
Abstract
This work focuses on the hydrothermal synthesis of Na-P1 zeolite by using a kaolinite rock coming from Romana (Sassari, Italy). The kaolin is calcined at a temperature of 650 °C and then mixed with calculated quantities of NaOH. The synthesis runs are carried out at ambient pressure and at variable temperatures of 65 and 100 °C. For the first time compared to the past, the Na-P1 zeolite is synthesized without the use of additives and through a protocol that reduces both temperatures and synthesis times. The synthesis products are analysed by X-ray diffraction, high temperature X-ray diffraction, infrared spectroscopy, scanning electron microscopy and inductively coupled plasma optical emission spectrometry. The cell parameters are calculated using the Rietveld method. Density and specific surface area are also calculated. The absence of amorphous phases and impurities in synthetic powders is verified through quantitative phase analysis using the combined Rietveld and reference intensity ratio methods. The results make the experimental protocol very promising for an industrial transfer.
Similar content being viewed by others

Introduction
Zeolites are a group of tectosilicates of about 50 minerals with synthetic analogues. Their structure is made of three-dimensional networks of Al/Si tetrahedra arranged to form channels containing water and exchangeable alkaline or alkaline earth cations. Thanks to these peculiar structural characteristics, they are widely used in separation and refinery industries as catalysts, adsorbents and ion exchangers.
The zeolite P class has the typical oxide formula: M2/nO.Al2O3.1.80–5.00SiO2.5H2O where M is a n-valent cation, normally an alkali metal1. P zeolites are characterised by water-softening properties over zeolite A2 and are used for the formation of environmental- friendly detergents3. Na-P is also useful for gas separation applications4, 5 and for removal of toxic and radioactive waste species1, 6,7,8. Synthetic zeolite Na-P1 is also recognized as a better sorbent of Cr (VI) than Clinoptilolite9.
Despite having variable Si/Al ratios, all P zeolites are characterised by the same framework topology, which is the GIS net, gismondine, according to Meier and Olson10. Three polymorphs are recognized for Na-P: the cubic phase called Na-P1, refined in I411 and with a unit cell content of Na6Al6Si10O32.12H2O; the orthorhombic one, the so-called Na-P2 with formula Na4Al4Si12O32.14H2O, crystallizing in Pnma12; the tetragonal one for high silica variety of Na-P (Na3.6Al3.6Si12.4O32.14H2O), refined in I41/amd5. Another zeolite P with a strict Si/Al ratio of 1.0 is called aluminum P (MAP), whose structure was firstly refined by Albert et al.13 in the monoclinic system, space group C2/c (Na8Al8Si8O32. 15.2H2O). Nery et al.1 refined the structure of low-silica Cd-MAP, Mn-MAP, Ba-MAP, Sr-MAP and Pb-MAP.
Zeolite P can be synthesized from various methods such as the hydrothermal method1, 8, 12,13,14,15,16, the microwave technique17, the sol–gel process18 and the sonochemical method19. In order to reduce the costs of synthesis protocols, alternative synthesis precursors have been tested in place of expensive chemical reagents used in the past, like fly ash20,21,22, kaolinite15, 23, clays18, 24, rice husk silica25 and nuclear wastes26.
The scope of the present work is to test a natural rock, i. e. a kaolinitic rock coming from Romana (Italy) in the synthesis of monomineralic powders of Na-P1. Among clay minerals, in fact, kaolinite is the most common phyllosilicate involved in successful zeolitic synthesis because of its particularly ample/large supply and availability and the well-known reactivity of thermally treated kaolin clays (metakaolin) with alkali3, 27,28,29,30,31.
Lovat et al.23 just investigated the reaction of metakaolinite (calcination temperature 800 °C) in presence of fluoride ions at 85 °C founding peaks of zeolite P after long periods (60 days reaction time). More recently, Li et al.15 synthesized Na-P in 48 h at 180 °C starting from metakaolin (calcinated at 800 °C) by using NaF as structure-directing agent.
In this paper we present the results of a research carried out to define the most favorable conditions for the Na-P1 synthesis from metakaolin. The aim is to improve previous mineral synthesis attempts starting from kaolinite; i.e. by developing a synthesis protocol that does not include the use of a structure-directing agents like NaF15, and secondly working on the reduction of both the calcination and synthesis temperatures and the synthesis times. An aspect that we also want to deepen in this work is the definition of the degree of purity of the synthesized powders expressed in terms of absence of amorphous phase and impurities coming from the natural kaolinite sample. In the past, Li et al.15 defined the synthesized zeolite as “pure” by utilizing the main X-ray diffraction peak of the mineral, i.e., through the use of a formula that calculates the degree of crystallinity of a powder as the ratio between the peak area of product divided by the peak area of referent sample. On the other hand, we believe systematic samplings during the experimental run enable the progress in the crystallization of the mineral to be traced/followed and allows to determine the time at which the climax in the crystallization is reached due to the absence of other phases or amorphous phases. The degree of purity of the synthesized zeolite is here defined through a quantitative phase analysis approach using the combined Rietveld and reference intensity ratio methods.
