
Abstract
High night temperatures (HNT) are shown to significantly reduce rice (Oryza sativa L.) yield and quality. A better understanding of the genetic architecture of HNT tolerance will help rice breeders to develop varieties adapted to future warmer climates. In this study, a diverse indica rice panel displayed a wide range of phenotypic variability in yield and quality traits under control night (24 °C) and higher night (29 °C) temperatures. Genome-wide association analysis revealed 38 genetic loci associated across treatments (18 for control and 20 for HNT). Nineteen loci were detected with the relative changes in the traits between control and HNT. Positive phenotypic correlations and co-located genetic loci with previously cloned grain size genes revealed common genetic regulation between control and HNT, particularly grain size. Network-based predictive models prioritized 20 causal genes at the genetic loci based on known gene/s expression under HNT in rice. Our study provides important insights for future candidate gene validation and molecular marker development to enhance HNT tolerance in rice. Integrated physiological, genomic, and gene network-informed approaches indicate that the candidate genes for stay-green trait may be relevant to minimizing HNT-induced yield and quality losses during grain filling in rice by optimizing source-sink relationships.
Similar content being viewed by others

Introduction
Rice is a major source of calories for more than 50% of the world’s population and plays a key role in sustaining food security in Asia1, Africa, and the Americas2. Current global climate models predict a mean temperature increase by 1.0–3.7 °C by 21003,4, which would potentially increase the occurrence of heat stress events in rice-growing tropical and subtropical countries. Disaggregating the predicted mean temperature increase has allowed unraveling a rapid rise in night temperature compared to the maximum day temperature at the farm5,6, region7,8, and global9 scale. A rapid rise in night temperature is fast becoming a significant challenge for sustaining crop yields, including rice5,10,11,12,13, wheat (Triticum aestivum)14,15 and barley (Hordeum vulgare)16,17. Previous studies have shown that an average season-long increase in minimum night temperature by 1 °C reduced tropical irrigated rice (Oryza sativa L.) grain yield by 10%5. Additionally, field-based studies document the susceptibility of rice inbred cultivars or commercial hybrids exposed to HNT (28 or 29 °C), leading to an average grain yield reduction of ~ 15%13,18 to 90% when exposed to severe HNT (32 °C) under controlled environments19. An increase in night temperatures have resulted in a negative impact on grain yield and quality in the major rice-growing regions of South and Southeast Asia5,7 and the United States20. Therefore, breeding programs need to focus on enhancing high night temperature (HNT) stress tolerance in rice to minimize yield and grain quality losses.
Unlike the pre-flowering phase, the post-flowering phase is most affected by HNT, wherein the rate of senescence is advanced leading to lower grain-filling duration, resulting in lower grain yield and quality13,14. In response to high day temperature (HDT), rice can benefit from early morning flowering mechanism to escape or effectively employ transpiration cooling to avoid heat stress during the day21,22. However, rice is not equipped with similar physiological processes that can minimize the negative impact of HNT22,23,24. Interestingly, the physiological routes through which HDT and HNT stress induce yield losses are shown to differ, although both the stresses finally result in significant yield and quality losses22. Experiments under controlled environments25,26,27,28,29 and field conditions30,31 have convincingly demonstrated that HDT stress negatively impacts reproductive processs in rice, leading to spikelet sterility. However, induction of HNT from panicle initiation until physiological maturity under field conditions did not induce a significant reduction in spikelet sterility and had no loss in seed number11. The same response was captured even with different levels of nitrogen application11,32 and across a range of inbreds and hybrids13,18,33, providing strong support for the differential route through which HNT induced yield and quality losses in rice. These results make a strong case that progress achieved through genetic mapping for HDT stress in rice may not be directly translated to induce HNT tolerance in rice22.
Rice exposed to HNT stress during the grain-filling stage significantly reduces whole plant and panicle carbohydrate content due to increased night respiration (carbon loss) resulting in lower biomass, lower grain yield (due to lower 100-seed weight), and enhanced chalkiness11,13,15,34. Grain yield and quality, specifically chalkiness, are key traits that drive market value and are considered significant aspects for assessing HNT impact in rice10,13. Despite the increasing knowledge of the metabolomic35 and transcriptomic36,37 responses of rice to HNT, our understanding of the genetic basis of rice response to HNT tolerance is mostly unexplored. Hence, to the best of our knowledge, this is the first study to identify genetic loci and putative candidate genes controlling HNT response in rice that will help in developing HNT tolerant varieties and hybrids for current and future warmer climates.
To fill the above knowledge gaps, an indica panel was phenotyped for grain yield, 100-seed weight, harvest index, grain size and chalkiness, under HNT using controlled environment walk-in chambers and field-based heat tents. The diversity panel consisting of improved and traditional genotypes with sub/tropical adaptation was phenotyped to (1) identify genomic regions associated with phenotypic traits in control and HNT in controlled environments and field conditions; (2) compare the extent of co-localization of the genetic loci across treatments under controlled environment and field conditions, and (3) identify putative candidate genes associated with enhanced HNT tolerance in rice using gene network analysis.
Materials and methods
Plant materials
The indica rice diversity panel (PRAY—Phenomics of Rice Adaptation and Yield) involving cultivars from tropical and subtropical rice-growing regions were assembled as a part of the Global Rice Phenotyping Network (GRPN, http://ricephenonetwork.irri.org). A sub-set of two-hundred and nine genotypes was chosen based on the uniformity in flowering phenology, using data generated under the International Rice Research Institute (IRRI) farm conditions38. Two independent experiments were conducted during 2015 (Experiment 1—controlled environment facility) and 2016 (Experiment 2—field-based heat tents) at IRRI, Los Baños (14° 11′ N, 121° 15′ E, 21 masl), Philippines. In Exp. 1, the 209 genotypes with 3 replicates and two treatments resulted in 1254 pots. In Exp. 2, the same 209 genotypes with each grown in a row (each row having 15 plants) and two treatments resulted in about 6270 plants (Supplementary Fig. S1). Plants in both experiments were grown under fully flooded conditions to study the effect of HNT stress starting from panicle initiation until physiological maturity. Unlike short episodes of high day heat stress, HNT is acknowledged to persist for longer growth periods encompassing different developmental stages, leading to yield and quality losses11,13. Previous studies using multiple genotypes with HNT stress imposed in the same field-based tents concluded that the impact of HNT was significant when coincided with panicle initiation and lasting until physiological maturity, but not during the early vegetative stage33.
Experiment 1 (walk-in chamber facility)
Crop management
Seed dormancy of rice genotypes was broken by exposing them to 50 °C for three days and were then hand sown in seeding trays. Fourteen-day-old seedlings were transplanted into plastic pots (25 cm in height; 26 and 20 cm diameter at the top and bottom, respectively), filled with 6 kg of clay loam farm soil and 2.0 g ammonium sulfate [(NH4)2SO4], 1.0 g single superphosphate (SSP) and 1.0 g muriate of potash (KCl). An additional 2.5 g (NH4)2SO4 was top-dressed at 30 days after transplanting. One plant per pot was grown under fully flooded conditions and was grown in the greenhouse conditions before imposing stress at the targeted stages (panicle initiation until physiological maturity). The 209 genotypes were grouped into five batches based on flowering time information from Kadam et al.38. The five batches were stagger sown with a 5-day interval between each batch (Fig. 1a) and transplanted with the same intervals to ensure best possible synchrony in panicle initiation and flowering. Representative plants for each batch were monitored and used to determine the panicle initiation stage by destructive inspection39.

