
Abstract
The aberrant expression of microRNAs (miRs) has been linked to several human diseases. A promising approach for targeting these anomalies is the use of small-molecule inhibitors of miR biogenesis. These inhibitors have the potential to (i) dissect miR mechanisms of action, (ii) discover new drug targets, and (iii) function as new therapeutic agents. Here, we designed Förster resonance energy transfer (FRET)-labeled oligoribonucleotides of the precursor of the oncogenic miR-21 (pre-miR-21) and used them together with a set of aminoglycosides to develop an interbase-FRET assay to detect ligand binding to pre-miRs. Our interbase-FRET assay accurately reports structural changes of the RNA oligonucleotide induced by ligand binding. We demonstrate its application in a rapid, qualitative drug candidate screen by assessing the relative binding affinity between 12 aminoglycoside antibiotics and pre-miR-21. Surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) were used to validate our new FRET method, and the accuracy of our FRET assay was shown to be similar to the established techniques. With its advantages over SPR and ITC owing to its high sensitivity, small sample size, straightforward technique and the possibility for high-throughput expansion, we envision that our solution-based method can be applied in pre-miRNA–target binding studies.
Similar content being viewed by others


Introduction
MicroRNAs (miRs) are short (typically 22 nt) single-stranded RNAs that have important cellular functions and are dysregulated in a variety of human diseases, including cancers, viral infections, cardiovascular disease, and inflammatory diseases1. Currently, 1,917 human miR precursor genes, which are processed into 2,675 mature miRs, have been annotated in the miRBase database2 and are thought to regulate one-third of the human proteome3. Genes encoding miRs are transcribed as long RNA precursors, which are processed by the Drosha nuclear microprocessor complex to yield approximately 70 nt hairpin precursor miRs (pre-miRs)4. Pre-miRs are then transported to the cytoplasm for further processing by RNase III Dicer to form a double-stranded RNA (dsRNA) molecule. One of its strands, the mature miR, is loaded into the RNA-induced silencing complex, which binds the complementary mRNA target through Watson–Crick base pairing and mediates gene silencing. Studies have demonstrated that numerous miRs act as tumor suppressors or oncogenes (also termed ‘onco-miRs’)5. Only a few onco-miRs have been well characterized so far, one of these being miR-216. The identification of small molecules that bind to onco-miR precursors (such as the pre-miR-21 presented herein) could be a viable approach to inhibit the biogenesis of miRs that are involved in cancer development and could lead to mechanistically novel cancer therapies7. Furthermore, miRs have now been thoroughly validated as a therapeutic target8.
The most commonly employed method to target disease-associated miRs is based on the use of antisense technologies9,10. However, this approach primarily relies on modified oligonucleotide structures; these structures suffer from poor cell permeability and cellular distribution due to their intrinsic anionic character11. In contrast, small molecules offer the advantage of having good absorption, distribution, and oral bioavailability. Furthermore, rather than using sequence complementarity for binding, small molecules have the potential to recognize and bind to pre-miRs through RNA secondary or tertiary structural motifs, such as bulges, internal loops, hairpin loops, junctions, pseudoknots, or higher-order structural elements12.
Several cellular and non-cellular reporter-based assays have previously been applied to identify small-molecule inhibitors of pre-miR-mediated gene silencing7. Unfortunately, these assays all have their limitations. In general, small-molecule microarrays13,14, molecular beacons15,16,17, fluorescence polarization18,19, and catalytic enzyme-linked click chemistry assays20 all utilize reporter probes that are either large or amphiphilic and potentially interfere with the assay and/or are distant from the binding site of interest. Small-molecule binding assays, such as fluorescence indicator displacement21 require either selectively binding fluorophores or the displacement of more promiscuous probes, such as ethidium bromide, which can cause false negatives if the mode of binding differs. The hits from such assays have typically been verified by examining the maturation level of the pre-miR after adding the Dicer enzyme16 and in several cases they have been furthered characterized using additional biophysical techniques such as isothermal titration calorimetry (ITC)13, differential scanning calorimetry (DSC)14 or surface plasmon resonance (SPR)22.
Biophysical techniques such as ITC and SPR are well suited for resolving direct pre-miR engagement and were employed as benchmark methods in the current study. A significant advantage of ITC is that it can be readily applied to almost any RNA–ligand complex without the need to label both molecules. Additionally, it can be performed under a broad range of pH values, temperatures, and ionic concentrations. The major drawbacks of ITC, namely, the limited throughput and a significant sample size, is partly mitigated by SPR. However, SPR also has disadvantages, as it cannot easily discriminate between specific and non-specific interactions with the sensor surface.
