBC1D24 was proposed using the following procedure. First, homologs of the human TBC1D24 protein (GenBank id: BAH16654.1) were identified in a PSI-Blast34 search (E-value threshold of 0.01, 4 iterations) performed against the NCBI non-redundant protein sequence database. Next, sequences from chordates were selected and their domain organization was predicted with hmmscan35. As the TBC1D24 sequence is characterized by the presence of the TBC domain followed by the TLDc domain13 only proteins with such domain organization were retained for further analysis. Finally, selected sequences were clustered with cd-hit36 using a 90% sequence identity threshold. MSA was constructed with the mafft program using the L-INS-I strategy37. The procedure was carried out independently for TBC and TLDc domains. Proteins with “hypothetical” or “low quality” descriptions were filtered out before clustering.
The homology model of the TBC domain was prepared using the following approach. The crystal structure of the protein from D. melanogaster (pdb|5hjn) was selected as a template after analyzing the results of the GeneSilico Metaserver38. The sequence-to-structure alignment was built using the consensus alignment approach and 3D assessment39 based on the FFAS, HHSearch and Psi-Blast results.The multiple sequence alignment of the family was also taken into consideration. The 3D model of the protein was built with MODELLER40. A model quality assessment was carried out using the MolProbity webserver41. Due to the presence of a nearly 40-residue long insertion in the human TLDc domain compared to the template (pdb|6r82), the model was proposed using the I-TASSER server42 rather than by MODELLER. To predict the putative conformation of TBC-TLDc complex in D. melanogaster, crystal structures of TBC and TLDc domains were used (pdb|5jhn and pdb|6r82, respectively). Protein–protein docking was carried out with the Hdock server using its default parameters43. For the TLDc domain, amino acid conservation was calculated using the ConSurf Server44 and the manually curated MSA for this domain. Visualization was done with Pymol (www.pymol.org).
Informed consent
Written informed consent was obtained from all participants.
Consent for publication
Participants consented to publication of nonidentifiable data.
Results
Cochlea involvement in patients with TBC1D24-related ADHL
TBC1D24 variants causative for HL were identified in four probands (4/102), which corresponds to an approx. 4%-prevalence of TBC1D24-related ADHL in our cohort. All affected individuals from the four TBC1D24 families were diagnosed with bilateral, postlingual, progressive sensorineural HL. Mean HL onset was at the earliest in the second decade of life, i.e., 19.5 y.o. in Family 1, 16.7 y.o. in Family 2, 16.3 y.o. in Family 3 and 35 y.o. in Family 4. In all families hearing thresholds had a similar down-sloping pattern and mainly mid and high frequencies were affected. Most HL patients routinely use HAs (Table 1).
As shown by average ARTA (Fig. 2A) TBC1D24-related HL starts as mild at low and mid frequencies and moderate at high frequencies. It progresses with age (up to 80 y.o.) and becomes moderate to severe at low and mid frequencies and profound at high frequencies. The ATD in patients with TBC1D24 pathogenic variants is statistically significant at all frequencies (p < 0.001) and ranges from 0.61 (8 kHz) to 0.75 (0.25 and 0.5 kHz) dB/year (Fig. 2B).

Audiological characteristics of TBC1D24-related ADHL. (A) ARTA for patients with TBC1D24 pathogenic variants. (B) Rate of HL deterioration in PTA.
Analysis of PTA data of patients with the TBC1D24 p.Ser178Leu, p.Asp185Asn and p.His487Leu variants revealed variant-dependent differences in the HL degree. In patients with p.Ser178Leu and p.His487Leu, ARTA presents mild HL at the age of 20. Later in life, HL becomes moderate to severe (Fig. 3A). The ATD is significant at all analyzed frequencies (p < 0.001) and ranges from 0.88 (4 kHz) to 1.08 (1 kHz) dB/year for p.Ser178Leu and from 0.59 (4 kHz) to 1.03 (0.5 kHz) dB/year for p.His487Leu (Supplementary Fig. S2). In patients with the p.Asp185Asn variant, ARTA shows a more severe HL over the analyzed time interval. From the age of 50 HL becomes profound at mid and high frequencies (Fig. 3A). For the TBC1D24 p.Asp185Asn pathogenic variant, ATD is statistically significant at all frequencies (p < 0.001) and ranges from 0.83 (0.25 kHz) to 1.72 (2 kHz) dB/year (Supplementary Fig. S2). The observations were confirmed by comparing the threshold feature arrays, which revealed a statistically significant difference between HL pattern found in p.Asp185Asn patients compared to an average TBC1D24 ARTA (Fig. 3B).

