
Abstract
Recurrent miscarriages occur in about 5% of couples trying to conceive. In the past decade, the products of miscarriage have been studied using array comparative genomic hybridization (a-CGH). Within the last decade, an association has been proposed between miscarriages and single or multigenic changes, introducing the possibility of detecting other underlying genetic factors by whole exome sequencing (WES). We performed a-CGH on the products of miscarriage from 1625 Iranian women in consanguineous or non-consanguineous marriages. WES was carried out on DNA extracted from the products of miscarriage from 20 Iranian women in consanguineous marriages and with earlier normal genetic testing. Using a-CGH, a statistically significant difference was detected between the frequency of imbalances in related vs. unrelated couples (P < 0.001). WES positively identified relevant alterations in 11 genes in 65% of cases. In 45% of cases, we were able to classify these variants as pathogenic or likely pathogenic, according to the American College of Medical Genetics and Genomics guidelines, while in the remainder, the variants were classified as of unknown significance. To the best of our knowledge, our study is the first to employ WES on the products of miscarriage in consanguineous families with recurrent miscarriages regardless of the presence of fetal abnormalities. We propose that WES can be helpful in making a diagnosis of lethal disorders in consanguineous couples after prior genetic testing.
Similar content being viewed by others


Introduction
According to the American Society for Reproductive Medicine (ASRM) report, recurrent pregnancy loss is defined as two or more miscarriages1 and occurs in about 5% of couples trying to conceive2. Both maternal and fetal causes can lead to pregnancy loss3. Numerical chromosome abnormalities are the most common cause of miscarriage, particularly under 13 weeks4,5. However, for the 40–50% of miscarriages with a normal karyotype (euploid miscarriage), the etiology is unknown and the genetic etiology is uncertain2. Karyotyping of products of miscarriage as a routine test has its limitations including culture failure rates (10–40%)6,7,8, and maternal cell contamination7,9,10, and resolution (usually below 5–10 Mb) is unable to detect minor genomic changes. In the past decade, the products of miscarriage have been studied using chromosomal microarray (CMA) technology (array comparative genomic hybridization (a-CGH))5,8,11,12,13 and clinically relevant copy number variants (CNVs) have been reported in about 1.6–1.8% of miscarriages. CMA overcomes most issues concerning the quality or quantity of fetal samples that have proved problematic in routine chromosomal study. CMA has its limitations including its inability to detect polyploidy, low-grade mosaicism and balanced rearrangements7.
In 2013, Larsen et al. proposed an association between miscarriage and single or multigenic changes14, introducing the possibility of being able to detect other underlying genetic factors by exome sequencing. Today, next-generation sequencing (NGS) is a crucial tool for pathogenic variants discovery in research and diagnostic settings15,16. Exome sequencing has a diagnostic yield of about 25–40% in patients with suspected Mendelian diseases in western populations17,18,19,20. Carss et al. performed exome sequencing on 30 non-aneuploid fetuses and neonates with diverse structural abnormalities detected by prenatal ultrasound. They identified candidate pathogenic variants and concluded that exome sequencing may substantially increase the detection rate of underlying etiologies of prenatal abnormalities. In 3 out of 30 fetuses, they found highly likely causative variants15. In 2018, Mengu Fu et al. performed exome sequencing on 19 products of miscarriage of unrelated couples and reported 36 rare variants associated with miscarriage21.
Studies of this kind have led to the use of NGS in prenatal diagnosis for the detection of pathogenic and causal genetic variants below the resolution of CMA15,22. The application of NGS in identifying the causes of lethal or abnormal prenatal development, including miscarriages, has been reported since 2012. An estimate of 30% of all mammalian genes are vital for life. Dickinson et al. (2016) identified 410 lethal genes in mice. In general, the diagnosis of lethal genes is challenging for the following reasons: the difficulties of phenotype–genotype correlation, the many potential genes, and the variable phenotypes associated with the same genetic causes23. There is controversy as to whether there is any correlation between recurrent pregnancy loss (RPL) and consanguinity. In studies conducted in the Middle East, where consanguineous marriage is culturally prevalent, some showed that the prevalence of RPL is higher in consanguineous couples. Kuntla et al. (2013) concluded that the occurrence of spontaneous miscarriages is higher among women in related vs. unrelated couples in India. In 2010, Rad conducted a study on recurrent miscarriage in Iran and concluded that the prevalence of RPL was higher in the consanguineous group24. On the other hand, in 2002, Saad and Jauniaux reported no association between consanguinity and recurrent miscarriage in Qatar and concluded that this finding could be because autosomal recessive alleles are uncommon in the Qatari population or because of the absence of any relationship between consanguinity and recurrent miscarriage25. In 2011, Gowri et al. reported that consanguinity does not appear to play a significant role in the etiology of recurrent miscarriage and is not related to recurrent miscarriage based on a study conducted in Oman26.