Förster resonance energy transfer (FRET) is a non-radiative process in which energy is transferred between donor and acceptor chromophores23. The efficiency of this process is highly dependent on both distance and orientation between the chromophores. Consequently, information about the relative position of different donor- and acceptor-labelled biomolecules and thereby the structure of the biomolecule can be obtained24. FRET has been used in the molecular beacon (MB) assay format to measure the inhibition of Dicer-mediated pre-miR hairpin cleavage15,16,17. The compounds tested in previous studies have typically been ranked relative to each other using the fluorescence increase resulting from the Dicer RNase activity. A potential drawback of MB pre-miR-assays is that the large amphiphilic FRET probes are attached to the ends of the oligoribonucleotide; this can significantly impact the results because Dicer recognizes both the 3′- and 5′-end of the pre-miR15,25. Unfortunately, no counter-screen has been performed to determine the detailed binding parameters; thus, the developed MB assay could not be benchmarked against established techniques. While a FRET MB assay can identify hits for Dicer inhibition, it cannot resolve subtle changes in ligand binding, such as local conformational changes in the RNA structure26.
In an attempt to combine a high-information screening technique with a potential high-throughput screening method, we aimed to mitigate the shortcomings of MB assays and direct pre-miR engagement assays such as SPR. FRET between conformationally restricted, non-perturbing, fluorescent nucleobase analogs (FBAs), called interbase FRET, can be utilized to measure distance and orientation between the involved donor and acceptor molecules27. This offers the possibility not only to measure Dicer inhibition more accurately from a ligand-binding event due to their non-perturbing nature but could also, if positioned wisely, be used to identify site-selective ligand binders. We have previously demonstrated that the emissive cytosine analogue 3-(β-d-ribofuranosyl)-3H-benzo[b]pyrimido[4,5-e][1,4]oxazin-2(10H)-one (tCO)39 (Fig. 1d) can be used together with the non-emissive 3-(β-d-ribofuranosyl)-7-nitro-3H-pyrimido[5,4-b][1,4]benzothiazin-2(10H)-one (tCnitro)28 (Fig. 1d), to create an interbase-FRET pair. This combination of cytosine analogues have been used to monitor the transition from A- to Z-form RNA with no significant perturbation of the natural RNA structure28.

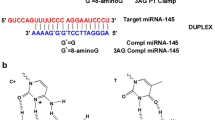
Design of the interbase-FRET assay. (a) The full-length pre-miR-21 (1) with miR-21 sequence in red and Dicer cleavage sites marked by the black line. (b) Ligand binding to the hairpin loop region of FRET-labelled pre-miR-21 construct (5, Table 1) causes a distance and/or orientation change between the fluorescent FRET donor (blue) and the non-fluorescent FRET acceptor (orange), thereby changing the FRET efficiency, and consequently, either an increase or a decrease in emission is observed. (c) Ligand binding to the stem FRET-labelled pre-miR-21 construct (7, Table 1). (d) The chemical structures of the fluorescent FRET donor tCO (blue) and the non-fluorescent FRET acceptor tCnitro (orange).
Previous work has demonstrated that aminoglycosides29 have a general affinity towards RNA30,31,32,33, including pre-miR-2113,22,34,35,36,37,38. However, the specificity of these binding events has yet to be fully elucidated38. Aminoglycosides have also been used to construct bifunctional aminoglycoside conjugates that consist of a pre-miRNA binding unit that is connected by a linker to a Dicer inhibiting unit that binds pre-miR-21 and regulates miR-21 maturation19. From the typical molecular structures of aminoglycosides (Figure S2), they are expected to be non-fluorescent and not to absorb light in the spectral region relevant for tCO–tCnitro FRET (i.e., above 350 nm, Figure S1)28,39. Hence, aminoglycosides will not interfere with the energy transfer process of our FRET pair, and we therefore decided to employ them as test compounds in this work to validate our pre-miR-21 FRET-based ligand-binding assay, which utilizes a tCO–tCnitro RNA FRET pair28.
Small molecules contained in screening libraries can themselves be fluorescent or act as a quencher, leading to potential false results through interference with the tCO–tCnitro RNA FRET pair. However, this potential problem can be reduced by eliminating library compounds with larger conjugated aromatic systems. Furthermore, counter assays can be utilized to identify compounds that can potentially interfere with the detection method.