Synchronization of the reproductive stage by stagger sowing (A) for phenotyping indica panel under HNT stress (B) in walk-in chambers (Exp. 1) and field-based heat tents (Exp. 2). DFF days to 50% flowering, S seeding, T transplanting, PI panicle initiation, FL flowering, GF grain filling, PM physiological maturity. Amplitude is calculated as a difference between average day temperature and average night temperature for each treatment separately. VPD vapor pressure deficit, RH relative humidity and SD standard deviation. Schematic representation of materials and methods followed to impose HNT stress is visualized in Supplementary Fig. S1.
Walk-in chambers and HNT stress imposition
Six walk-in chambers, each with a total area of 10.6 m2 (3.3 m [length] × 3.2 m [width] × 2.7 m [height]) were used in the study (Supplementary Fig. S1a). Each chamber was fitted with six independent units of 1 kW high-intensity discharge lamps, providing a photosynthetic photon flux density (PPFD) of ~ 650 µmol m−2 s−1 at 1.2 m from the base of the chamber. A photoperiod of 11 h (13 h of dark) and a constant relative humidity of ~ 70% was maintained throughout the stress period. The HNT temperature in the chambers was maintained at 29 °C for 11 h during the night (18:30 to 05:30 h), with an optimum of 31 °C during the day (06:30 to 17:30 h) starting from panicle initiation until physiological maturity. A linear change in temperature from day to night and vice versa was programmed between 17:30 and 18:30 h; and 05:30 and 06:30 h. MINCERs (Micrometeorological Instrument for Near Canopy Environment of Rice, developed by the National Institute of Agrobiological Sciences, Japan) was placed in the middle of the chambers with temperature sensors above the plant canopy. MINCERs equipped with data loggers (RX-350TH, As One Co., Osaka, Japan) were used to record ambient air temperature and relative humidity at 15 min intervals both in the greenhouse and in the walk-in chambers. Three replicate plants for each genotype were transferred into three different HNT chambers at panicle initiation. Simultaneously, a similar number of plants (three plants per genotype) were maintained under greenhouse conditions throughout the crop growth cycle and considered as absolute controls. In the Exp. 1, temperatures in control (greenhouse) and HNT treatments (walk-in chambers) were 26.6 °C (standard deviation, SD = 0.4) and 29.3 °C (SD = 0.61 °C), respectively. The relative humidity during the treatment period was 82.2% (SD = 1.2%) in the control treatment, 80.5% (SD = 1.3%) in the HNT treatment (controlled environment experiment; Fig. 1b). Amplitude (temperature difference between day and night) during the HNT stress treatment was 2.3 °C higher during the night (Fig. 1b).
Experiment 2 (Field-based heat tents)
Crop management
This experiment was carried out during the dry season (January to May of 2016) at the Experimental Farm, IRRI. The pre-germinated seeds of 209 cultivars were staggered sown [similar to Exp. 1] in a seeding tray filled with the same clay loam farm soil and fourteen-day-old seedlings were hand transplanted at a spacing of 20 × 20 cm. Each row had 15 plants (rows were not replicated, due to limitation in space), in an augmented design. All the genotypes were grown under fully flooded conditions, with about 2 cm of standing water at transplanting and about 5 cm from crop establishment until physiological maturity. Nitrogen (N), phosphorus (P), potassium (K) [150:30:40, N:P:K kg ha−1] and micronutrient zinc were applied as urea (150 kg ha−1), single superphosphate (30 kg ha−1) and muriate of potash (40 kg ha−1) along with zinc sulfate heptahydrate (5 kg ha−1). All the fertilizers were incorporated into the plots as a basal dose (1 day before transplanting) except for urea, which was applied in four splits of 3:2:3:2 at basal, mid-tillering, panicle initiation, and heading stage, respectively. All the agronomic and crop management practices were followed, similar to Shi et al.11 and Bahuguna et al.13.
HNT stress using field-based tents
All genotypes were exposed to two-night temperature treatments i.e., control night (CNT; 24 °C) and high night temperatures (HNT; 29 °C) with a common day-time temperature across both treatments. Plants were exposed to 24 °C (CNT) or 29 °C (HNT) night temperature by using field-based tents between (18:00 to 06:00 h) and during the day-time (06:00 to18:00 h) the tents were open, exposing the plants to natural conditions [Supplementary Fig. S1]. To start the HNT stress, the sides of the tents were manually rolled down at 18:00 h every evening and re-opened at 6:00 h in the morning, exposing the plants to12 h of photoperiod. Each tent was outfitted with an air conditioner, which was programmed to automatically regulate control (24 °C) and stress (29 °C) temperatures at night. Heat tents were equipped with temperature and relative humidity sensors and were connected to data loggers (HOBO, Onset Computer Corp., Bourne, MA). These were fixed in each tent above the canopy to record environmental conditions at 15-min intervals. Average night (18:00–06:00 h) temperature during stress treatment (from panicle initiation until maturity) was 24.3 °C (SD = 0.7) for the control and 28.3 °C (SD = 0.53) with HNT in Exp. 2 (Field experiment; Fig. 1b). Amplitude (the difference between day and night) temperatures during the stress treatment was 5.8 °C (Fig. 1b).
Phenotypes and statistical analysis
At physiological maturity, about three (Exp. 1) to twelve plants (Exp. 2) were harvested separately to determine the impact of HNT on the grain yield per plant (YPP) and 100-grain weight (HWT), harvest index (HI), and chalkiness (CHALK). Panicles were hand-threshed to estimate grain yield per plant and 100-grain weight. The harvest index was calculated as the ratio of grain yield to the plant's total dry matter. In Exp. 2, representative samples from each genotype were analyzed for grain size and grain chalkiness at the Grain Quality and Nutrition Center, IRRI, Philippines, following the ISO 17025 certified protocols of the IRRI GQNSL (http://www.knowledgebank.irri.org/ricebreedingcourse/bodydefault.htm#Grain_quality.htm). All phenotypic traits were subjected to statistical analysis to detect the effects of genotype and treatment using the library ("agricolae") RStudio 3.6.1 (https://rstudio.com/). Least square difference (LSD) was used to compare the mean values differences between control and HNT. Pearson’s correlation coefficient was carried out between all traits using the library ("corrplot") in RStudio 3.6.1. The relative performance of a genotype under stress was determined using the following equation.
Genome-wide association analysis (GWAS)
One hundred and seventy-three genotypes in Exp. 1 (panicles did not emerge in 36 genotypes under HNT), and 206 genotypes in Exp. 2 had complete phenotypic data. However, the genotypic data for twenty genotypes were missing, and hence only those genotypes that had both genotypic and phenotypic data (158 genotypes in Exp. 1 and 188 in Exp. 2) were used for GWAS analysis. GWAS was performed to estimate the association between marker-trait using the Genomic Association and Prediction Integrated Tool40. A mixed-model approach was implemented in the Compressed Mixed-Model Association (CMLM), which controls the familial relatedness and population structure and kinship (as a random effect to control population structure) to minimize false-positive associations41. A total of 45, 200 single nucleotide polymorphisms (SNPs; minor allele frequency > 0.05) that were generated under the Global Rice Phenotyping Network (http://ricephenonetwork.irri.org) were used for performing GWAS38,42,43. Estimated absolute phenotypic or relative values along with PC and Kinship were used to discover marker-trait associations (MTAs). The Bonferroni correction had a more stringent threshold; therefore it did not result in many significant associations, so we have used a significant (–log(p) = 3.5) threshold to discover MTAs similar to several previous studies42,44. Manhattan plots were constructed using the library ("qqman") in RStudio 3.6.1. (https://cran.r-project.org/web/packages/qqman/vignettes/qqman.html). The SNP with the lowest p-value within 100 kb is considered as a candidate marker-trait association (cMTA) to represent that locus. Thus, the SNP with minimum p‐value for each locus was defined as cMTA. Then we compared cMTAs/peak SNP to detect shared loci and unique loci across treatments and experiments. Candidate genes were searched in ± 50 kb from the cMTAs in rap-db on IRGSP-1.0 (http://rapdb.dna.affrc.go.jp/).