Since 2010, about 1625 cases with pregnancy loss (recurrent or single) have been studied in our center using quantitative fluorescent polymerase chain reaction (QF-PCR) and a-CGH, in which about 20% of clinically relevant CNVs and aneuploidies were detected. About 35% of the cases were from consanguineous couples. The inbreeding coefficients in consanguineous couples were between F = 0.125 and F = 0.0156. We realized that the detection rate in unrelated couples was higher. The difference between the frequency of imbalances in related vs. unrelated couples was significant (χ2 = 11.4926, P < 0.001)27. We proposed that it is plausible through the same mechanism by which single gene disorders have a higher prevalence of manifesting disease in consanguineous couples, they can cause lethal genetic disorders leading to pregnancy loss. As miscarriage is etiologically heterogeneous, the selection criteria for the evaluation of this postulate are very important. Whole exome sequencing (WES) on cases that present with a strong Mendelian inheritance pattern is more likely to be successful28. Here we have used WES to detect the causes of miscarriage in consanguineous Iranian families with recurrent miscarriages, in whom oligoarray CGH was normal and the maternal causes of miscarriage had been ruled out.
Materials and methods
Signed informed consent was obtained from couples for participation in this study and publication of data. The research was performed under the National Institute for Medical Research Development, Tehran, Iran (IR.NIMAD.REC.1396.355).
Subjects
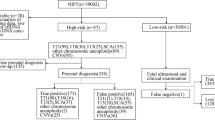
Twenty consanguineous Iranian couples were selected from a pool of 1625 couples in whom oligoarray CGH was normal and maternal causes of miscarriage had been ruled out by Kariminejad–Najmabadi Pathology & Genetics Center. The couples had a history of two or more pregnancy losses (RPL) and the results of previous a-CGH and QF-PCR were normal. Five of the cases had fetal autopsy reports.
The cases were selected from consanguineous couples with spontaneous loss of a pregnancy at less than 20 weeks of gestation and whose fetuses had sufficient quantity of DNA with appropriate quality. To minimize the effect of maternal causes of miscarriage, we chose families who had a history of one normal child, or one pregnancy beyond the second trimester or whose fetus showed evidence of fetal abnormalities (non-immune hydrops fetalis (NIHF), cardiac anomalies, etc.)29. None of the cases had induced abortion or prior abnormality detected in ultrasound. (Table 1).
All experimental protocols were approved by National Institute for Medical Research Development committee.
Whole exome sequencing
We used archival genomic DNA from the products of spontaneous miscarriage and extracted DNA from the peripheral blood of parents. All DNA was extracted using the conventional salting out method30. For fetuses or the products of miscarriage, DNA was extracted from chronic villi of the products of miscarriage and where fetal tissue was available, from the quadriceps muscle. Where possible, we saved some fetal tissue in a freezer at –80 °C. We checked the quality and quantity of DNA using a Nanodrop spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA) and gel electrophoresis. Samples were enriched using the Agilent SureSelectXT Human All Exon V6 platform (Agilent Technologies Inc, Santa Clara, CA, USA). Whole exome sequencing was performed using an Illumina HiSeq2000 platform (Illumina Inc., San Diego, CA, USA)31. All methods were carried out in accordance with ACMG guidelines and regulations.
Data analysis
Sequence reads were aligned to human GRCH37 using the Burrows–Wheeler Aligner (BWA) with the MEM algorithm32. Data processing and variant (SNPs and indels) discovery were performed according to the Genome Analysis Toolkit (GATK) best practices workflow33,34. Variant annotation was performed using ANNOVAR software35. To refine the list of causative variants, additional filtering was applied as follows. We kept exonic, splicing, UTR3 and UTR5 regions with the following criteria:
1. Rarely seen in populations (cut of 0.01) based on the Genome Aggregation Consortium (http://gnomad.broadinstitute.org/), 1000 genome project (http://www.internationalgenome.org/data), The Exome Aggregation Consortium (http://exac.broadinstitute.org/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) and Iranome databases (http://www.iranome.ir/)36,37,38,39.
2. Predicted to have functional consequences based on prediction scores such as SIFT (http://sift.bii.a-star.edu.sg/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org/), M-CAP (http://bejerano.stanford.edu/mcap/) and CADD (https://cadd.gs.washington.edu/)40,41,42,43,44,45,46,47,48.
We confined our analysis to the primary list of genes reported to be lethal in animal models, lethal genes from earlier articles, and lists of genes incorporated in NIHF and fetal development collected by examining earlier publications and databases. In cases where no candidate variants were found, we then used Tru Sight’s gene list for inherited diseases40 and finally, we used all Online Mendelian Inheritance in Man (OMIM) genes (https://omim.org/downloads/).