Results and discussion
FRET-labelled oligoribonucleotide design
Mature miRs do not contain secondary structural elements that allow specific and high-affinity binding by small molecules. Consequently, miR-regulating strategies utilizing small molecules are often based on pri- or pre-miR biogenesis. The pre-miRs are approximately 70 nt-long hairpins containing discrete secondary and tertiary structures that are susceptive to specific binding by drug-like molecules. An examination of the predicted secondary structure of the full-length pre-miR-21 hairpin (1, Table 1, Fig. 1a)40 and the available solution-NMR structure of the pre-miR-21 hairpin segment34 revealed that the majority of the secondary structures to which a ligand could potentially bind (mismatch bulges and a hairpin loop) were located in proximity to the hairpin loop and the functional Dicer processing site (Fig. 1a). From a chemical synthesis point of view, long, modified RNA sequences are challenging to prepare, which led us to truncate the full-length pre-miR-21 sequence to a 39 nt hairpin. We next added two extra GC pairs to mitigate end-fraying effects, yielding the 43 nt truncated pre-miR-21 oligoribonucleotide (2, Table 1). This construct was used as a starting point for the development of FRET-labelled oligoribonucleotides (5 and 7, Table 1, Fig. 1b,c) and corresponding donor-only oligoribonucleotides (6 and 8, Table 1) for our interbase-FRET assay. Because of this oligoribonucleotide design, our assay has the potential to probe structural changes within pre-miRs using a highly sensitive fluorescence-based readout (Fig. 1b,c).
We envisioned that by replacing a pair of C residues in oligoribonucleotide 2 with the tCO–tCnitro RNA FRET pair (Fig. 1d), we would be able to monitor changes in the FRET efficiency that arise from ligand binding (Fig. 1b,c). Furthermore, using sequences 5 and 7 in parallel (which report on different regions of the pre-miR-21) also enables us to identify where the ligand binding takes place. Based on previous work regarding the tCO–tCnitro inside the RNA duplexes28, we anticipated that inserting this FRET pair would have a minimal effect on the pre-miR-21 structure. The corresponding 5′-biotinylated sequences (3 and 4, Table 1), enabling attachment of the oligoribonucleotide to the streptavidin-coated surface, were prepared for immobilisation onto a streptavidin-coated SPR chip. Sequence 3 contains no donor or acceptor while sequence 4 contains the tCO-tCnitro FRET pair (Table 1) to verify if the incorporation of our labelles has an impact on ligand binding readout when using the SPR technique.
ITC and SPR interaction reference measurements
ITC was used to explore experimental conditions for the aminoglycoside neomycin binding to unmodified truncated pre-miR-21 (2, Table 1), including optimization of the buffer conditions. Ultimately, we employed a sodium cacodylate buffer (20 mM sodium cacodylate, 80 mM NaCl, 100 mM total Na+, pH 7.2) that was previously used in studies of aminoglycosides binding to the HIV-1 RNA dimerization initiation site41. This buffer resulted in excellent reproducibility of the ITC binding isotherms and a dissociation constant (Kd) of 5.2 ± 0.8 µM for the interaction between pre-miR-21 2 and neomycin (Fig. 2a,b).

Neomycin binding to pre-miR-21. (b) ITC data showing heat evolution upon titrating neomycin to oligoribonucleotide 2 (10 µM). (b) Integrated peak areas from panel (a) versus neomycin:2 molar ratio for the successive additions and the fitted curve (solid line, assuming a 1:1 binding) yielding a Kd of 5.2 ± 0.8 µM. (c) Neomycin binding to oligoribonucleotide 3 at 10 different concentrations using SPR, where the green marked region shows steady-state conditions from which Kd values are determined. (d) The fitted curve (solid line, assuming an equivalent affinity for all binding sites) from panel (c) yielding an average Kd of 3.9 ± 2.8 µM.
To investigate whether the tCO–tCnitro RNA FRET pair had any impact on the binding affinity between aminoglycosides and our FRET-labelled oligoribonucleotides, a comparative SPR measurement was performed using the biotinylated, FRET pair-containing pre-miR-21 (4, Table 1) and its FRET pair-free counterpart (3, Table 1). The Kd values of neomycin for sequences 3 and 4 were determined to be 3.9 ± 2.8 µM (Fig. 2c,d) and 2.7 ± 1.7 µM (Table 2), respectively. These results agree well with the ITC data (Fig. 2a,b). The difference in affinity of neomycin for 3 and 4 was minor and within the experimental error of the SPR measurements. This strongly suggests that the FRET pair does not perturb the overall secondary structure of the pre-miR-21 construct nor the ligand–pre-miR interaction.