Audiological characteristics of HL observed in patients with a particular TBC1D24 pathogenic variant. (A) ARTA for patients with p.Ser178Leu, p.Asp185Asn and p.His487Leu pathogenic variants. (B) Threshold feature array for HL pattern observed for a particular TBC1D24 pathogenic variant. The graph represents the distribution of ARTA data points counted for different classes of HL degree and frequency (prepared based on the model proposed by Huygen et al. 2003)19. lf low frequencies, mf mid frequencies, hf high frequencies.
Results of additional audiological and neurotological examinations revealed cochlear involvement in the TBC1D24-related ADHL. No TEOAEs were recorded in the tested probands, and no or increased thresholds of stapedial muscle reflexes were observed. The vestibulocochlear nerve function measured by ABR was in line with the PTA results. No vestibular dysfunction or anatomical abnormalities of the cochleovestibular system were found. All probands reported sporadic or chronic tinnitus, one patient had sporadic vertigo (Table 1).
Identification of TBC1D24 pathogenic variants
After performing next-generation sequencing (NGS) in four tested families very rare heterozygous variants of the TBC1D24 gene were selected. In Families 1 and 2 were identified, respectively, a heterozygous c.533C>T, p.Ser178Leu variant and a heterozygous c.553G>A, p.Asp185Asn variant, both located in exon 2 of the TBC1D24 gene. In Families 3 and 4, a heterozygous c.1460A>T, p.His487Leu variant and a heterozygous c.1461C>G, p.His487Gln variant were found in TBC1D24 exon 7 (Fig. 4, Supplementary Fig. S3). All selected variants were either very rare or not present in population databases. Most computational algorithms predicted their damaging role (Table 2). No impact of the analyzed variants on TBC1D24 RNA splicing was predicted. Family studies confirmed variants segregation with HL (Fig. 1A–D). Based on the applicable standards and guidelines, detected TBC1D24 variants were classified as likely pathogenic32,33. All variants have been submitted to the Global Variome shared LOVD. No other pathogenic or likely pathogenic variants related to isolated or syndromic hereditary HL were found.

Schematic representation of TBC1D24 gene and protein organization. Gene and protein structure are depicted based on the canonical transcript NM_001199107.1 and reference protein sequence NP_001186036.1. Previously reported TBC1D24 pathogenic variants involved in the development of nonsyndromic ARHL (DFNB86) are written in black. Variants causative for ADHL (DFNA65) are shown in blue; variants identified in this study are bolded. Domain boundaries (TBC residues 21–314; TLDc residues 339–555) were determined based on the sequence to structure alignment between human TBC1D24 protein and Skywalker/TBC1D24 protein from D. melanogaster.
The c.533C>T variant identified in Family 1 has already been described in two ADHL families1,11. It was the first, and for a long time, the only known TBC1D24 variant causally involved in the development of ADHL. The second identified variant (c.553G>A) is located seven amino acids downstream of this known variant; both are placed in the TBC domain of the TBC1D24 protein. Variants identified in Families 3 and 4 are nucleotide substitutions in adjacent positions within the same codon of the TBC1D24 sequence, resulting in an amino acid change from histidine respectively to glutamine or leucine. The latter variants are the first ADHL-related genetic alterations located in the TLDc domain of TBC1D24.