Confirmation of findings
All candidate variants, suspected by exome sequencing of DNA in fetuses from miscarriages, were confirmed by Sanger sequencing of the mutated point and flanking sequences in parents, using BIG Dye Terminators (Applied Biosystems 3130 Genetic Analyzer; Applied Biosystems, Foster City, CA, USA). The sequencing primers were designed using Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) and Oligoanalyzer 3.1 (http://eu.idtdna.com/analyzer/applications/oligoanalyzer/). The Sanger sequencing results were analyzed by CodonCode Aligner 5.0.1 (CodonCode Corp., Dedham, MA, USA).
Results
We collected DNA from the products of miscarriage from 20 Iranian women in consanguineous marriages and with a history of recurrent pregnancy loss. QF-PCR and a-CGH had previously been performed and reported as normal. None of the women had a specific diagnosis for pregnancy loss. As all cases were of loss of a pregnancy at less than 20 weeks of gestation, most (70%, n = 14) had no phenotype other than lethality. An autopsy was available in five cases and clinical data for these five cases are shown in Table 2. WES positively identified relevant alterations in 65% of cases (n = 13). In 45% (n = 9) of cases, we were able to classify these variants as pathogenic or likely pathogenic, while in the remaining 20% (n = 4), the candidate variants were classified as variants of unknown significance (VUS) according to the American College of Medical Genetics and Genomics guidelines (ACMG)49. In contrast, we were able to reach a precise diagnosis in all 5 cases (100%) with autopsies. In total, 13 rare variations including 2 de novo heterozygous, 9 inherited homozygous, and 2 inherited compound heterozygous variants were found in 11 genes; 2 genes were autosomal dominant (COL1A1, SCN5A). In silico predictions of the sequence variants are included in Supplementary Data I.
In the five cases with autopsies, pathogenic/likely pathogenic variants were identified in genes PIEZO1, POMT1, FRAS1, COL1A1 and DIS3l2 (Tables 2, 3 and 4).
-
1.
PIEZOs are large transmembrane ion channel proteins, and mutations in this gene have recently been reported to be a cause of hydrops fetalis50. PIEZO1 has a role in urine osmolarity regulation, blood pressure control and blood vessel formation50,51,52,53,54. Mutations of this gene are associated with two diseases: (1) dehydrated hereditary stomatocytosis with/without pseudohyperkalemia and/or perinatal edema (DHS; OMIM 194380); this is an autosomal dominant disease caused by gain of function mutations; (2) hereditary lymphedema III, the generalized lymphatic dysplasia of Fotiou with non-immune fetal hydrops (GLDF; OMIM 616843), an autosomal recessive disease caused by loss of function mutations50. In a non-immune hydrops fetalis sample (case 82169), we detected a homozygous c.30_31delAC alteration in PIEZO1. This variant is a frame shift mutation in the first exon that causes nonsense-mediated decay (NMD) in translated mRNA. In this case, the biallelic loss of function of PIEZO1 and the fetal phenotype give credence to GLDF. The parents were both carriers of this variant. According to ACMG, this variant is considered pathogenic.
-
2.
Mutations in FRAS1 are associated with Fraser syndrome, a rare autosomal recessive disorder. Thomas et al. (1986) proposed criteria for clinical diagnosis: major features are cryptophthalmos, syndactyly, siblings with cryptophthalmos and abnormal genitalia, and minor features include congenital malformations of the nose, ear and larynx, skeletal defects, umbilical hernia, renal agenesis and mental retardation. For diagnosis, two major or one major and four minor criteria are needed55. A male fetus (case 90377) with syndactyly, dysplastic ears, right kidney agenesis, club foot, flexion contracture of the hip, and atretic external auditory canals was diagnosed with a homozygous alteration in FRAS1 (c.404C > T). The parents were confirmed to be carriers of the inherited variant. This finding was considered likely pathogenic by ACMG guidelines.
-
3.
De novo variants in COL1A1 gene (17q21.33) were found in two families (Families 6 and 9): p.G1169S in exon 47 (case 93926), and p.E1249G in exon 48 (case 76452). Type I collagen is encoded by the COL1A1 and COL1A2 genes. Collagen I is the major protein in bone, skin, and other tissue. COL1A1 includes repeats of the Gly-X–Y triplet. Missense mutation of a conserved Gly generally leads to osteogenesis imperfecta (OI). OI is a group of skeletal dysplasia with clinical heterogeneity ranging from mild to lethal phenotype56. In case 93926 with mild rhizomelia, osteopenia, several fractures and a coronal cleft, and metaphyseal chondrodysplasia, a de novo mutation was detected in COL1A1 (c.3505G > A) (p.G1169S). This mutation has been reported in the HGMD database (CM070692). Residues that substitute for glycine cause either severely debilitating or lethal phenotypes, according to Bodian et al. (2008). In this case, it may be lethal56.