Dissociation constants of the pre-miR-21-aminoglycoside interaction determined using SPR
To benchmark our interbase-FRET binding assay (vide infra), we investigated the binding affinities of a series of 12 aminoglycosides (Tables 2 and S2; see Figure S2 for structures) to pre-miR-21 3 and 4 using SPR. The obtained dissociation constants ranged from approximately 3 μM to 300 μM for both pre-miR-21 3 and 4, and these values were used to determine the SPR binding rank (Table 2). Because of the similar binding affinities for pre-miR-21 3 and 4, we concluded that the introduction of our FRET pair did not significantly alter the mode or strength of binding or the overall interaction between pre-miR and this type of ligand. Our results agree with a previous study by Yan et al. in which a fluorescence polarization assay was used to identify neomycin, netilmicin, sisomicin, and tobramycin as particularly potent binders to pre-miR-2119. Notably, the aminoglycosides exhibited a binding stoichiometry that varied between 4 and 5 depending on the aminoglycoside used, indicating that they bind non-specifically across pre-miR-21. Nearly identical binding stoichiometries were obtained for both 3 and 4 (unlabeled vs. FRET-labeled, respectively) (Table S3).
Interbase-FRET binding assay design
Our FRET-based assay for studying small-molecule binding to pre-miRs is based on evaluating the integrated emission of the FRET pair-labeled (tCO and tCnitro incorporated, donor–acceptor, DA) pre-miR sequence before and after addition of the ligand in relation to the same being performed for the FRET donor-containing (tCO incorporated, donor only, D) pre-miR sequence (see Materials and Methods Eqs. 1–4). The ligand is added to the FRET-labeled pre-miR, and the change in emission for the DA strand relative to the corresponding D strand is measured. The FRET efficiency is calculated as the fraction of the integrated emission from the DA and D sequences (Eq. 1). The difference in FRET before and after ligand addition (ΔFRET, Eq. 2) is then normalized between the two different ligand binding experiments (vide infra) to obtain ΔFRETnorm (Eq. 3, Tables S4 and S5). The average ΔFRETnorm for the two different sets of FRET-labeled oligoribonucleotides (averaged ΔFRETnorm, Eq. 4) is used to rank the ligands (FRET rank), ranging from potent to less potent binders for the screened set of small molecules (Tables 2 and S6).
Aminoglycoside affinity rank for pre-miR-21 determined by interbase-FRET assay
To obtain a significant change in FRET, the aminoglycosides (Table 2 and Figure S2) were added to sequences 5–8 (Table 1) at a concentration corresponding to a 90% degree of pre-miR-21 complexation. Higher degree of complexation would require non-feasible concentrations or volumes of the aminoglycoside stock solutions. The hairpin loop-labeled oligonucleotide (5, Table 1) showed a range of 2–5% with a 3% average increase in FRET efficiency (Figure S4), whereas the stem-labeled oligonucleotide (7, Figure S6) showed an average increase of 5% (except for hygromycin, which induced a decrease in FRET efficiency upon binding; Figure S6). For the donor-only control sequences (6 and 8, Table 1) small changes in FRET efficiency were observed (Figure S5 and S7), indicating that the local microenvironment of the donor changes upon the addition of aminoglycosides. Taken together, the results indicate that the aminoglycosides bind to the hairpin loop and to the stem parts of the FRET-labeled pre-miR constructs (5 and 7) and cause only small changes in the relative orientation/distance of the donor and acceptor. Furthermore, these results indicate that none of the aminoglycosides bind selectively to the hairpin loop or the stem parts of the labeled pre-miR constructs (5 and 7) but rather across the whole pre-miR. This is in agreement with the aminoglycoside:oligoribonucleotide binding ratio of 4–5:1 measured by SPR. It is also in line with work from the groups of Arenz and Maiti, showing that aminoglycoside antibiotics, such as kanamycin and streptomycin, generally inhibit Dicer-mediated miRNA processing by non-specific binding to the Dicer substrate, the pre-miR15,16.
The second set of ligand-binding experiments was designed to investigate whether our FRET-based assay could distinguish relative binding affinities of the aminoglycosides (Table 2, Figure S2). Furthermore, we wanted to investigate if ΔFRET for the aminoglycosides would correlate with the SPR affinity values.