Modeling the functional role of TBC1D24 variants
The homologs of the human TBC1D24 protein were collected and clustered using the procedure described in the “Materials and methods” section. For the TBC domain, it resulted in 55 proteins that represent the sequence variability of the TBC domain from the TBC1D24 family. Analysis of the multiple sequence alignment (MSA for selected residues and species is presented in Fig. 5C) allowed us to identify that the amino acids crucial for interactions with IP3 in D. melanogaster, i.e. Lys75, Arg79, Lys277, Arg281, Arg335, Gly336 and Thr339, are either unchanged or substituted with positively charged residues—Lys36, Arg40, Lys238, Arg242. Arg293, Arg297, Lys298 (residues are numbered according to the human sequence). This might lead to even stronger interactions between the protein and IP3 in humans compared to D. melanogaster. Additionally, the MSA analysis revealed that both residues involved in ADHL, i.e., Asp185 and Ser178, have a different conservation level. In only one case Asp185 is replaced with glutamic acid, preserving the negatively charged residue. However, Ser178 is present only in 25 out of 55 sequences and is often replaced by other small residues like alanine or threonine. A similar analysis was independently performed for the TLDc domain, resulting in 118 protein sequences. According to the MSA, His487 is a part of a long loop (between β5 and β6) with variable length across different Chordata (MSA for selected residues and species is presented in Fig. 6C). In the TBC1D24 homolog from D. melanogaster, this loop is 18 residues long (from Pro511 to Phe528), while, in humans, it spans 56 residues (from Glu443 to Phe498). Despite high sequence variability observed in the middle of this loop, its termini are conserved across Chordata. His487 is localized near the C-terminus of this loop and is observed in 81 sequences. Since the MSA reflects the sequence variability of TLDc domains, the remaining 37 sequences that lacked H487 were verified manually. It turned out that these sequences originated from 36 organisms. The majority of them were either a partially solved sequence or represented one of many isoforms of the TLDc domain present in that species. Only for ten species we could not find an isoform of TBC1D24 with His at position 487. It can be therefore concluded that His487 is a conserved residue among Chordata.

Structure of the human TBC domain from TBC1D24. (A) The homology model of TBC1D24 protein was superimposed over the IP3-Skywalker complex to obtain the structure of the human protein—IP3 complex. The binding sites constitute of several, conserved, mostly positively charged amino acids. (B) The C-terminal helix is stabilized by polar interactions between N307, E308 with T182, D185 and S178. The latter two amino acids are changed to N and L, respectively, in patients with ADHL (this study). (C) Multiple sequence alignment of the human TBC domain from TBC1D24 and its homologs from selected organisms. Only regions comprising the binding site residues and two helices harboring S178, T182, D185, N307 and E308 are shown. D185 is conserved among Chordata, while S178 is present only in a subset of organisms, suggesting its auxiliary role in stabilizing the C-terminal helix. Residues highlighted in grey are involved in recognizing the membrane by the TBC domain, while residues highlighted in cyan are involved in the stabilization of conformation of the C-terminal helix of the TBC domain.

Structure of the human TLDc domain from TBC1D24. (A) The residues conservation for Chordata TBC1D24 mapped onto the surface of pdb|6r82. Two faces of the protein are shown. Conserved residues are colored blue, while variable ones are colored red. (B) The possible orientation of TBC and TLDc domains in TBC1D24 protein as proposed by HDock. The TLDc domain is colored using the same scheme as in (A). TBC domain is presented as cartoon. The dotted sphere represents the putative position of the loop harboring H487. (C) Multiple sequence alignment of the TLDc domain from TBC1D24 protein and its homologs from selected organisms. Only the loop harboring His487 is shown. Both termini (including His487 highlighted in cyan) of this loop are conserved among Chordata, while the middle of the loop shows the significant sequence and structural variability. Residues highlighted in gray are conserved across Chordata (a specific residue is present in more than 80% of analyzed sequences).
To analyze the putative role of p.Asp185Asn and p.Ser178Leu mutations, a homology model of the human TBC domain from TBC1D24 was constructed. Asp185 is located outside the active site on the 6th helix and does not interact directly with IP3 (Fig. 5A). The distance to the closest Cα atom of a residue from the binding site (Lys238) is nearly 14 Å. According to our model Asp185 forms a hydrogen bond with Asn307 (Fig. 5B) located in the C-terminal helix of the TBC domain. Asn307, like Asp185, is also conserved among Chordata (with only two exceptions where histidine is present). The C-terminal helix itself is a part of the IP3 binding site, as its N-terminal fragment harbors Ser296, Arg297, and Lys298. Thus, it is plausible that the Asp185-Asn307 bond is a part of a network of interactions responsible for maintaining a proper geometry of the 6th and C-terminal helices, which influences the position of Arg297 and Lys298 within the binding site. Close inspection of our model revealed that two additional polar interactions are part of this network. First, Asn307 forms an additional hydrogen bond with Thr182 residue, also conserved among Chordata. The second interaction in this model involves Ser178 and Glu308 (Fig. 5B). However, as both Ser178 and Glu308 are present only in a subset of chordates’ TBC1D24, this hydrogen bond may perform only an auxiliary role. Intriguingly, p.Ser178Leu was already linked with ADHL1,11 and based on the model proposed by Parzefall et al., Ser178 interacts with Asn30712. Considering the close proximity of both Asn307 and Glu308 residues, Ser178 interaction with either of these two residues is equally probable.