-
4.
A stop-gain variant found in exon 7 of POMT1 gene (mapped to chromosome 9q34.13) in Family 7 (case 91414) was predicted to be pathogenic by ACMG guidelines. POMT1 encodes protein O-mannosyltransferase and is associated with muscular dystrophy-dystroglycanopathy. POMT1 and protein O-mannosyltransferase 2 (POMT2) make up the protein O-mannosyltransferase (POMT) enzyme complex57. Both genes are necessary to have a functioning enzyme complex58. This complex is abundant in fetal brain, skeletal muscles and testes57. Case 91414 with four ventricular hydrocephaly, atrophic cortex, hypoplastic cerebellum, severe general cortical dysplasia type II (lissencephaly type II), cerebellar cortex dysplasia and clenched hands was found to have a homozygous variation in POMT1 (c.490C > T). This variation has been reported in the HGMD database (CM022978) and is a stop-gain resulting in NMD. Disruption of this gene in mice is lethal59. This variation was considered to be pathogenic.
-
5.
Another homozygous variant in intron 3 of DIS3L2 gene (2q37.1) in case 97406 (Family 13) was predicted to be likely pathogenic from ACMG guidelines. DIS3L2 is associated with Perlman syndrome (#267000), an autosomal recessive disorder. Perlman syndrome is a congenital overgrowth syndrome60 characterized by distinctive facies, visceromegaly, abdominal wall hypoplasia, bilateral renal hamartomas, nephroblastomatosis, Wilms tumor and neonatal lethality60,61. A 15-week fetus (case 97406) with limb atrophy, body weight at the 91 percentile, abdominal muscle agenesis, mild hydrocephaly, choroid plexus cysts, muscular and corticospinal tract atrophy , and kidney weight at the 99 percentile (observed to expected ratio of kidney weight to heart weight: 3.38, 1.99 in that order) was found to have a homozygous alteration in DIS3L2 (c.211-1G > A) which is predicted to disrupt the highly conserved acceptor splice site of axon 4. This alteration was classified as likely pathogenic according to ACMG guidelines.
In the other 15 cases, we have no phenotype other than lethality, and possible justifying variants were found in eight cases. (Table 3) In four cases, we could classify the variant as pathogenic/likely pathogenic. These variants were found in genes SCN5A, FUCA1, GBE1 and COL1A1 (Table 5).
-
1.
SCN5A encodes a membrane protein, which is an α-subunit of the predominant cardiac sodium channel isoform. This protein is responsible for the initial upstroke of the action potential in the heart. In case 94947, a homozygous alteration was detected in SCN5A (c.3749C > T). The parents were both carriers with a history of cardiac events in the family. This variant has been reported to cause long QT syndrome 3 (LQT3) (#603830) in the heterozygous state62,63.
Homozygous mutations in SCN5A in mice cause intrauterine lethality mostly during organogenesis due to heart defects64.
-
2.
FUCA1 expresses a lysosomal enzyme. Fucosidosis (#230000), an autosomal recessive disorder, is a lysosomal storage disease caused by homozygous or compound heterozygous mutations in FUCA1. Cardinal features are coarse facies, neurological signs, visceromegaly, intellectual disability and dysostosis multiplex. There are two types of fucosidosis: type 1 is more severe and signs are seen around 6 months of age with a lifespan of a decade; type 2 is milder with longer survival65. In case 90202, a homozygous mutation in FUCA1 was detected (c.404C > T). Both parents were carriers of this variant which was considered likely pathogenic.
-
3.
GBE1 encodes the glycogen branching enzyme. Mutations in this gene are associated with Glycogen storage disease IV (GSD IV) (#232500), an autosomal recessive metabolic disorder. GSD IV is a heterogeneous disease, which is known to have hepatic and neuromuscular features. The prenatal manifestations are fetal hydrops, polyhydramnios and decreased fetal movement66. Case 90759, a 13-week fetus with hydrops fetalis observed in ultrasonography, was found to have two heterozygous variants in GBE1 gene (trans): c.467G > A and c.-35_-54del GCTCAGGCCCCACTCGACCC.
-
4.
Case 76452 was the second case with a mutation in COL1A1 (c.3746A > G). Mutations in this gene are associated with several OMIM diseases: Caffey disease, OMIM #114000, autosomal dominant (AD); Ehlers–Danlos syndrome, arthrochalasia type, 1, OMIM #130060, AD; Osteogenesis imperfecta, type I, OMIM #166200, AD; Osteogenesis imperfecta, type II, OMIM #166210, AD; Osteogenesis imperfecta, type III, OMIM #259420, AD; Osteogenesis imperfecta, type IV, OMIM #166220, AD. This mutation is in exon 68 and does not affect the triplet repeats and Glu has been replaced by Gly at the amino acid level. It was classified as likely pathogenic.