Aminoglycosides were added to 1 µM FRET-labeled pre-miRs (5 and 7; Table 1) and their corresponding donor-only references (6 and 8; Table 1) at a total aminoglycoside concentration of 15 µM, chosen from SPR Kd as a middle-point between potent and non-potent binders (Table 2, Figure S8-S11). The data clearly show that high-affinity aminoglycosides such as neomycin (avg. SPR Kd = 3.3 µM, Table 2) caused a relatively large change in emission (avg. ΔFRET = 0.043 for 5 and 7, Table 2). By contrast, aminoglycosides with lower affinity such as streptomycin (avg. SPR Kd = 124 µM, Table 2) caused minimal changes in emission (avg. ΔFRET = 0.006 for 5 and 7, Table 2, for further details see Table 2). Overall, this experiment enabled us to distinguish the most potent binders with ΔFRET of 0.02–0.04 from the less potent binders with ΔFRET of 0.000–0.02.
By comparing the averaged ΔFRETnorm values based on both types of FRET experiments (adding aminoglycoside to reach a total concentration of 15 µM and adding aminoglycoside to reach a 90% degree of complexation; the latter representing the almost saturated binding event and hence a maximum change in FRET) and sorting them from high to low, the aminoglycosides were ranked based on FRET (Table 2, FRET Rank, Eq. 4). Overall, the FRET rank matched well with the SPR rank within a given class of ligand affinity (high, medium, and low), and the experimental errors of the two techniques were low. As in SPR, neomycin, sisomicin, and tobramycin were found to have the highest affinity for pre-miR-21 (avg. Kd < 10 µM, Table 2) using the FRET assay. Amikacin, gentamicin, apramycin, netilmicin, and kanamycin A all belong to the medium affinity group of pre-miR-21 ligands (avg. 15–45 µM, Table 2). A good correlation was observed when plotting the log Kd from SPR of 4 against the averaged ΔFRETnorm (R2 = 0.82, Figure S4). Finally, ribostamycin, geneticin, streptomycin, and hygromycin were found to be low-affinity binders in SPR (avg. Kd > 84 µM) and in our FRET assay. Thus, our interbase-FRET binding assay discriminates between high-, medium- and low-affinity binders equally well as the established SPR technique.
Conclusion
In summary, we present a novel pre-miR binding assay based on interbase-FRET between a fluorescent tCO donor and a non-fluorescent tCnitro acceptor that incorporates into a pre-miR hairpin. Measuring the FRET efficiency changes enabled determination of the binding affinities between an onco-miR precursor (pre-miR-21, used here as a model pre-miR) and aminoglycoside antibiotics as small-molecule ligands. Initially, we used SPR to prove that the introduction of our FRET pair does not perturb the binding affinity. Importantly, changes in the FRET efficiency in our new established FRET-based assay correlated well with the Kd ranking of the aminoglycoside binding to pre-miR-21, as determined by the established SPR technique. We envisage that our assay will be useful for investigating ligand binding, including drug molecules and peptides, to various pre-miRs for the modulation of miR biogenesis and disease pathway modulation. Considering the low ligand and pre-miR amounts required and the sensitivity of our method, this assay should be compatible with high-throughput screening. We are currently investigating site-selective pre-miR binders using our novel interbase-FRET assay. Additionally, to increase its versatility, we have already expanded the RNA interbase-FRET methodology to include modifications of adenine positions41.
Methods
Synthesis of RNA oligonucleotides
The pre-miR-21 and truncated pre-miR-21 oligoribonucleotide sequences used in this study are listed in Table 1. Sequences 3 and 5–8 were purchased from ATDBio Ltd, Southampton, UK and Eurogentec, Liège, Belgium, respectively. Oligoribonucleotides 5 and 7 contained a FRET pair, while 6 and 8 contained a FRET donor only. Oligoribonucleotides 2 and 4 were synthesized in-house; see the Supplementary Information for details on their synthesis and characterization.
General information on photophysical measurements and sample preparation
Sodium cacodylate buffer (20 mM sodium cacodylate, 80 mM NaCl, 100 mM total Na+, pH 7.2) was used for all measurements unless otherwise stated. Absorption spectra of the oligoribonucleotides were recorded between 200 and 550 nm on a Cary 4000 or Cary 5000 (Varian Technologies) at a scan rate of 600 nm/min and spectral bandwidth of 2.0 nm. The absorption at 260 nm was used to calculate the oligoribonucleotide concentration, and the molar absorptivity at 260 nm was taken as the linear combination of the molar absorptivities of the individual bases at this wavelength, multiplied by 0.9 to account for the effect of base stacking. The values used for the molar absorptivity of each base at 260 nm were: ε(T) = 9300 M–1 cm–1, ε(C) = 7400 M–1 cm–1, ε(G) = 11,800 M–1 cm–1, ε(A) = 15,300 M–1 cm–1, ε(tCO) = 11,000 M–1 cm–1, and ε(tCnitro) = 9700 M–1 cm–1. All spectroscopic measurements were carried out in a 0.3 cm pathlength quartz cuvette with a 45 µL chamber volume (Hellma). Pre-miR annealing was achieved by heating the solution rapidly (approximately 5–10 °C/min) to 90 °C, holding for 2.0 min, then cooling to 5.0 °C at approximately 1 °C/min. Aminoglycosides (Figure S2 in the Supporting Information) were purchased from various chemical vendors. Stock solutions (10 mM) were prepared gravimetrically and further diluted to the desired concentrations.