A natural, cost effective starting material makes this route especially attractive when expanded to an industrial scale as long as the material properties of the synthesized products remain satisfactory, in terms of chemical composition and purity of powders.
Materials and methods
The kaolin used in this study was collected in a mine located in Romana (Sassari province, Italy). For the chemical composition of kaolin and its mineralogical, morphological and spectroscopic characterization, see Novembre et al.30. The kaolin was triturated, and the sandy fraction was separated by retention in a sieve. The fraction below 90 μm was then collected, suspended in distilled water, sonicated, and centrifuged for separation of the silt fraction and collection of the clay fraction. Preliminary calcination of kaolin was carried out using the following procedure: aliquots of kaolin were placed in open porcelain crucibles which were heated in a Gefran Model 1200 furnace (Gefran Spa, Brescia, Italy) to the calcination temperature (650 °C) at a pressure of 1 atm. The heating rate of the sample was 1.5 °C s−1. Once the calcination temperature was reached, the crucibles were left in the furnace for 2 h and then removed and cooled at room temperature. The NaOH used in the synthesis protocol were purchased from Riedel-de Haën (Honeywell Riedel-de Haën, Bucharest, Romania). The purity of the reagent was of 99%. 2 g of metakaolinite have been dissolved in 20 ml of a NaOH (8%) solution. The initial mixture had the composition: 6.25 SiO2–1.00 Al2O3–3.6 Na2O. The mixture was homogenized for two hours with a magnetic stirrer. Then was put inside a stainless-steel hydrothermal reactor and heated at 10 °C/min until the desidered temperature (65 and 100 °C) and kept for different times. Synthesis products were sampled periodically from the reactor, filtered with distilled water and dried in an oven at 40 °C for a day.
Metakaolinite and products of synthesis were analysed by powder X-ray diffraction (XRPD); the instrument was a Siemens D5000 operating with a Bragg–Brentano geometry (CuKα = 1.518 Å, 40 kV, 40 mA, 2°–45°, 2°–90° 2theta scanning interval, step size 0.020° 2theta). Metakaolinite revealed an amorphous character by the XRD analysis. Identification of Na-P and relative peak assignment was performed with reference to the following JCPDS code: 00-039-0219. Both the crystalline and amorphous phases in the synthesis powders were estimated using quantitative phase analysis (QPA) applying the combined Rietveld and reference intensity ratio (RIR) methods; corundum NIST 676a was added to each sample, amounting to 10% (according to the strategy proposed by Novembre et al.32 and the powder mixtures were homogenized by hand-grinding in an agate mortar. Data for the QPA refinement were collected in the angular range 5–120° 2theta with steps of 0.02° and 10 s step−1, a divergence slit of 0.5° and a receiving slit of 0.1 mm.
Data were processed with the GSAS software33 and the graphical interface34 starting with the structural models proposed by Albert et al.13 for Na-P1 and Gatta et al. for nepheline35. The following parameters were refined: background parameters, zero shift, cell parameters and peak profiles.
Thermal stability and phase transformations were studied using high-temperature X-ray diffractometry with a PANalytical X’Pert PRO MPD (CuKα = 1.518 Å, 45 kV, 40 mA, X’Celerator Detector with active length of 2.122°, θ/2θ scan from 5° to 50° 2θ with step size of 0.017° and measuring time of 100 s per step), equipped with a high temperature camera Anton Paar HTK1200N (thermocouple Pt 10% RhPt). The sample holder was a platform with a 16 mm diameter equipped with a ceramic cup (0.8 mm deep and 14 mm inner diameter) for holding powder. The analyses were taken at different temperatures: from 28 °C up to 1000 °C, every 100 °C. Slope was 10 °C/min. The program package GSAS—EXPGUI was used for the calculation of cell parameters, using the Rietveld full-profile method starting with the structural models proposed by Albert et al.13 for Na-P1 and Gatta et al.35 for nepheline.
Morphological analyses were obtained by means of scanning electron microscopy (JEOL JSM-840 served by a LINK Microanalysis EDS system, with operating conditions of 15 kV and window conditions ranging from 18 to 22 mm)36.
Induced coupled plasma optical emission spectroscopy technique (ICP-OES, Perkin Elmer Optima 3200 RL) was conducted on synthesized powders through previous fusion (Pt meltpot) in lithium meta-tetra borate pearls and subsequent acid solubilisation and analytical determination37.
Zeolite density was calculated by He-picnometry using an AccuPyc 1330 pycnometer. The specific surface and porosity were obtained by applying the BET (Brunauer–Emmett–Teller) method with N2 using a Micromeritics ASAP2010 instrument (operating from 10 to 127 kPa)38.
The infrared analysis was performed with a spectrometer FTLA2000, served by a separator of KBr and a DTGS detector; the source of IR radiation was a SiC (Globar) filament. Samples were treated according the method of Novembre et al.39, 40 using powder pressed pellets (KBr/sample ratio of 1/100, pressure undergone prior determination 15 t/cm2); spectra were processed with the program GRAMS-Al (GRAMS/AI Spectroscopy Software, Thermo Scientific Company).
Differential thermal analysis (DTA) and thermogravimetry (TG) were performed on the zeolitic powder using a Mettler TGA/SDTA851e instrument (10°/min, 30–1100 °C, sample mass of ~ 10 mg, Al2O3 crucible) (Mettler Toledo, Greifensee, Switzerland)41.
Results
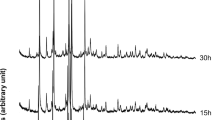
Results of XRPD analyses performed on the two synthesis runs conducted at 65 °C and 100 °C are illustrated in Figs.1, 2, respectively.