Prioritizing gene candidates
Part of this study was aimed to reduce the inherent bias in selecting candidate genes based on existing literature by incorporating alternate ways to identify the top genes of interest45. To the 638 candidate genes identified in the 100-kb window surrounding the unique 51 cMTAs, three potential gene prioritization approaches were applied: (1) Comparison to genes previously identified as responsive to HNT via differential expression analysis, (2) identification of genes highly connected to transcriptional regulators through a gene regulatory network, and (3) genes orthologous to genes previously identified as associated with traits through QTG-Finder246. These three comparisons integrate data from different experimental approaches to narrow down a list of potential candidate genes.
Prioritization based on differential expression analysis
This approach used altered expression in response to HNT as a filter to prioritize candidate genes. A prior experiment performed RNA-Sequencing analysis on the O. sativa genotype IR64, grown under similar experimental conditions at IRRI, Philippines (data accessible at NCBI GEO database37, accession GSE159073). RNA-Seq data from this prior time course study from flag-leaf tissue was normalized, mapped, and expression count per gene obtained as described in Desai et al.37. Only 354 genes, 223 in the genes identified from the absolute analysis and 131 from the relative analysis, were in the candidate gene list from trait loci and had detectible expression levels in the initial transcriptomic analysis. The expression data were analyzed using the R package edgeR47 to identify genes differentially expressed at any time point. Candidate genes were prioritized when identified as significantly differentially expressed (FDR adjusted p-value < 0.05) for any of the comparisons between HNT and control.
Prioritization based on connectedness in a gene regulatory network
We used a previously published gene regulatory network created using expression data from different rice developmental stages37. The connectedness of transcriptional regulators and their target genes in the network was visualized and examined using the R package networkD348. In this network, the confidence that a transcription factor interacts with a target gene is based on the strength of the association. Interaction scores ranged from 0 (the transcription factor is unlikely to interact with the target) to 871 (a high-confidence interaction). The full network was subset to include only interaction scores greater than or equal 200 to focus on transcription factors and target genes with strong associations. Only 0.3% of the interactions passed this stringent filter, leaving 45,879 interactions between transcription factors and target genes. The 638 annotated genes identified in the GWAS were compared to these networks. The gene candidates were evaluated based on the strength and number of interactions it had with transcriptional regulators.
Orthologous genes
The last approach used to prioritize candidate genes used the relationship to known causal genes to determine the best candidate genes. Lin et al.46 developed a machine learning approach to prioritizing association-identified loci called QTG-Finder. QTG-Finder takes a list of QTL loci and their associated potential candidate genes from a locus (or loci) and ranks each gene in order from the most to least likely to be a causal gene for each respective locus. This approach has been tested and works well in O. sativa and A. thaliana46. This has recently been extended in QTG-Finder2 to use orthologous genes to identify causal genes in Setaria and Sorghum based on rice and Arabidopsis studies46. The list of candidate genes from the GWAS study was submitted to QTG-Finder2 to rank the genes per locus. The genes were translated from RAP_ID to MSU_ID format to be compatible with the software. This reduced the total to 523 genes, 333 candidate genes from 34 absolute loci and 190 genes from the 20 relative loci. QTG-Finder2 could not be run on two loci due to an error we could not resolve. Of the remaining loci, the top-ranked genes for each locus were considered to be a prioritized candidate gene. Top-ranked genes were the top two genes for each locus ranked by QTG-Finder2.
Results
Phenotypic diversity and correlations among traits
All the traits examined recorded significant differences (p ≤ 0.05) under control and HNT conditions both in the walk-in chamber (Exp. 1, Fig. 2a–c) and field experiments (Exp. 2, Fig. 2d–f). A wide range in phenotypic variability was observed in the diversity panel for yield per plant, 100-seed weight, and harvest index (Fig. 2). Marker-based heritability values were low to intermediate, ranging from 0.15 (100-seed weight in Exp. 1 under HNT and grain yield in Exp. 2 under control) to 0.64 (chalkiness under HNT in Exp. 2). Genotypes under HNT stress resulted in significantly lower phenotypic values compared to control (Fig. 2), with reductions ranging from 4.7% (100-seed weight in Exp. 2, Fig. 2e) to 69% (grain yield in Exp. 1, Fig. 2a). Yield (69%), 100-seed weight (16%), and harvest index (63.3%) decreased more significantly under HNT compared to control, in the controlled-environment study (Exp. 1, Fig. 2a–c). An increase in night-time temperature from 24 to 29 °C in the field-based heat tents resulted in a two-fold increase in chalkiness.

Boxplots showing the phenotypic responses of traits (A, D yield; B, E 100-seed weight; C, F harvest index) under control and high night temperature stress in Exp. 1 (n = 173) and Exp. 2 (n = 206). In Exp. 1 (walk-in chambers; A-C), temperature treatments included control night temperature (CNT) and high night temperature (HNT) starting from panicle initiation to physiological maturity. In Exp. 2 (D-F), genotypes were exposed to control night (CNT) and high night temperature (HNT) with common day-time ambient temperature across treatments. The solid and dotted line represents the median and means of the data. The closed circles at the boundary show outliers at 90th percentile. The marker-based heritability (h2) was obtained in the Genomic Association and Prediction Integrated Tool. Different letters indicate statistically significant least square difference (LSD) for treatments at the level of p < 0.05.
Yield per plant (YPP, r = 0.21, p < 0.01) and 100-seed weight (HWT, r = 0.56, p < 0.001) were correlated significantly between the two treatments in Exp. 1 (Fig. 3a). Similarly, all the measured traits (YPP, r = 0.74; HWT, r = 0.85; and HI, r = 0.68, p < 0.001), including chalkiness (CHALK, r = 0.59, p < 0.001) were strong and positively correlated between the two treatments in Exp. 2 (Fig. 3b). The yield under control condition (in Exp. 1) showed positive correlations with yield and 100-seed weight, under both treatments with Exp. 2 (Fig. 3c). Strong significantly positive correlations were observed between Exp. 1 and Exp. 2 across treatments for the 100-grain weight (Fig. 3c). Yield under HNT in Exp. 1 was negatively correlated (r = − 0.24, p < 0.01) with the harvest index in Exp. 2 (Fig. 3c). Grain width (r = 0.80, p < 0.001; Fig. 4a) and grain length (r = 0.85, p < 0.001) were strongly correlated across treatments in Exp. 2 (Fig. 4b). Nonetheless, the correlations between the grain width and length were consistent for both absolute (r = − 0.65, p < 0.001) under HNT and relative value (r = − 0.50, p < 0.001).

Pearson’s correlation coefficients between the traits in walk-in chambers (173 genotypes in Exp. 1; A) field-based heat tents (206 genotypes in Exp. 2; B) and between experiments (Exp. 1 vs. Exp. 2, C). In experiment 1 (walk-in chambers), temperature treatments included control night temperature (CNT) and high night temperature (HNT), and in experiment 2, two different night temperature treatments i.e., control night (CNT) and high night temperature (HNT) were imposed with common day-time ambient temperature across treatments. YPP- grain yield (g per plant); HWT-100-seed weight (g); HI Harvest index and CHALK chalkiness (%). *p < 0.05, **p < 0.01, ***p < 0.001 indicate significant correlations among traits.