Isothermal titration calorimetry (ITC) measurements
ITC was performed on a MicroCal iTC200 from Malvern Panalytical. A 206 µL aliquot of 10 µM oligoribonucleotide was loaded into the sample cell of the ITC using a gastight syringe. Aminoglycoside (100–300 µM) was added to the ITC titration syringe, and the cell was kept at 25 °C. The initial delay time was set to 120 s and the stirring speed of the syringe to 750 rpm. In each addition, a volume of 2.0 μL was injected over the course of 2.0 s from the titration syringe into the cell. The total number of additions was 20, administered with 180 s intervals. The aminoglycoside–oligoribonucleotide binding data were corrected for the heat of ligand and oligoribonucleotide dilution by subtracting the average heat per injection of aminoglycoside to buffer and buffer to oligoribonucleotide, respectively. The Kd was determined using the Origin-based MicroCal iTC200 instrument software by non-linear curve fitting to a 1:1 binding model.
Surface plasmon resonance (SPR) measurements
Biotinylated oligoribonucleotide was immobilized on a streptavidin-coated biosensor chip (XanTec Bioanalytics GmbH). Capture levels were 300–500 response units. Each aminoglycoside sample was injected over the surface in a 10 concentration–response series. Sensorgrams were fitted with a steady-state model (region shown in Fig. 2c) using the SPR module of the Genedata Screener software package to obtain the Kd values and assuming equivalent affinity for all binding sites. The immobilization step was repeated to ensure a fresh surface prior to each run. The stoichiometry was obtained by dividing the observed maximum response (Rmax) by the theoretical Rmax. The theoretical Rmax was obtained from the molecular weight ratio for each aminoglycoside vs. oligoribonucleotide multiplied by the total immobilization level.
Fluorescence measurements and interbase-FRET binding assay
Steady-state fluorescence emission spectra were recorded on a Spex Fluorolog 3 (JY Horiba) with excitation at 365 nm and the excitation slit width set to 1.5 nm. The initial concentration of the oligoribonucleotide sample was 1.0 µM in all measurements, and the initial sample volume was 60 µL. Emissions were recorded between 380 and 700 nm at a scan rate of 600 nm min–1, with the emission slit width set to 5.0 nm. All spectra were recorded in duplicate, which were averaged prior to further evaluation.
For experiments where the aminoglycoside was added to reach a 90% degree of FRET-labeled oligoribonucleotide (5–8) complexation by the aminoglycoside, the dissociation constant obtained from SPR was used to calculate the required amount of aminoglycoside. SPR revealed a binding stoichiometry of 4–5:1 (aminoglycoside:oligoribonucleotide). However, owing to the simple binding dynamics observed (Fig. 2d), as well as nearly saturating the oligoribonucleotide with the aminoglycoside before measuring the emission spectrum, a 1:1 binding model could be used.
Reaching a 90% degree of FRET-labeled oligoribonucleotide (5–8) complexation by the aminoglycoside was achieved by preparing 60 µL of the desired oligoribonucleotide at a concentration of 1 µM in a reduced volume quartz cuvette and adding 5 µL of varying concentrations of aminoglycoside solution (Table S7). Emission spectra of the FRET-labeled oligoribonucleotide (5 and 7) were recorded and compared before and after ligand addition. This procedure was then repeated for the corresponding oligoribonucleotide containing only the FRET donor (6 and 8, Table 1).
For experiments where the aminoglycoside was added to reach a final concentration of 15 µM in the cuvette, 60 µL of 1 µM oligoribonucleotide was mixed with 1 µL of 915 µM aminoglycoside. An identical protocol to the one presented above was executed to obtain the emission spectra and calculate the FRET efficiency.
The averaged and normalized ΔFRET (averaged ΔFRETnorm) resulting in the ranking presented in Table 2 was calculated by Eqs. (1–4), shown below.