As mentioned above, His487 is part of a long flexible loop that hinders the probability of correctly identifying its position in relation to the rest of the TLDc domain. I-Tasser, employed to predict the possible orientations of that fragment, produces several, equally likely and quite diverse conformations. However, in most cases, residue conservation indicates amino acid importance in maintaining the proper 3D structure of a protein, its function or ability to bind to other protein. We investigated the possible role of His487 in the binding of the TBC domain. This hypothesis was put forward as we observed that the putative position of the loop harboring His487 is close to a patch of several conserved, solvent-exposed residues (Fig. 6A). To obtain a putative conformation of the TBC-TLDc complex, we performed a protein–protein docking using available crystal structures of these domains from D. melanogaster. Analysis of the top ten conformations revealed that the TLDc domain interface involves residues from this conserved patch (Fig. 6B). No such consistency was observed for the TBC domain. Considering the His487 proximity to this region, p.His487Gln and p.His487Leu might be responsible for weakening the interface between both domains of the TBC1D24 protein.
Discussion
Since the first association of TBC1D24 with isolated HL in 2014, the number of its identified variants causing a recessive form of HL has been steadily growing, currently reaching as many as ten. The case has been different for TBC1D24 variants involved in ADHL development. The first ADHL-related TBC1D24 pathogenic variant (p.Ser178Leu) was also identified in 2014, found in parallel in a European and a Chinese family1,11. Before 2020, no other TBC1D24 alteration has been associated with this condition. The second TBC1D24-related ADHL variant (p.Asn307His) has been reported recently in a study of two unrelated HL families from Austria and the UK12 (Table 3). Altogether, the data suggested that the involvement of TBC1D24 in ADHL might be considered relatively rare and exceptional.
Our study shows that the role of TBC1D24 in ADHL may be underestimated. After testing 102 multigenerational families fulfilling strict criteria for the autosomal dominant inheritance of HL, the pathogenic TBC1D24 variant was detected in almost 4% (4/102). The involvement of TBC1D24 in ADHL can be even more significant than that. There are two additional ADHL families in our cohort whose probable underlying cause of HL is a defective TBC1D24 (data not shown). As further investigations are required to unequivocally confirm the TBC1D24 pathogenic potential, the families have not been presented here. We can also speculate that there were other patients with TBC1D24-related ADHL who have been excluded from our analysis because (i) their pattern of HL inheritance was mimicking a mitochondrial mode of HL inheritance, or (ii) the variants arose de novo, and the family history of HL was negative.
It is important to note that the contribution of one particular gene to ADHL development does not exceed a dozen percent. Among the currently known 50 genes implicated in ADHL development (https://hereditaryhearingloss.org/dominant-genes; accessed 03/2021), there is no single major gene responsible for ADHL. It varies among populations, but pathogenic variants in TECTA, KCNQ4, WFS1, or MYO6 are more frequent than other ADHL genes45,46. Pathogenic variants in MYO6, KCNQ4, WFS1, GSDME (DFNA5) or TECTA were also repeatedly found in our ADHL cohort (unpublished data). While MYO6 pathogenic variants explained 11% of HL causes in the studied ADHL families (data not shown), the remaining genes did not exceed 4%, as observed for TBC1D24.
The TBC1D24 gene (OMIM *613577) has five transcripts (www.ensembl.org; accessed 02/2021) encoding different isoforms found in multiple human tissues. The highest expression of the TBC1D24 gene was observed in the brain, but it was also found in testis, skeletal muscle, heart, kidneys, lung, and liver47. In the human auditory system, TBC1D24 mRNA and protein were detected in hair cells and spiral ganglion neurons48. Clinically, our patients presented typical features of isolated sensorineural HL with cochlea involvement, impaired hair cell function, normal function of the auditory nerve and normal anatomical ear structures. Their HL was progressing slowly and was accompanied by tinnitus. Some differences in the HL progression rate were mutation-specific and the highest progression rate was observed for p.Asp185Asn. It was significantly higher than the average TBC1D24 progression rate calculated based on all available data.