In four cases, we found variations that are classified as variants of uncertain significance (VUS) because of the lack of phenotypic information. (Table 6) These variations were present in genes BBS12, STIL, COG6 and PIEZO1.
-
1.
A mutation in BBS12 causes Bardet-Biedl syndrome 12 (OMIM #615989). Bardet-Biedl is a heterogeneous ciliopathy disorder with an autosomal recessive pattern of inheritance. The BBS12 mutation is more common in the Iranian population than reported in other populations67. Case 84904 was found to have a homozygous mutation in BBS12 gene (c.2014G > A). Bardet-Biedl 12 is associated with obesity and mental impairment that cannot be detected in fetal stages. This alteration was considered to be of uncertain clinical significance.
-
2.
STIL is expressed in proliferating cells during early embryonic development and is necessary for mitotic spindle organization in the human cell cycle68,69,70,71. Alteration of this gene is associated with Primary microcephaly (#612703), an autosomal recessive disorder71. Izraeli et al. disrupted STIL in mice and the homozygous mutant caused death with neural tube defects, holoprosencephaly and left–right development abnormalities during embryonic development70. In case 93272, a homozygous mutation was present in STIL (c.1012C > T) and this was considered to be of uncertain clinical significance.
-
3.
Mutations in COG6 are associated with two diseases: congenital disorder of glycosylation III (#614576) and Shaheen syndrome (#615328). Shaheen syndrome is an autosomal recessive disorder characterized by intellectual disability and microcephaly. Congenital disorder of glycosylation III (CDG2L) is also autosomal recessive, characterized by intrauterine growth retardation, gastrointestinal abnormalities, infection and hematologic abnormalities. CDG2L usually results in death in infancy72. In case 94162, a homozygous mutation with uncertain clinical significance was detected in COG6 (c.1884 T > G).
-
4.
Case 95136 was found to have two mutations in PIEZO1 (c.C6584T and c.G2764T). As discussed earlier, mutations in this gene are associated with GLDF and DHS. In this case, we have no phenotype to correlate it with our findings and thus it was considered to be of uncertain clinical significance.
Discussion
Whole exome sequencing can detect the underlying genetic causes of disease in 25–40% of patients (pediatric and adult) with clinical indications such as congenital anomalies or developmental delay74. Before WES, array CGH and targeted genetic testing were performed in sporadic cases. The Middle East has the highest rate of consanguineous marriage globally due to socio-cultural factors. The prevalence of consanguineous marriage in Iran is about 38.6%75. Consanguineous marriage is preferred in Arab countries, particularly first cousin marriages with a rate of 20–50%76.
Since 2012, the possible benefit of WES for detection of the cause of miscarriage has been discussed and the results so far are promising29. A number of studies have been published with sample sizes of over 7 fetuses (7 to 84 cases) and these have reported a 10% to 54.5% detection rate of variants using WES in prenatal and fetal samples. In these studies, 30% to 70% of detected variants were autosomal recessive15,74,77,78,79,80,81,82,83. In our study, 84% of variants were autosomal recessive. These studies have variable inclusion criteria such as (1) single versus multiple anomalies or specific organ anomalies, or (2) singleton WES vs. trio WES. The highest diagnostic yield and pathogenic variants reported are in a study where fetuses with specific organ anomalies were studied83. In other studies, it was reported that trio WES increases the detection rate84,85. The advantages of trio WES have been reported in postnatal and prenatal studies. Trio WES allows reviewing variants with a higher probability of being pathogenic and detecting de novo variants83. In these studies, all fetuses had anomaly/anomalies and most cases were from unrelated couples.
To the best of our knowledge, our study is the first to employ WES on the products of miscarriage in consanguineous families with recurrent miscarriages regardless of fetal anomalies. The main difference between our study and others is in the inclusion criteria. We chose consanguineous families with spontaneous recurrent pregnancy loss (RPL) and studied the products of loss of a pregnancy at less than 20 weeks of gestation. In most cases, we had no phenotype other than lethality, and in 65% of cases, we found a possible variant. In only 45% of cases could we classify the variant as pathogenic/likely pathogenic according to ACMG guidelines. Since all fetuses were under 20 weeks gestation and some were under 10 weeks, there was no phenotype, and this made it more challenging to interpret the findings. Although all couples had RPL, we only had one product of miscarriage per family so segregation analysis was not possible due to lack of DNA samples from the other affected cases in each family. What makes interpretation of WES findings on the products of miscarriage more challenging is that Mendelian disorders in severe cases can be present as embryonic lethality, out of which many have no prenatal phenotype so mutations may not be suspected in fetuses85 (80).