Rice grain width (mm, A) and grain length (mm, B) in response to control night (CNT; 24 °C, covered tents) and high night temperatures (HNT; 29 °C, covered and heated) under common day-time temperature under field conditions (Exp. 2; Supplementary Fig. S1). Manhattan plots of genome-wide association results for rice mature grain width (C–E) and length (F–H) in the study. The marker-based heritability (h2) was obtained in the Genomic Association and Prediction Integrated Tool. The previously characterized major genes controlling grain shape traits (yellow bar) such as the grain width (GW5, C,D) on chromosome 5 and grain length (GS3, F,G) genes on chromosome 3 are labeled. r—Pearson’s correlation coefficient between treatments. ***p < 0.001 indicate significant correlation of traits between CNT and HNT.
Genome-wide association study
Experiment and treatment specific MTAs
We identified genomic regions associated with different traits under control (representing controlled non-stress night temperature), HNT (representing high night temperature) and relative (representing genotype response as a ratio of HNT to CNT), by CMLM (Supplementary Figs. S2 and S3). A summary of GWAS results is given in Table 1, and the list of peak SNPs or candidate MTAs (cMTAs) for each locus are presented in Table 2. Significant MTAs varied between traits (absolute and relative values), from a minimum of five to a maximum of 25 (Table 1). The phenotypic variance (R2) explained by each marker ranged from 0.07 (relative yield in Exp. 2) to 0.37 for chalkiness (in Exp. 2) under HNT (Table 1). A total of 75 MTAs were associated with measured phenotypic traits under HNT, of which 24 were highly significant (p ≤ 7E−06 to 9E−05), 20 were significant at p value = 1E−04, and the rest met a suggestive threshold (p ≤ 2E−04 to 6E−04) (Supplementary Table S1). Sixty-one MTAs were detected (Supplementary Table S1) for relative values from two experiments (Supplementary Figs. S2 and S3).
Candidate MTAs across treatments
A total of 18, 20 and 19 cMTAs (SNP with a minimum p‐value for each locus) were identified for the traits under control, HNT and for relative values, respectively (Table 2). Twenty candidates MTAs were identified for the yield and quality-related traits i.e., yield (5), 100-seed weight (3), harvest index (6), chalkiness (4) and one each for grain width and grain length under HNT across experiments (Table 2). We also noticed that the identified genetic loci for grain width (Chr5pos5366489.1) and grain length (Chr3pos16794259.1) co-localized on the well-characterized, previously identified genes across treatments (Fig. 4). Three co-localizing cMTAs were detected between relative and absolute values for grain yield (Chr1pos22255654.1, Chr2pos34468097.1) and harvest index (Chr11pos3631830.1).
We detected three (Q5–Q7) and two (Q8–Q9) loci for grain yield under HNT, in Exp.1 and Exp.2, respectively. Minor alleles of five grain yield loci (Q5–Q9) revealed small (− 4.53 g) to medium (12.02 g) allelic effects in HNT conditions (Table 2). The allelic variation explained by the SNP markers of loci Q5-Q7 (i.e., Chr-1pos22255654.1, Chr2pos34468097.1 and Chr11pos23922165.1) showed that accessions carrying alleles ‘C, G and C’ had a higher grain yield under HNT (in Exp. 1) compared to accessions carrying ‘G, T and T’ alleles, respectively (Table 2). In Exp. 2, the minor allele of Q8 (Chr2pos31666003.1) had a negative effect on yield, while the minor allele of Q9 (Chr8pos1149355.1) had a positive impact on yield (Table 2). Three loci (Q2, Q3, and Q8; different SNPs but falls within the same LD block) on chromosome 2 clustered within 0.49 Mb region and were commonly associated with yield across treatments (Table 2). Three cMTAs were identified for relative grain yield (Table 2). Of the three cMTAs, two SNPs (Chr1pos22255654.1 and Chr2pos34468097.1) were shared between the grain yield under HNT and relative grain yield in Exp. 1 (Table 2, Supplementary Fig. S2).
Test weight (HWT; 100-seed weight) is a key yield component and is shown to be highly positively correlated with yield (Fig. 3) and significantly affected by HNT. We identified three loci (Q10–Q12) for 100-seed weight under control. The minor alleles of all three loci had a negative effect on 100-seed weight in both Exp. 1 and Exp. 2 (Table 2). Three cMTAs (one on chromosome 2 [Q13] in Exp. 1; one on chromosome 3 [Q14], and 4 [Q15] in Exp. 2) were associated with the 100-seed weight under HNT (Table 2). Similarly, the minor alleles of all three loci (Q13, Q14, and Q15) had a negative effect on 100-seed weight under HNT. Based on the relative 100-seed weight, five cMTAs were detected (2 in Exp. 1 and 3 in Exp. 2). Of the five cMTAs, minor alleles of three loci (Chr4pos33782238.1-‘G’; Chr5pos29593994.1-‘A’; Chr8pos25495868.1-‘G’) from Exp. 2 had negative effects on relative 100-seed weight (Table 2).
Harvest index has been used as a potential proxy for partitioning capacity between source and sink both under non-stress and stress conditions. Six loci (Q16-Q21) were detected for harvest index under control. Minor alleles of the loci associated with harvest index affecting the trait negatively under control (Table 2), except locus Q17 (Chr4pos15947167.1) (Table 2). We detected four and two genetic loci for harvest index in Exp. 1 (Q22–Q25) and Exp. 2 (Q26 and Q27) under HNT, respectively. Four markers, Chr2pos23789929.1, Chr6pos15730222.1, Chr7pos6689342.1, and Chr11pos3631830.1, were associated with relative harvest index across experiments (Table 2). For harvest index, cMTAs under HNT were located on chromosomes 1, 2, 4, and 11, and the most significant favorable allele associated with the Chr2pos25528306.1 (Q22, T) positively influenced the trait by 14% (p = 2E−04) in Exp. 1 and Chr1pos4312404.1 (Q26, C) influenced the same trait by 12% (p = 3E−05) in Exp. 2, under HNT (Table 2). In Exp. 2 the minor allele of Q26 had a negative impact on the harvest index, while the minor allele at Q27 affected the same trait positively under HNT (Table 2). Genomic regions (Q22–Q27) associated with harvest index under HNT were in close proximity to the region of genes controlling plant nutrient status (phloem sucrose transport, nitrogen metabolism, photoperiodic control of flowering, osmotic stress response, and lipid homeostasis) (Supplementary Tables S2 and S3). In Exp. 1, GWAS identified a key region on chromosome 2 (34.46 Mb) that was commonly associated with harvest index (Q23) and yield (Q6) under HNT (Table 2). Out of four cMTAs detected across experiments for relative harvest index, one candidate SNP (Chr11pos3631830.1) was shared between absolute and relative harvest index in Exp. 1 (Table 2).
Chalkiness is one of the primary factors determining rice milling and appearance qualities, both under control and stress conditions11. Eleven cMTAs (3 in control, 4 in HNT and 4 for relative) were identified for chalkiness in Exp. 2 (Table 2, Supplementary Fig. S3). Under control conditions, GWAS identified locus (Q30, Chr11pos22646338.1) on chromosome 11 (22.6 Mb) that was highly associated with chalkiness (Supplementary Fig. S3b), but not for HNT (Supplementary Fig. S3c). The minor allele of the lead SNP (Chr3pos1662112.1) underlying the peak Q33 had a negative effect (− 17.8%) on chalkiness (Table 2). Another cMTA (Q31) for chalkiness had the highest positive allele effect (39.6%) under HNT, detected on chromosome 1. A minor locus (Q34) on chromosome 4 (Chr4pos20331507.1) induced chalkiness (effect = 24.3%) under HNT. Also, one genomic region on chromosome 4 (19.9–20.3 Mb) for chalkiness was found to be co-localized both under control and HNT conditions (Supplementary Fig. S3b, c). Among the identified loci for relative chalkiness in Exp. 2, a SNP (Chr2pos18751285.1) on chromosome 2 was strongly (p = 6E−07) associated with relative chalkiness (Table 2). The Chr2pos18751285.1 SNP marker's minor allele had a positive effect on relative chalkiness (Table 2).