The FRET efficiency (E) between the donor and acceptor within an oligoribonucleotide was calculated according to Eq. (1):
where ID and IDA are the integrated (over wavelength) steady-state emission spectra for the donor only and donor–acceptor samples, respectively. The change in FRET efficiency (ΔFRET) was calculated according to Eq. (2):
where E0 and EL are the FRET efficiencies before and after addition of the ligand, respectively. The normalized change in FRET efficiency (ΔFRETnorm) was calculated according to Eq. (3):
where \({\Delta FRET}_{15\mathrm{ \mu M}}\) is the change in FRET efficiency upon adding 15 µM ligand, and \({\Delta FRET}_{90\%}\) is the change in FRET efficiency upon adding ligand at a concentration corresponding to a 90% degree of complexation (based on the Kd from SPR and assuming 1:1 binding). Finally, the \({\mathrm{Averaged }\Delta FRET}_{\mathrm{norm}}\) was calculated according to Eq. (4):
where \({\Delta FRET}_{\mathrm{norm},5}\) and \({\Delta FRET}_{\mathrm{norm},7}\) are the normalized change in FRET efficiencies for oligoribonucleotides pairs 5 and 7, respectively.
References
Li, Z. & Rana, T. M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 13, 622–638 (2014).
Kozomara, A. & Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68-73 (2014).
Hammond, S. M. An overview of microRNAs. Adv. Drug Deliv. Rev. 87, 3–14 (2015).
Bartel, D. P. MicroRNAs: Target recognition and regulatory functions. Cell 136, 215–233 (2009).
Di Leva, G., Garofalo, M. & Croce, C. M. MicroRNAs in cancer. Annu. Rev. Pathol. 9, 287–314 (2014).
Selcuklu, S. D., Donoghue, M. T. & Spillane, C. (2009) miR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 37, 918–925 (2009).
Wen, D., Danquah, M., Chaudhary, A. K. & Mahato, R. I. Small molecules targeting microRNA for cancer therapy: Promises and obstacles. J. Control. Release 219, 237–247 (2015).
Matsui, M. & Corey, D. R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 16, 167–179 (2016).
Davis, S., Lollo, B., Freier, S. & Esau, C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 34, 2294–2304 (2006).
Stenvang, J., Petri, A., Lindow, M., Obad, S. & Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 3, 1 (2012).
Juliano, R. L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548 (2016).
Costales, M. G., Childs-Disney, J. L., Haniff, H. S. & Disney, M. D. How we think about targeting RNA with small molecules. J. Med. Chem. 63, 8880–8900 (2020).
Chirayil, S., Chirayil, R. & Luebke, K. J. Discovering ligands for a microRNA precursor with peptoid microarrays. Nucleic Acids Res. 37, 5486–5497 (2009).
Connelly, C. M., Boer, R. E., Moon, M. H., Gareiss, P. & Schneekloth, J. S. Discovery of inhibitors of MicroRNA-21 processing using small molecule microarrays. ACS Chem. Biol. 12, 435–443 (2017).
Davies, B. P. & Arenz, C. A homogenous assay for Micro RNA maturation. Angew. Chem. Int. Ed. 45, 5550–5552 (2006).
Bose, D., Jayaraj, G. G., Kumar, S. & Maiti, S. A molecular-beacon-based screen for small molecule inhibitors of miRNA maturation. ACS Chem. Biol. 8, 930–938 (2013).
Vo, D. D. et al. Targeting the production of oncogenic MicroRNAs with multimodal synthetic small molecules. ACS Chem. Biol. 9, 711–721 (2014).
Tan, G. S. et al. Small molecule inhibition of RISC loading. ACS Chem. Biol. 7, 403–410 (2012).
Yan, H., Bhattarai, U., Guo, Z.-F. & Liang, F.-S. Regulating miRNA-21 biogenesis by bifunctional small molecules. J. Am. Chem. Soc. 139, 4987–4990 (2017).
Garner, A. L. cat-ELCCA: Catalyzing drug discovery through click chemistry. Chem. Commun. 54, 6531–6539 (2018).
Maiti, M., Nauwelaerts, K. & Herdewijn, P. Pre-microRNA binding aminoglycosides and antitumor drugs as inhibitors of Dicer catalyzed microRNA processing. Biorg. Med. Chem. Lett. 22, 1709–1711 (2012).
Garner, A. L. et al. Tetracyclines as inhibitors of pre-microRNA maturation: A disconnection between RNA binding and inhibition. ACS Med. Chem. Lett. 10, 816–821 (2019).