WES on the products of miscarriage is helpful to verify lethal genes, and genes essential for embryonic development, and it expands our knowledge of prenatal phenotypes of many Mendelian disorders.
This study and others of this kind show that WES can assist in the diagnosis of the cause of miscarriage. Positive results and having a diagnosis can be useful in preconception genetic counselling for future successful pregnancies. Preimplantation genetic testing may be possible and the results may provide families with closure. After diagnosis, it is important to advise families on the risks of recurrence and their options for future pregnancies78,86. Identification of the cause of miscarriage will determine the risk for future pregnancies, and enable prenatal diagnosis or preimplantation genetic diagnosis for the given family. In addition, we will be able to identify lethal genes and their role in pregnancy loss. This and other studies of this kind will provide information that can assist in the assessment of repeated pregnancy loss.
Conclusion
WES can be helpful in making a diagnosis of lethal disorders (especially autosomal recessive disorders) in consanguineous couples after prior genetic testing (QF-PCR and a-CGH).
References
Medicine, P. C. o. t. A. S. f. R. Definitions of infertility and recurrent pregnancy loss: A committee opinion. Fertil. Steril. 99, 63 (2013).
Sierra, S. & Stephenson, M. in Seminars in Reproductive Medicine. 017-024 (Copyright© 2006 by Thieme Medical Publishers, Inc.).
Stephenson, M. & Kutteh, W. Evaluation and management of recurrent early pregnancy loss. Clin. Obstet. Gynecol. 50, 132–145 (2007).
Kolte, A. et al. Terminology for pregnancy loss prior to viability: a consensus statement from the ESHRE early pregnancy special interest group. Hum. Reprod. 30, 495–498 (2014).
Qiao, Y. et al. Whole exome sequencing in recurrent early pregnancy loss. MHR: Basic Sci. Reprod. Med. 22, 364–372 (2016).
Lomax, B. et al. Comparative genomic hybridization in combination with flow cytometry improves results of cytogenetic analysis of spontaneous abortions. Am. J. Hum. Genet. 66, 1516–1521 (2000).
Schaeffer, A. J. et al. Comparative genomic hybridization–array analysis enhances the detection of aneuploidies and submicroscopic imbalances in spontaneous miscarriages. Am. J. Hum. Genet. 74, 1168–1174 (2004).
Donaghue, C. et al. Efficient and cost-effective genetic analysis of products of conception and fetal tissues using a QF-PCR/array CGH strategy; five years of data. Mol. Cytogenet. 10, 12. https://doi.org/10.1186/s13039-017-0313-9 (2017).
Bell, K. A., Van Deerlin, P. G., Haddad, B. R. & Feinberg, R. F. Cytogenetic diagnosis of “normal 46, XX” karyotypes in spontaneous abortions frequently may be misleading. Fertil. Steril. 71, 334–341 (1999).
Robberecht, C., Schuddinck, V., Fryns, J.-P. & Vermeesch, J. R. Diagnosis of miscarriages by molecular karyotyping: Benefits and pitfalls. Genet. Med. 11, 646 (2009).
Rajcan-Separovic, E. et al. Identification of copy number variants in miscarriages from couples with idiopathic recurrent pregnancy loss. Hum. Reprod. 25, 2913–2922 (2010).
Rajcan-Separovic, E. et al. Genomic changes detected by array CGH in human embryos with developmental defects. MHR: Basic Sci. Reprod. Med. 16, 125–134 (2009).
Bagheri, H., Mercier, E., Qiao, Y., Stephenson, M. D. & Rajcan-Separovic, E. Genomic characteristics of miscarriage copy number variants. Mol. Hum. Reprod. 21, 655–661 (2015).
Larsen, E. C., Christiansen, O. B., Kolte, A. M. & Macklon, N. New insights into mechanisms behind miscarriage. BMC Med. 11, 154 (2013).
Carss, K. J. et al. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum. Mol. Genet. 23, 3269–3277 (2014).
Ku, C. S. et al. Exome sequencing: dual role as a discovery and diagnostic tool. Ann. Neurol. 71, 5–14 (2012).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511 (2013).
Sawyer, S. et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: Time to address gaps in care. Clin. Genet. 89, 275–284 (2016).
Retterer, K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696 (2016).
Matos, C. M., Alonso, I. & Leão, M. Diagnostic yield of next-generation sequencing applied to neurological disorders. J. Clin. Neurosci. (2019).
Fu, M. et al. Whole-exome sequencing analysis of products of conception identifies novel mutations associated with missed abortion. Mol. Med. Rep. 18, 2027–2032 (2018).
Talkowski, M. E. et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N. Engl. J. Med. 367, 2226–2232 (2012).