Grain shape/size (GS) is usually evaluated by grain width (GW)and grain length (GL), which are the key grain quality traits that influence the rice consumer preferences. Interestingly, a highly significant association (p = 1E−06 and p = 7E−05) was detected for grain width (5.3 Mb) on chromosome 5 across treatments (CNT, and HNT, respectively) (Fig. 4c,d), which is in the region of a well-characterized gene, qSW5/GW5 (encoding a calmodulin-binding motif family protein) that regulates grain width in rice49,50. No significant common loci were detected for grain width between absolute and relative values (Fig. 4e). Similar to absolute grain width, the SNP (Chr3pos16794259.1) associated with grain size (GS3)51,52, was detected as most significantly associated locus with absolute grain length across treatments (Fig. 4f,g). No loci were shared between the absolute and relative grain length. However, one SNP (Chr3pos33471970.1) on chromosome 3 was significantly associated with relative grain length but not co-located with the GS3 locus (Fig. 4h).
Overall, we found two prominent GWAS loci peaks on chromosome 2 (region 1: 31.4–31.9 Mb and region 2: 34.6–34.7) influencing grain yield and harvest index in rice (Supplementary Fig. S2). Region 1 (31.4–31.9 Mb) is associated with Q2, Q3, Q8, and Q12 that control yield and 100-seed weight either under control or HNT (Table 1, Supplementary Fig. S2). Similarly, region 2 (34.5 on chromosome 2; Q6 and Q23) commonly associated with absolute grain yield and harvest index under HNT and relative grain yield and relative harvest index in Exp. 1 (Table 2, Supplementary Table S1). A SNP marker associated with Chr1pos22255654.1 (Q5) co-located with the relative grain yield in Exp. 2 (Table 2).
Prioritizing gene candidates
An in-silico search for all annotated genes in a 100-kb window surrounding the unique 51 candidate SNPs yielded 638 (396 for absolute, 209 for relative and 33 shared between absolute and relative) annotated genes, ranging between 2 (Q20) and 23 (Q19) genes (Table 2, Supplementary Table S2). We found that these genes encode several classes of protein with known or predicted functions in abiotic and grain development pathways, including helix-loop-helix DNA-binding domain-containing proteins, similar to starch synthase, GRAS transcription factor domain-containing protein, a calcium-binding protein, mitogen-activated protein kinase 4, ROS scavenging, sugar signaling and metabolism, and auxin-responsive protein (Supplemental Table S2). Further, a search for characterized genes within candidate loci yielded 127 functional candidate genes (http://funricegenes.ncpgr.cn/) with a range of 1–17 candidate genes per locus (Supplementary Table S3).
A common approach is prioritizing candidates for each locus based on functional roles from prior literature and coding variants. However, the negative effects of HNT manifest through different mechanisms than HDT, and little is known about the underlying genes driving the genetic architecture of these traits under HNT. Therefore, previous literature may not cover the genes involved in HNT response regulation. Moreover, since non-coding variants can play critical roles in trait expression, prioritizing coding variants ignores significant non-coding regulatory variants. Therefore, three independent approaches were applied to prioritize candidate causal genes driving the traits' genetic architecture under both control and HNT conditions. The annotated genes in the 100-kb window surrounding each locus were examined for overlap in three ways: (1) with genes differentially expressed in response to HNT, (2) strength of association in a gene regulatory network, and (3) genes orthologous to prior causal genes. This approach reduces the bias by prioritizing candidate genes based on other data sets.
The first approach, which examined if candidate genes were differentially expressed in HNT, found 34 significant candidate genes, 22 of the genes from loci identified in the absolute analysis and 12 from the relative loci. The gene regulatory network approach prioritized 76 genes based on their interaction with transcription regulators, 50 from the absolute candidates and 26 from the relative candidates (Fig. 5a,b). The final method, which prioritized candidate genes based on their orthogonality to known causal genes, ranked 97 genes as top priority candidates, 66 absolute and 34 relative. To further prioritize and select the most promising candidates, each approach's selected genes were crosschecked for their performance in the other two techniques. Candidates were considered highly promising if they had two methods supporting them. Additionally, since many of the genes could not be tested in one or more of the approaches, candidates could still be considered highly promising if supported by one method. However, they could not be tested in both methods. For example, Os02g0635600 ranked highest in Q22 (harvest index in absolute HNT) based on ortholog to known causal genes using QTG-Finder2, but this gene was not present in the full gene regulatory network and was not detected in the transcription data (Table 3). Therefore, we only penalized against genes that did not perform well where they could be tested. A total of 56 genes are identified as the “high-priority” gene candidates based on this method, 35 genes from the loci identified in the absolute HNT or control and 21 genes from the relative analysis (Supplementary Table S4). Seventeen candidate genes were identified as strong candidates in two of the three evaluations, 12 in the absolute and 5 in the relative analysis. An additional 23 candidate genes were selected as priority candidates in absolute HNT or control based on one criterion but could not be tested in either of the other two methods. Similarly, 16 additional priority candidates identified by one method, but not testable in the other two were identified from the loci associated with the relative change between control and HNT (Supplementary Table S4). A detailed list of the transcription factor (TF) for each candidate gene is given in Supplementary Table S5.

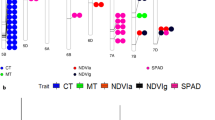
Gene regulatory networks are made up of top candidate genes from the GWAS of absolute (A) and relative trait values (B) and candidate genes with strong transcription factor interaction levels (> 200). A list of transcription factors for each candidate is given in Supplementary Table S5.
Discussion
Increasing tolerance to HNT while simultaneously minimizing yield and quality losses is an emerging goal in rice breeding. A night temperature of > 27 ℃ has been used to determine differential physiological, biochemical, and agronomic responses both under controlled19,53 and field experiments11,13,32, using a limited number of genotypes [two11 to 3633]. Here, we carried out stage-targeted phenotyping of indica diversity panel both under controlled-environmental and field conditions (Supplementary Fig. S1), which allowed us to identify genetic loci associated with key phenotypic traits that determine HNT responses (Table 2).
Phenotypic variation explains underlying trait correlations and physiological responses under HNT
Our study involving rice diversity panel (entries from different rice-growing regions) recorded a significant decline in yield, 100-seed weight, and increased chalkiness under prolonged HNT stress (Fig. 2), in both controlled environment and field studies, supporting previous findings5,11,13. A more significant reduction in grain yield and 100-seed weight, at least in part, is known to be related to reduced carbohydrate availability due to higher night-time respiration or reduced assimilates supply to sink or a combination of both, under HNT in rice13. HNT stress during grain development terminates or reduces the activity of a series of enzymes involved in starch synthesis in the endosperm, negatively affecting grain size under HNT in rice13,35 and other crops14. For example, a significant correlation between changes in the fructose-6-phosphate content of the panicles and the yield reduction in rice emphasized the significant metabolic changes in the sink during grain development in response to HNT35. Thus, minimizing yield losses and quality deterioration caused by HNT during grain filling is becoming extremely important to sustain global rice production and end use quality.