Hildebrandt, N. How to apply FRET: From experimental design to data analysis. In FRET—Förster Resonance Energy Transfer (eds Medintz, I. & Hildebrandt, N.) (Wiley, 2013).
Algar, W. R., Hildebrandt, N., Vogel, S. S. & Medintz, I. L. FRET as a biomolecular research tool—Understanding its potential while avoiding pitfalls. Nat. Methods 16, 815–829 (2019).
Park, J. E. et al. Dicer recognizes the 5’ end of RNA for efficient and accurate processing. Nature 475, 201–205 (2011).
Shajani, Z., Deka, P. & Varani, G. Decoding RNA motional codes. Trends Biochem. Sci 31, 421–424 (2006).
Bood, M., Sarangamath, S., Wranne, M. S., Grøtli, M. & Wilhelmsson, L. M. Fluorescent nucleobase analogues for base-base FRET in nucleic acids: synthesis, photophysics and applications. Beilstein J. Org. Chem. 14, 114–129 (2018).
Füchtbauer, A. F. et al. Interbase FRET in RNA: from A to Z. Nucleic Acids Res. 47, 9990–9997 (2019).
Krause, K. M., Serio, A. W., Kane, T. R. & Connolly, L. E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 6, a027029 (2016).
Thomas, J. R. & Hergenrother, P. J. Targeting RNA with small molecules. Chem. Rev. 108, 1171–1224 (2018).
Carter, A. P. et al. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407, 340–348 (2000).
Fourmy, D., Recht, M. I., Blanchard, S. C. & Puglisi, J. D. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 274, 1367–1371 (1996).
Xie, Y., Dix, A. V. & Tor, Y. FRET enabled real time detection of RNA-small molecule binding. J. Am. Chem. Soc. 131, 17605–17614 (2009).
Diaz, J. P. et al. Association of a peptoid ligand with the apical loop of pri-miR-21 inhibits cleavage by Drosha. RNA 20, 528–539 (2014).
Bose, D. et al. The tuberculosis drug streptomycin as a potential cancer therapeutic: Inhibition of miR-21 function by directly targeting its precursor. Angew. Chem. Int. Ed. Engl. 51, 1019–1023 (2012).
Shortridge, M. D. et al. A macrocyclic peptide ligand binds the oncogenic MicroRNA-21 precursor and suppresses dicer processing. ACS Chem. Biol. 12, 1611–1620 (2017).
François, B. et al. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 33, 5677–5690 (2005).
Lim, D., Byun, W. G., Koo, J. Y., Park, H. & Park, S. B. Discovery of a small-molecule inhibitor of protein–MicroRNA interaction using binding assay with a site-specifically labeled Lin28. J. Am. Chem. Soc. 138, 13630–13638 (2016).
Füchtbauer, A. F. et al. Fluorescent RNA cytosine analogue—An internal probe for detailed structure and dynamics investigations. Sci. Rep. 7, 2393 (2017).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Wypijewska del Nogal, A. et al. Getting DNA and RNA out of the dark with 2CNqA: a bright adenine analogue and interbase FRET donor. Nucleic Acids Res. 48, 7640–7652 (2020).
Acknowledgements
We thank Dr. Laurent Knerr (AstraZeneca, Gothenburg) for discussions on study design and choice of ligands, Dr. Anders Foller Füchtbauer (Department of Chemistry and Molecular Biology, University of Gothenburg) for fruitful discussions on FRET experiment design and SSL (AstraZeneca, Gothenburg) for assistance in setting up oligoribonucleotide purification protocols used herein. This work was supported by the Swedish Foundation for Strategic Research (SSF, grants No. ID14-0036, IS14-0041 and IRC15-0065) and the Swedish Research Council (VR, grant No. 2013-4375). This work is a part of a project that has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Actions Individual Fellowship (H2020-MSCA-IF-2016, grant agreement No. 753595 to A.W.d.N.).
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
RNA synthesis and purification were performed by M.B. The photophysical measurements were performed by M.B., A.W.d.N. and F.E., and were analyzed by M.B., A.W.d.N., F.E. and J.N. L.M.W., A.D., M.L. and M.G. supervised the project, analyzed data and contributed in the manuscript writing. M.B. primarily wrote the manuscript, but all authors contributed to the discussion of the results and commented on and adjusted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bood, M., del Nogal, A.W., Nilsson, J.R. et al. Interbase-FRET binding assay for pre-microRNAs. Sci Rep 11, 9396 (2021). https://doi.org/10.1038/s41598-021-88922-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88922-0
- Springer Nature Limited