Ellard, S. et al. An exome sequencing strategy to diagnose lethal autosomal recessive disorders. Eur. J. Hum. Genet. 23, 401 (2015).
Rad, I. A. The impact of consanguinity on fetal loss. Med. J. Islamic World Acad. Sci. 18, 151–154 (2010).
Saad, F. A. & Jauniaux, E. Recurrent early pregnancy loss and consanguinity. Reprod. Biomed. Online 5, 167–170 (2002).
Gowri, V., Udayakumar, A. M., Bsiso, W., Al Farsi, Y. & Rao, K. Recurrent early pregnancy loss and consanguinity in Omani couples. Acta Obstet. Gynecol. Scand. 90, 1167–1169 (2011).
Najafi, K. et al. Chromosomal aberrations in pregnancy and fetal loss: Insight on the effect of consanguinity, review of 1625 cases. e820 (2019).
Filges, I. & Friedman, J. M. Exome sequencing for gene discovery in lethal fetal disorders–harnessing the value of extreme phenotypes. Prenat. Diagn. 35, 1005–1009 (2015).
Shamseldin, H. E., Swaid, A. & Alkuraya, F. S. Lifting the lid on unborn lethal Mendelian phenotypes through exome sequencing. Genet. Med. 15, 307–309. https://doi.org/10.1038/gim.2012.130 (2013).
Miller, S., Dykes, D. & Polesky, H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215 (1988).
Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Esteghamati, A. et al. Trends of diabetes according to body mass index levels in Iran: results of the national Surveys of Risk Factors of Non-Communicable Diseases (1999–2007). Diabet. Med. 27, 1233–1240 (2010).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010).
Van der Auwera, G. A. et al. From FastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11.10. 11–11.10. 33 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Consortium, G. P. A global reference for human genetic variation. Nature 526, 68 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285 (2016).
Esmaeli, B. & Sniegowski, M. in Orbital Tumors 145–153 (Springer, 2015).
Akbari, M.R.F.Z., Beheshtian, M., Mohseni, M., Poustchi, H., Sellars, E., Nezhadi, H., Amini, A., Arzhangi, S., Jalalvand, K., Jamali, P., Davarnia, B., Nikuei, P., Oladnabi, M., Mohammadzadeh, A., Zohrehvand, E., Shamsi-Gooshki, E., Börno, S., Timmermann, B., Najafipour, R., Khorram Khorshid, H.R., Kahrizi, K., Najmabadi, H. A human genome variation database of eight major ethnic groups that live in Iran and neighboring countries in the Middle East. in ASHG Annual Meeting (2017).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073 (2009).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Ng, P. C. & Henikoff, S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 12, 436–446 (2002).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Ng, P. C. & Henikoff, S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 7, 61–80 (2006).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11, 361 (2014).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310 (2014).
Jagadeesh, K. A. et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48, 1581 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405 (2015).
Martin-Almedina, S., Mansour, S. & Ostergaard, P. Human phenotypes caused by PIEZO1 mutations; one gene, two overlapping phenotypes?. J. Physiol. 596, 985–992 (2018).
Martins, J. R. et al. Piezo1-dependent regulation of urinary osmolarity. Pflügers Archiv-Eur. J. Physiol. 468, 1197–1206 (2016).
Wang, S. et al. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 126, 4527–4536 (2016).
Li, J. et al. Piezo1 integration of vascular architecture with physiological force. Nature 515, 279 (2014).
Ranade, S. S. et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. 111, 10347–10352 (2014).
Thomas, I. et al. Isolated and syndromic cryptophthalmos. Am. J. Med. Genet. 25, 85–98 (1986).
Bodian, D. L., Madhan, B., Brodsky, B. & Klein, T. E. Predicting the clinical lethality of osteogenesis imperfecta from collagen glycine mutations. Biochemistry 47, 5424–5432 (2008).
Health, U. N. I. o. (2012).
Manya, H. et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. 101, 500–505 (2004).
Willer, T. et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc. Natl. Acad. Sci. 101, 14126–14131 (2004).
Astuti, D. et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat. Genet. 44, 277 (2012).
Alessandri, J. L. et al. Perlman syndrome: report, prenatal findings and review. Am. J. Med. Genet. A 146, 2532–2537 (2008).
Wattanasirichaigoon, D. et al. Sodium channel abnormalities are infrequent in patients with long QT syndrome: Identification of two novel SCN5A mutations. Am. J. Med. Genet. 86, 470–476 (1999).
Kapplinger, J. D. et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Heart Rhythm 6, 1297–1303 (2009).
Papadatos, G. A. et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. 99, 6210–6215 (2002).
Kousseff, B. G. et al. Fucosidosis type 2. Pediatrics 57, 205–213 (1976).