Unlike HDT stress at anthesis, post-flowering HNT reduces grain yield and quality by lowering grain weight caused by heat-induced premature senescence14 and quicker break‐down of stored assimilates during the night13,32,35. In particular, heat stress-accelerated chlorophyll degradation, inhibition of chlorophyll biosynthesis54, and loss of carbon already fixed by daily photosynthesis55 are key aspects that limit the source capacity during grain filling. The depletion of carbohydrates or imbalance with the source (decrease in photosynthesis and increase in respiration) during grain filling exhausts the assimilate reserves that are particularly needed for grain development under heat stress14,34,56. Similar to the impacts of HNT stress, low solar radiation also interferes with starch accumulation during grain filling, which significantly affects the seed weight and market value (quality) of rice57. An increase in average night-time VPD seems to be coupled with warmer night and lower relative humidity (Fig. 1b). Such an increase in night VPD has been shown to impact the plant productivity58. Hence, further studies are needed to disentangle the HNT vs. low solar radiation, differential VPD levels under HNT scenarios and their combined effect on yield and grain quality losses in rice.
Interestingly, canopy functional stay-greenness is known to have the potential to enhance radiation use efficiency and photo-assimilates supply by improving the longevity of photosynthetic machinery during post-anthesis. We hypothesize that the canopy's extended ability to generate additional assimilates under HNT could potentially fill the carbon gap created by increased carbon losses due to higher night respiration in response to HNT. Therefore, exploring the relevance of functional stay green in combination with an efficient transfer of photo-assimilates (water-soluble carbohydrates) from source to sink is proposed to minimize the HNT-induced yield and quality losses during grain filling in rice.
Comparison of GWAS revealed common loci for phenotypic traits across treatments
Grain size/shape and chalkiness are two crucial components that determine rice grain quality59, with increased chalkiness having a significant negative impact on the economic returns20. Both grain size and chalkiness are polygenic quantitative traits60. The high genetic and phenotypic correlations among measured traits across treatments, particularly for seed weight and grain size, indicate strong potential surrogate traits for breeding HNT tolerance in rice. A weak correlation was observed between controlled environment and field for some traits, which might be due to one or more factors (thermal amplitude [Fig. 1b], low light condition, relative humidity) differing between these environments23,30. In spite of these differences, several GWAS loci were in the genic region or close to previously reported genes controlling grain shape traits, e.g., GS351,52 and GW5/qGSW549,50,61 across treatments. This indicates that (1) GWAS is an effective way to identify putative genes/target regions for HNT tolerance in rice and (2) exhibited the presence of strong and common genetic regulation across control and HNT in rice (Fig. 4). For example, a strong association for grain width was identified on chromosome 5 under both control and HNT conditions, which is in the region of a well-characterized gene, GW5 (Os05g0187500, encoding a calmodulin-binding motif family protein) that regulates grain width in rice50 (Fig. 4c,d). Another prominent peak on chromosome 3 (Chr3pos16794259.1) associated with the gene, GS3 (Os03g0407400, a regulator of grain size and organ size), was detected for grain length across treatments (Fig. 4f,g). Co-localization of GWAS loci for grain size (length or width) variations under control and warmer nights suggest that tolerance to HNT stress for grain size is likely to have common regulators in rice (Fig. 4). Another cMTA was identified on chromosome 3 (Q28) for grain chalkiness, which is also near the GS3, indicating that the molecular mechanisms of grain chalkiness in rice might be rather complex than the grain size59. Therefore, further functional studies are needed to explore the association of grain size and chalkiness genes in regulating rice grain weight and quality under heat stress conditions.
For instance, one genomic region on chromosome 2 (31.4–31.9 Mb) associated with Q2, Q3, Q8, and Q12 was shown to control yield and 100-seed weight either under control or HNT (Supplementary Fig. S2). Loci (Q3 and Q8) identified under control and HNT grain yield were close (25 kb) to the trehalose-6-phosphate phosphatase gene (TPP7, Os02g0753000), which is shown to display significant improvement in abiotic stress tolerance in plants62. In Maize, the overexpression of trehalose-6-phosphate phosphatase in-ears increased yield by increasing both kernel set and harvest index in drought stress conditions63. On the other hand, increased trehalose concentration was recorded after four hours of heat stress (at 40 °C) in Arabidopsis thaliana64. The locus (Q8) associated with yield under HNT was close (32.2 kb, Os02g0753800) to a calcium-binding protein (Supplementary Table S3), which is found to enhance heat stress tolerance by modulating the reactive oxygen species (ROS) production, particularly H2O2 and reproductive organ development under different abiotic stresses65,66. A similar family gene (OsANN10) has been mapped for the heat stress tolerance index in rice67. A defined role for these genes in combating heat stress at reproductive and grain-filling has not been fully explored.
Despite the variable number of genotypes and different experimental conditions, our phenotypic correlations and co-localized GWAS loci exhibited the presence of strong and common genetic regulation across treatments in rice. A genomic region associated with the relative grain length on chromosome 3 (33.47) co-localized with locus influencing absolute spikelet fertility under high temperature68 and relative spikelet fertility69. Two cMTAs for absolute grain yield under HNT on chromosome 1 (Q5, Chr1pos22255654.1) and chromosome 2 (Q6, Chr2pos34468097.1) were related to genomic regions for relative grain yield in experiment 1 (Table 2). Similarly, a common MTA was observed between harvest index under HNT (Q25, Chr11pos3631830.1) and relative harvest index in experiment 1 (Table 2). Therefore, further characterization of loci (31.46 to, 34.46 Mb on chromosome 2 and 33.47 on chromosome 3) would be interesting to decipher the role of these pleiotropy regions for enhancing heat stress resilience (both day-time and night-time) in rice. Relatively little is known about the interaction between high day and night temperature stress on grain yield and quality70. Identifying genomic regions associated with combined stress tolerance would set a stage to breed rice with improved heat stress tolerance.
A limited number of shared loci between relative and absolute phenotype were observed across treatments suggesting that mapping genetic loci based on stress index (stress effects relative to control) may not always reflect the true stress tolerance68. While comparing the relative change in phenotypes between control and HNT allows the evaluation of sensitivity to HNT stress across all phenotypic ranges. In addition, evaluating the candidate genes in these loci may provide firsthand targets for pathways that alter adaptation to HNT stress. Nevertheless, quantifying stress tolerance by combining both absolute and relative traits would help develop rice accessions with greater productivity and adaptability under stress. The identified loci can be used as a starting point for future candidate gene validation and breeder-friendly markers development to enhance HNT tolerance in rice.
Refinement of candidate genes by available molecular data
Although the possible genetic determinants of phenotypic trait variations in response to stress have been effectively determined based on GWAS in crops, detecting their functional relevance or prioritizing candidate genes is a severe bottleneck46. Using available prior data, we refined the possible candidate genes associated with the traits for each genomic region through three independent methods: evaluating the transcriptional response of the genes in each locus to HNT, determining the connectedness of each gene to transcriptional regulators in a gene regulatory network, and determining the orthology to genes previously identified as causative in other studies. Validation of the causal gene for each GWAS-identified locus is a key component to move forward. However, it is not practical to experimentally test all the genes that overlay the genomic regions. Therefore, there is a need to prioritize the target genes. Previous genetic mapping or GWAS studies have routinely used currently available literature to decide on the potential candidates to validate based on annotations and known functions. This approach, although widely used, introduces bias at different levels45,46. Hence, developing new approaches that reduce this bias would be beneficial. The three methods used to find top candidates from this study demonstrate integrating multiple ways to prioritize candidates for further validation. One limitation of this approach is that not all candidate genes could be tested for many of these datasets. For example, genes not on the Agilent rice 4 × 44 K microarray were not included in the gene regulatory network. Likewise, genes not expressed in the tissue evaluated for HNT response were not included in the transcriptional analysis. However, as data resources extend and more data becomes available in accessible formats, these approaches will become even more reliable for narrowing down the candidate genes to test for causality.