The majority of our patients benefit from using hearing aids, even throughout their lifetime. An exception in this regard was the proband from Family 3. Unlike other patients with TBC1D24-related ADHL, including her family members, she was diagnosed with HL at 8 and received a cochlear implant at 22. In this patient we could not identify any additional genetic or environmental factors that could aggravate her HL. Generally, the age of HL onset in our patients ranged between the second and the fourth decade of life, which was in line with the data reported previously1,11,12.
All of the ADHL-related TBC1D24 pathogenic variants, identified so far represent missense changes that based on bioinformatic predictions, do not affect TBC1D24 transcript formation and are scattered throughout the gene. It is in line with the observation that the type and location of the TBC1D24 variant cannot predict the associated phenotype49. Out of three novel variants identified here, the p.Asp185Asn localizes in the N-terminal protein region seven amino acids away from p.Ser178Leu, both within the TBC domain. The other two alterations (p.His487Gln and p.His487Leu) localize in the more C-terminal part of the protein corresponding to the TLDc domain. Interestingly, a frameshift variant involving this codon (p.His487Glnfs*71) was found in a patient with the DOORS syndrome50.
Both Ser178 and Asp185 appear to serve a similar structural role in establishing the proper conformation of the TBC1D24 C-terminal helix and, consequently, the IP3 binding site. Considering their structural proximity, both mutations identified in this work, i.e., p.Ser178Leu and p.Asp185Asn should have a common molecular mechanism leading to HL. In our opinion, both mutations would most likely influence the conformation of the C-terminal helix. That would affect the orientation of binding site residues, i.e., Arg293, Arg297 and Lys298, which could result in the disruption of some of the interactions between IP3 and the TBC1D24 protein. Consequently, the decreased binding affinity would be responsible for impaired vesicular traffic that could lead to HL. The mutations affecting the TLDc domain, target conserved His487 amino acid. This residue is located in the long loop near the putative interface between TBC and TLDc domains. Thus, these mutations can destabilize the formation of this complex.
In summary, our data revealed that TBC1D24 should be considered an important contributor to ADHL. As ADHL shows similarities with presbyacusis, TBC1D24 can be proposed as a candidate gene for the age-related HL. Similar to other TBC1D24-associated disorders, variants causative for ADHL can be found in different parts of the gene, which makes the studies of its functional role even more challenging. Based on the in silico analysis of homology models, we assume that the positions of amino acids involved in ADHL development are responsible for maintaining a proper geometry of the TBC1D24 protein, which is crucial for its interactions with phosphoinositides.
Data availability
Results of the bioinformatic analysis can be found in a GitHub repository: https://github.com/SFGLab/TBC1D24.
Abbreviations
- ABRs:
-
Auditory brainstem responses
- ARTA:
-
Age-related typical audiogram
- ATD:
-
Annual threshold deterioration
- ADHL:
-
Autosomal dominant hearing loss
- OAE:
-
Otoacoustic emissions
- nHL:
-
Normal hearing level
- cVEMP:
-
Cervical vestibular evoked myogenic potentials
- oVEMP:
-
Ocular vestibular evoked myogenic potentials
References
Azaiez, H. et al. TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum. Mutat. 35, 819–823. https://doi.org/10.1002/humu.22557 (2014).
Ozieblo, D. et al. First confirmatory study on PTPRQ as an autosomal dominant non-syndromic hearing loss gene. J. Transl. Med. 17, 351. https://doi.org/10.1186/s12967-019-2099-5 (2019).
Kremer, H. Hereditary hearing loss; about the known and the unknown. Hear Res. 376, 58–68. https://doi.org/10.1016/j.heares.2019.01.003 (2019).
Muona, M. et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat. Genet. 47, 39–46. https://doi.org/10.1038/ng.3144 (2015).
Balestrini, S. et al. TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features. Neurology 87, 77–85. https://doi.org/10.1212/WNL.0000000000002807 (2016).
Rehman, A. U. et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am. J. Hum. Genet. 94, 144–152. https://doi.org/10.1016/j.ajhg.2013.12.004 (2014).