Alegria, A. et al. Glycogen storage disease type IV presenting as hydrops fetalis. J. Inherit. Metab. Dis. 22, 330–332 (1999).
Fattahi, Z. et al. Mutation profile of BBS genes in Iranian patients with Bardet-Biedl syndrome: genetic characterization and report of nine novel mutations in five BBS genes. J. Hum. Genet. 59, 368 (2014).
Pfaff, K. L. et al. The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Mol. Cell. Biol. 27, 5887–5897 (2007).
Izraeli, S. & Colaizzo-Anas, T. Expression of the SIL gene is correlated with growth induction and cellular proliferation. Leukemia 3, 4 (1997).
Izraeli, S. et al. The SIL gene is required for mouse embryonic axial development and left–right specification. Nature 399, 691 (1999).
Kumar, A., Girimaji, S. C., Duvvari, M. R. & Blanton, S. H. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am. J. Hum. Genet. 84, 286–290 (2009).
Rymen, D. et al. Key features and clinical variability of COG6-CDG. Mol. Genet. Metab. 116, 163–170 (2015).
Shamseldin, H. E. et al. Molecular autopsy in maternal–fetal medicine. Genet. Med. 20, 420 (2018).
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879. https://doi.org/10.1001/jama.2014.14601 (2014).
Saadat, M., Ansari-Lari, M. & Farhud, D. J. A. o. h. b. Short report consanguineous marriage in Iran. 31, 263–269 (2004).
Tadmouri, G. O. et al. Consanguinity and reproductive health among Arabs. Reprod. Health. 6, 17 (2009).
Drury, S. et al. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat. Diagn. 35, 1010–1017. https://doi.org/10.1002/pd.4675 (2015).
Alamillo, C. L. et al. Exome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies. Prenat. Diagn. 35, 1073–1078. https://doi.org/10.1002/pd.4648 (2015).
Vora, N. L. et al. Prenatal exome sequencing in anomalous fetuses: New opportunities and challenges. Genet. Med. 19, 1207–1216. https://doi.org/10.1038/gim.2017.33 (2017).
Wapner, R. et al. 8: Whole exome sequencing in the evaluation of fetal structural anomalies: A prospective study of sequential patients. Am. J. Obstet. Gynecol. 216, S5–S6 (2017).
McMullan, D., Eberhardt, R. & Rinck, G. Exome Sequencing of 406 Parental/Fetal Trios with Structural Abnormalities Revealed By Ultrasound in the UK Prenatal Assessment of Genomes and Exomes (PAGE) Project (European Society of Human Genetics, 2017).
Yadava, S. M. & Ashkinadze, E. 125: Whole exome sequencing (WES) in prenatal diagnosis for carefully selected cases. Am. J. Obstet. Gynecol. 216, S87–S88 (2017).
Yates, C. L. et al. Whole-exome sequencing on deceased fetuses with ultrasound anomalies: Expanding our knowledge of genetic disease during fetal development. Genet. Med. 19, 1171–1178. https://doi.org/10.1038/gim.2017.31 (2017).
Best, S. et al. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat. Diagn. 38, 10–19 (2018).
Jelin, A. C. & Vora, N. Whole exome sequencing: Applications in prenatal genetics. Obstet. Gynecol. Clin. N. Am. 45, 69–81 (2018).
Soden, S. E. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Translat. Med. 6, 265ra168–265ra168 (2014).
Funding
This study was funded by the National Institute for Medical Research Development (Grant Number: 963279).
Author information
Authors and Affiliations
Contributions
K.N. analyzed data, gathered cases and wrote part of the text. Z.M. analyzed data. F.A. performed sanger sequencing and DNA preparation. S.G. autopsied the fetuses. A.K. genotype–phenotype correlation and consulted patients. R.K. prepared the experimental setup and wrote a part of the text. H.N. devised and supervised the project, and wrote part of the text.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Najafi, K., Mehrjoo, Z., Ardalani, F. et al. Identifying the causes of recurrent pregnancy loss in consanguineous couples using whole exome sequencing on the products of miscarriage with no chromosomal abnormalities. Sci Rep 11, 6952 (2021). https://doi.org/10.1038/s41598-021-86309-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86309-9
- Springer Nature Limited
This article is cited by
-
Miscarriage risk assessment: a bioinformatic approach to identifying candidate lethal genes and variants
Human Genetics (2024)
-
Towards solving the genetic diagnosis odyssey in Iranian patients with congenital anomalies
European Journal of Human Genetics (2024)
-
Evaluation of platelet parameters, coagulation markers, antiphospholipid syndrome, and thyroid function in palestinian women with recurrent pregnancy loss
BMC Pregnancy and Childbirth (2023)
-
Essential genes: a cross-species perspective
Mammalian Genome (2023)