Conclusion
The current study is the first report investigating the natural variation of rice genotypes to HNT using walk-in chambers and field-based heat tents. The observed significant positive correlations (Fig. 3) and common GWAS loci across treatments (Fig. 4) for grain size indicate that selection for higher phenotypic values from the control conditions could be a proxy for high yields under HNT. Advanced gene sorting methods combined with GWAS allowed us to narrow down the candidate region to potential genes controlling yield and related traits across control and HNT. Exploring the genetic regulation of functional stay green and source-sink carbohydrate relationships under night temperature is an exciting area for further research. Our findings provide insights towards integrating multi-disciplinary approaches such as physiological, genomic, and network algorithms to identify possible causal loci/genes. The identified favorable alleles for HNT tolerance would help develop molecular markers to support ongoing heat stress breeding programs in rice.
References
Wassmann, R. et al. Regional vulnerability of climate change impacts on Asian rice production and scope for adaptation. in Advances in Agronomy vol. 102 91–133 (Academic Press, 2009).
Muthayya, S., Sugimoto, J. D., Montgomery, S. & Maberly, G. F. An overview of global rice production, supply, trade, and consumption: Global rice production, consumption, and trade. Ann. N. Y. Acad. Sci. 1324, 7–14 (2014).
Intergovernmental Panel on Climate Change. Climate Change 2014 Mitigation of Climate Change: Working Group III Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. (Cambridge University Press, 2014). doi:https://doi.org/10.1017/CBO9781107415416.
Intergovernmental Panel on Climate Change. IPCC. Global Warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty. (2018).
Peng, S. et al. Rice yields decline with higher night temperature from global warming. Proc. Natl. Acad. Sci. 101, 9971–9975 (2004).
Nagarajan, S. et al. Local climate affects growth, yield and grain quality of aromatic and non-aromatic rice in northwestern India. Agric. Ecosyst. Environ. 138, 274–281 (2010).
Rao, B. B., Chowdary, P. S., Sandeep, V. M., Pramod, V. P. & Rao, V. U. M. Spatial analysis of the sensitivity of wheat yields to temperature in India. Agric. For. Meteorol. 200, 192–202 (2015).
Welch, J. R. et al. Rice yields in tropical/subtropical Asia exhibit large but opposing sensitivities to minimum and maximum temperatures. Proc. Natl. Acad. Sci. 107, 14562–14567 (2010).
Vose, R. S., Easterling, D. R. & Gleason, B. Maximum and minimum temperature trends for the globe: An update through 2004. Geophys. Res. Lett. 32, L23822 (2005).
Cooper, N. T. W., Siebenmorgen, T. J. & Counce, P. A. Effects of nighttime temperature during kernel development on rice physicochemical properties. Cereal Chem. J. 85, 276–282 (2008).
Shi, W. et al. Source-sink dynamics and proteomic reprogramming under elevated night temperature and their impact on rice yield and grain quality. New Phytol. 197, 825–837 (2013).
Ambardekar, A. A., Siebenmorgen, T. J., Counce, P. A., Lanning, S. B. & Mauromoustakos, A. Impact of field-scale nighttime air temperatures during kernel development on rice milling quality. Field Crops Res. 122, 179–185 (2011).
Bahuguna, R. N., Solis, C. A., Shi, W. & Jagadish, K. S. V. Post-flowering night respiration and altered sink activity account for high night temperature-induced grain yield and quality loss in rice (Oryza sativa L.). Physiol. Plant. 159, 59–73 (2017).
Impa, S. M. et al. High night temperature induced changes in grain starch metabolism alters starch, protein, and lipid accumulation in winter wheat. Plant Cell Environ. 43, 431–447 (2020).
Hein, N. T. et al. Integrating field-based heat tents and cyber-physical system technology to phenotype high night-time temperature impact on winter wheat. Plant Methods 15, 41 (2019).
García, G. A. et al. Warm nights in the Argentine Pampas: Modelling its impact on wheat and barley shows yield reductions. Agric. Syst. 162, 259–268 (2018).
García, G. A., Serrago, R. A., Dreccer, M. F. & Miralles, D. J. Post-anthesis warm nights reduce grain weight in field-grown wheat and barley. Field Crops Res. 195, 50–59 (2016).
Shi, W. et al. Grain yield and quality responses of tropical hybrid rice to high night-time temperature. Field Crops Res. 190, 18–25 (2016).
Mohammed, A.-R. & Tarpley, L. Impact of high nighttime temperature on respiration, membrane stability, antioxidant capacity, and yield of rice plants. Crop Sci. 49, 313–322 (2009).
Lyman, N. B., Jagadish, K. S. V., Nalley, L. L., Dixon, B. L. & Siebenmorgen, T. Neglecting rice milling yield and quality underestimates economic losses from high-temperature stress. PLoS ONE 8, e72157 (2013).
Ishimaru, T. et al. A genetic resource for early-morning flowering trait of wild rice Oryza officinalis to mitigate high temperature-induced spikelet sterility at anthesis. Ann. Bot. 106, 515–520 (2010).
Jagadish, S. V. K., Murty, M. V. R. & Quick, W. P. Rice responses to rising temperatures-challenges, perspectives and future directions. Plant Cell Environ. 38, 1686–1698 (2015).
Bheemanahalli, R. et al. Temperature thresholds for spikelet sterility and associated warming impacts for sub-tropical rice. Agric. For. Meteorol. 221, 122–130 (2016).
Xu, J., Henry, A. & Sreenivasulu, N. Rice yield formation under high day and night temperatures-A prerequisite to ensure future food security. Plant Cell Environ. 43, 1595–1608 (2020).
Matsui, T. Rice (Oryza sativa L.) cultivars tolerant to high temperature at flowering: anther characteristics. Ann. Bot. 89, 683–687 (2002).
Jagadish, S., Craufurd, P. & Wheeler, T. High temperature stress and spikelet fertility in rice (Oryza sativa L.). J. Exp. Bot. 58, 1627–1635 (2007).
Jagadish, S. V. K., Craufurd, P. Q. & Wheeler, T. R. Phenotyping parents of mapping populations of rice for heat tolerance during anthesis. Crop Sci. 48, 1140 (2008).
Jagadish, S. V. K. et al. Physiological and proteomic approaches to address heat tolerance during anthesis in rice (Oryza sativa L.). J. Exp. Bot. 61, 143–156 (2010).
Yoshida, S., Satake, T. & Mackill, D. S. High-temperature stress in rice [study conducted at IRRI, Philippines] (IRRI Res. Pap. Ser, 1981).
Bahuguna, R. N. et al. Physiological and biochemical characterization of NERICA-L-44: a novel source of heat tolerance at the vegetative and reproductive stages in rice. Physiol. Plant. 154, 543–559 (2015).
Bheemanahalli, R. et al. Is early morning flowering an effective trait to minimize heat stress damage during flowering in rice?. Field Crops Res. 203, 238–242 (2017).
Shi, W., Xiao, G., Struik, P. C., Jagadish, K. S. V. & Yin, X. Quantifying source-sink relationships of rice under high night-time temperature combined with two nitrogen levels. Field Crops Res. 202, 36–46 (2017).
Zhang, Y. et al. Effects of high night temperature on yield and agronomic traits of irrigated rice under field chamber system condition. Aust. J. Crop Sci. 7, 7 (2013).