Bakhchane, A. et al. Recessive TBC1D24 mutations are frequent in Moroccan non-syndromic hearing loss pedigrees. PLoS ONE 10, e0138072. https://doi.org/10.1371/journal.pone.0138072 (2015).
Danial-Farran, N. et al. Genetics of hearing loss in the Arab population of Northern Israel. Eur. J. Hum. Genet. 26, 1840–1847. https://doi.org/10.1038/s41431-018-0218-z (2018).
Safka Brozkova, D. et al. Spectrum and frequencies of non GJB2 gene mutations in Czech patients with early non-syndromic hearing loss detected by gene panel NGS and whole-exome sequencing. Clin. Genet. 98, 548–554. https://doi.org/10.1111/cge.13839 (2020).
Xiang, Y. B. et al. Next-generation sequencing identifies rare pathogenic and novel candidate variants in a cohort of Chinese patients with syndromic or nonsyndromic hearing loss. Mol. Genet. Genomic Med. 8, e1539. https://doi.org/10.1002/mgg3.1539 (2020).
Zhang, L. et al. A dominant mutation in the stereocilia-expressing gene TBC1D24 is a probable cause for nonsyndromic hearing impairment. Hum. Mutat. 35, 814–818. https://doi.org/10.1002/humu.22558 (2014).
Parzefall, T. et al. A novel variant in the TBC1D24 lipid-binding pocket causes autosomal dominant hearing loss: Evidence for a genotype-phenotype correlation. Front. Cell Neurosci. 14, 585669. https://doi.org/10.3389/fncel.2020.585669 (2020).
Fukuda, M. TBC proteins: GAPs for mammalian small GTPase Rab?. Biosci. Rep. 31, 159–168. https://doi.org/10.1042/BSR20100112 (2011).
Pan, X., Eathiraj, S., Munson, M. & Lambright, D. G. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature 442, 303–306. https://doi.org/10.1038/nature04847 (2006).
Fischer, B. et al. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat. Struct. Mol. Biol. 23, 965–973. https://doi.org/10.1038/nsmb.3297 (2016).
Finelli, M. J. & Oliver, P. L. TLDc proteins: New players in the oxidative stress response and neurological disease. Mamm. Genome 28, 395–406. https://doi.org/10.1007/s00335-017-9706-7 (2017).
Audiology, B. S. O. Recommended procedures for pure-tone audiometry using a manually operated instrument. Br. J. Audiol. 15(3), 213–216. https://doi.org/10.3109/03005368109081440 (1981).
Oonk, A. M. et al. Progressive hereditary hearing impairment caused by a MYO6 mutation resembles presbyacusis. Hear Res. 299, 88–98. https://doi.org/10.1016/j.heares.2012.12.015 (2013).
Huygen, P. L., Pennings, R. J. & Cremers, C. W. Characterizing and distinguishing progressive phenotypes in nonsyndromic autosomal dominant hearing impairment. Audiol. Med. 1, 37–46. https://doi.org/10.1080/16513860310003049 (2003).
Jerger, J. Clinical experience with impedance audiometry. Arch. Otolaryngol. 92, 311–324 (1970).
Piker, E. G., Jacobson, G. P., McCaslin, D. L. & Hood, L. J. Normal characteristics of the ocular vestibular evoked myogenic potential. J. Am. Acad. Audiol. 22, 222–230. https://doi.org/10.3766/jaaa.22.4.5 (2011).
Zapala, D. A. & Brey, R. H. Clinical experience with the vestibular evoked myogenic potential. J. Am. Acad. Audiol. 15, 198–215. https://doi.org/10.3766/jaaa.15.3.3 (2004).
Ozieblo, D., Pazik, J., Stepniak, I., Skarzynski, H. & Oldak, M. Two novel pathogenic variants confirm RMND1 causative role in perrault syndrome with renal involvement. Genes (Basel). https://doi.org/10.3390/genes11091060 (2020).
Oldak, M. et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J. Transl. Med. 15, 25. https://doi.org/10.1186/s12967-017-1129-4 (2017).
Robinson, J. T., Thorvaldsdottir, H., Wenger, A. M., Zehir, A. & Mesirov, J. P. Variant review with the integrative genomics viewer. Cancer Res. 77, e31–e34. https://doi.org/10.1158/0008-5472.CAN-17-0337 (2017).