Abdelrahman, M., Burritt, D. J., Gupta, A., Tsujimoto, H. & Tran, L.-S.P. Heat stress effects on source–sink relationships and metabolome dynamics in wheat. J. Exp. Bot. 71, 543–554 (2020).
Schaarschmidt, S. et al. Season affects yield and metabolic profiles of rice (Oryza sativa) under high night temperature stress in the field. Int. J. Mol. Sci. 21, 3187 (2020).
Liao, J.-L. et al. Transcriptome changes in rice (Oryza sativa L.) in response to high night temperature stress at the early milky stage. BMC Genomics 16, 18 (2015).
Desai, J. S. et al. Warm nights disrupt global transcriptional rhythms in field-grown rice panicles. https://doi.org/10.1101/702183 (2019).
Kadam, N. N., Struik, P. C., Rebolledo, M. C., Yin, X. & Jagadish, S. V. K. Genome-wide association reveals novel genomic loci controlling rice grain yield and its component traits under water-deficit stress during the reproductive stage. J. Exp. Bot. 69, 4017–4032 (2018).
Quinones, C., Mattes, N., Faronilo, J., Sudhir-Yadav & Jagadish, K. S. V. Drought stress reduces grain yield by altering floral meristem development and sink size under dry-seeded rice cultivation. Crop Sci. 57, 2098–2108 (2017).
Lipka, A. E. et al. GAPIT: Genome association and prediction integrated tool. Bioinformatics 28, 2397–2399 (2012).
Zhang, Z. et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360 (2010).
Rebolledo, M. C. et al. Combining image analysis, genome wide association studies and different field trials to reveal stable genetic regions related to panicle architecture and the number of spikelets per panicle in rice. Front. Plant Sci. 7, (2016).
Kikuchi, S. et al. Genome-wide association mapping for phenotypic plasticity in rice: Genome-wide association mapping and rice plasticity. Plant Cell Environ. 40, 1565–1575 (2017).
Moghimi, N. et al. New candidate loci and marker genes on chromosome 7 for improved chilling tolerance in sorghum. J. Exp. Bot. 70, 3357–3371 (2019).
Baxter, I. We aren’t good at picking candidate genes, and it’s slowing us down. Curr. Opin. Plant Biol. 54, 57–60 (2020).
Lin, F., Fan, J. & Rhee, S. Y. QTG-Finder: A machine-learning based algorithm to prioritize causal genes of quantitative trait loci in Arabidopsis and rice. G3 Genes Genomes Genet. 9, 3129–3138 (2019).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Allaire, J. J. et al. networkD3: D3 JavaScript Network Graphs from R. (2017).
Shomura, A. et al. Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 40, 1023–1028 (2008).
Weng, J. et al. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 18, 1199–1209 (2008).
Mao, H. et al. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. 107, 19579–19584 (2010).
Fan, C. et al. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 112, 1164–1171 (2006).
Coast, O., Šebela, D., Quiñones, C. & Jagadish, S. V. K. Systematic determination of the reproductive growth stage most sensitive to high night temperature stress in rice (Oryza sativa). Crop Sci. 60, 391–403 (2020).
Jespersen, D. & Huang, B. Physiological and biochemical mechanisms of plant tolerance to heat stress. in Handbook of Plant and Crop Physiology, Third Edition vol. 20144886 389–404 (CRC Press, 2014).
Liang, J., Xia, J., Liu, L. & Wan, S. Global patterns of the responses of leaf-level photosynthesis and respiration in terrestrial plants to experimental warming. J. Plant Ecol. 6, 437–447 (2013).
Impa, S. M. et al. Carbon balance and source-sink metabolic changes in winter wheat exposed to high night-time temperature. Plant Cell Environ. 42, 1233–1246 (2019).
Ishimaru, T. et al. High temperature and low solar radiation during ripening differentially affect the composition of milky-white grains in rice (Oryza sativa L.). Plant Prod. Sci. 21, 370–379 (2018).
Sadok, W. & Jagadish, S. V. K. The hidden costs of nighttime warming on yields. Trends Plant Sci. 25, 644–651 (2020).
Liu, C. et al. Rapid prediction of head rice yield and grain shape for genome-wide association study in indica rice. J. Cereal Sci. 96, 103091 (2020).
Gong, J. et al. Dissecting the genetic basis of grain shape and chalkiness traits in hybrid rice using multiple collaborative populations. Mol. Plant 10, 1353–1356 (2017).
Liu, J. et al. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 3, 1–7 (2017).
Li, H.-W., Zang, B.-S., Deng, X.-W. & Wang, X.-P. Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 234, 1007–1018 (2011).
Nuccio, M. L. et al. Expression of trehalose-6-phosphate phosphatase in maize ears improves yield in well-watered and drought conditions. Nat. Biotechnol. 33, 862–869 (2015).
Kaplan, F. et al. Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol. 136, 4159–4168 (2004).
Szalonek, M. et al. Potato Annexin STANN1 promotes drought tolerance and mitigates light stress in transgenic Solanum tuberosum L. plants. PLOS ONE 10, e0132683 (2015).
Xu, L. et al. Comprehensive analyses of the annexin gene family in wheat. BMC Genomics 17, 415 (2016).
Shanmugavadivel, P. et al. High resolution mapping of QTLs for heat tolerance in rice using a 5K SNP array. Rice 10, 28 (2017).
Jagadish, S. V. K. et al. Genetic analysis of heat tolerance at anthesis in rice. Crop Sci. 50, 1633–1641 (2010).
Yue, B. et al. Genetic basis of drought resistance at reproductive stage in rice: Separation of drought tolerance from drought avoidance. Genetics 172, 1213–1228 (2006).
Shi, W. et al. High day- and night-time temperatures affect grain growth dynamics in contrasting rice genotypes. J. Exp. Bot. 68, 5233–5245 (2017).
Acknowledgements
We thank the Federal Ministry for Economic Cooperation and Development, Germany, for financially supporting the heat stress research carried out by RB and CQ at IRRI. Contribution No. 21-087-J from Kansas Agricultural Research Station.
Funding
This work was supported by the USAID and BMGF- funded Cereal Systems Initiative for South Asia (Phase II) and by the Agriculture and Food Research Initiative competitive Grant #2015-67013-22814 of the USDA National Institute of Food and Agriculture. Partial support from The Federal Ministry for Economic Cooperation and Development, Germany (Contract No. 81141844; Project No. 11.7860.7-001.00).
Author information
Authors and Affiliations
Contributions
R.B., S.V.K.J., C.D.: designed and implemented the study; R.B., C.Q.: collected data; R.B.: conducted the phenotypic and GWAS analyses, review, writing-original draft, and data visualization; M.K., C.D.: carried out bioinformatics and network analysis; R.B., M.K., C.D., S.V.K.J.: interpreted the data, review, co-writing original draft; all authors edited and finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bheemanahalli, R., Knight, M., Quinones, C. et al. Genome-wide association study and gene network analyses reveal potential candidate genes for high night temperature tolerance in rice. Sci Rep 11, 6747 (2021). https://doi.org/10.1038/s41598-021-85921-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85921-z
- Springer Nature Limited
This article is cited by
-
New insights into QTNs and potential candidate genes governing rice yield via a multi-model genome-wide association study
BMC Plant Biology (2024)
-
Molecular mapping and characterization of QTLs for grain quality traits in a RIL population of US rice under high nighttime temperature stress
Scientific Reports (2023)
-
Deep learning based high-throughput phenotyping of chalkiness in rice exposed to high night temperature
Plant Methods (2022)