Ioannidis, N. M. et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. https://doi.org/10.1016/j.ajhg.2016.08.016 (2016).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. https://doi.org/10.1093/nar/gky1016 (2019).
Chun, S. & Fay, J. C. Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. https://doi.org/10.1101/gr.092619.109 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. https://doi.org/10.1038/nprot.2009.86 (2009).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. https://doi.org/10.1038/nmeth.2890 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613. https://doi.org/10.1002/humu.23630 (2018).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. https://doi.org/10.1093/nar/25.17.3389 (1997).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195. https://doi.org/10.1371/journal.pcbi.1002195 (2011).
Li, W. & Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. https://doi.org/10.1093/bioinformatics/btl158 (2006).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Kurowski, M. A. & Bujnicki, J. M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31, 3305–3307 (2003).
Ginalski, K. & Rychlewski, L. Protein structure prediction of CASP5 comparative modeling and fold recognition targets using consensus alignment approach and 3D assessment. Proteins 53(Suppl 6), 410–417. https://doi.org/10.1002/prot.10548 (2003).
Fiser, A. & Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491. https://doi.org/10.1016/S0076-6879(03)74020-8 (2003).
Chen, V. B. et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. https://doi.org/10.1107/S0907444909042073 (2010).
Roy, A., Kucukural, A. & Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. https://doi.org/10.1038/nprot.2010.5 (2010).
Yan, Y., Zhang, D., Zhou, P., Li, B. & Huang, S. Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 45, W365–W373. https://doi.org/10.1093/nar/gkx407 (2017).
Ashkenazy, H. et al. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350. https://doi.org/10.1093/nar/gkw408 (2016).
Hilgert, N., Smith, R. J. & Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics?. Mutat. Res. 681, 189–196. https://doi.org/10.1016/j.mrrev.2008.08.002 (2009).
Oka, S. I. et al. Clinical characteristics and in vitro analysis of MYO6 variants causing late-onset progressive hearing loss. Genes (Basel) https://doi.org/10.3390/genes11030273 (2020).
Falace, A. et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 87, 365–370. https://doi.org/10.1016/j.ajhg.2010.07.020 (2010).
Tona, R. et al. Mouse models of human pathogenic variants of TBC1D24 associated with non-syndromic deafness DFNB86 and DFNA65 and syndromes involving deafness. Genes (Basel). https://doi.org/10.3390/genes11101122 (2020).
Rehman, A. U., Friedman, T. B. & Griffith, A. J. Unresolved questions regarding human hereditary deafness. Oral Dis 23, 551–558. https://doi.org/10.1111/odi.12516 (2017).
Campeau, P. M., Hennekam, R. C. & DOORS Syndrome Collaborative Group. DOORS syndrome: Phenotype, genotype and comparison with Coffin-Siris syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 327–332. https://doi.org/10.1002/ajmg.c.31412 (2014).
Acknowledgements
We are grateful to the patients and their families for participation in this study.
Funding
This work was funded by the Institute of Physiology and Pathology of Hearing and Sonata BIS6 grant of the National Science Centre 2016/22/E/NZ5/00470 (M.O.). M.L. and D.P. were supported by 2019/35/O/ST6/02484 and 2020/37/B/NZ2/03757 from the National Science Center, Poland, and TEAM from the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (TEAM to D.P.). D.P. and M.L. research is funded by Warsaw University of Technology within the Excellence Initiative: Research University (IDUB) programme.
Author information
Authors and Affiliations
Contributions
D.O. and M.L.L. performed genotyping; D.O., A.S., G.T., K.K., H.S. and M.O. participated in phenotyping and clinical data collection; D.O., M.L. and D.P. performed computational analysis; D.O., M.O. and M.L. analyzed the data and wrote the manuscript; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oziębło, D., Leja, M.L., Lazniewski, M. et al. TBC1D24 emerges as an important contributor to progressive postlingual dominant hearing loss. Sci Rep 11, 10300 (2021). https://doi.org/10.1038/s41598-021-89645-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89645-y
- Springer Nature Limited
This article is cited by
-
Investigation of a novel TBC1D24 variation causing autosomal dominant non-syndromic hearing loss
Scientific Reports (2024)