X-ray diffractometric sequence of the synthesis run at 65 °C.

X-ray diffractometric sequence of the synthesis run at 100 °C.
Analyzing the two synthesis runs carried out at 65 and 100 °C, it is clear that the best results are obtained in the second case. In fact, in the synthesis conducted at 65 °C, the presence in the XRD spectrum of small reflections below 10° of 2 theta is observed, probably referable to intermediate metastable phases; in fact, these peaks tend to decrease considerably in intensity passing to 91 h, and this leads us to think that with the prolongation of the heat treatment these phases are eliminated. The synthesis run carried out at 100 °C, on the other hand, shows the absence of such peaks and leads us to think that the effect of raising the synthesis temperature from 65 to 100 °C is unfavorable to the formation of these phases. When analyzing the experiment performed at 65 °C, appearance of Na-P1 zeolite is evident at 24 h. The existence field of the NaP1 zeolite is very large; peaks grow in height until reaching the maximum intensity at 91 h. Results of the QPA analyses conducted on samples at 24, 57 and 91 h are illustrated in Table 1. The zeolitic percentage increases over time to the detriment of the amorphous component and reaches its climax at 91 h (91.98%). For the sample at 91 h the observed and calculated profiles and difference plots for Na-P1 and corundum NIST 676a are reported in Fig. 3a.

Rietveld refinement plot: observed (+) and calculated profiles and difference plot for Na-P1 zeolite and corundum NIST 676a with tick marks at the position of the Bragg peaks. From the bottom: Na-P1 zeolite, curundum NIST 676a. (a) Sample at 65 °C; (b) Sample at 100 °C.
The crystallization of Na-P1 zeolite is verified by the PXRD analyses (Fig. 2) in the time interval 8–54 h in the experiment performed at 100 °C. About 50% of the crystallization of the Na-P1 zeolite takes place in the first 22 h of the synthesis run (Table 1). The climax in the crystallization of the zeolite is reached at 54 h with the achievement of 92.7% of zeolitic phase. The observed and calculated profiles and difference plots for Na-P1 and corundum NIST 676a have been performed for the sample at 54 h (Fig. 3b).
Cell parameters of Na-P1, refined with orthorombic simmetry space group Pna21, remain constant within error as a function of the experimental run time both at 65 and 100 °C (Table 1). The results of the Rietveld refinements provide cell values that are in good agreement with the structural model proposed by Albert et al.13.
Figure 4a,b reports SEM images of Na-P1 crystals from experimental runs performed at 65° (91 h) and 100 °C (54 h), respectively. It results a ball-like morphology of the crystals and an average maximum length of crystals around 1–2 µm.

SEM image of Na-P1 zeolite crystals obtained at 91 h (65 °C) (a) and at 54 h (100 °C) (b) of the synthesis runs.
Chemical analysis performed on samples at 91 h (65 °C) and 54 h (100 °C) resulted in the stoichiometry of Na6.00Al5.94Si10.02O32 and Na6.02Al5.91Si10.03O32 respectively. The density of Na-P1 from samples at 91 h (65 °C) and 54 h (100 °C) was determined to be 2.031(5) and 2.042 (4) g/cm3, respectively, in good agreement with Breck42.
Further characterizations were carried out on the sample at 91 h (65 °C). Figure 5 illustrates the infrared spectrum of the sample. The significant broad peaks are located at 3408 and 1636 cm−1 for O–H stretching and bending, respectively. The band at 1094 is assigned to to the asymmetric stretching vibration due to external linkages between tetrahedra, structure sensitive, sensu Flaningen et al.43 and that at 977 cm−1 is assigned to the asymmetric stretching vibration caused by internal vibrations of the framework SiO4. The bands at 744 and 676 cm−1 are attributed to Si–O–Si symmetric stretching vibration of internal tetrahedron. The peak at 604 cm−1 is attributed to the double rings vibration. Data are coherent with those available in the literature8, 13, 43.

IR spectrum of the Na-P1 zeolite at 91 h (100 °C).
Thermogravimetric analysis conducted on sample at 91 h (65 °C) revealed a gradual and continuous water loss up to 1000 °C (Fig. 6). In particular it indicates a two-stage mass loss. At the first stage, c.a. 12% loss occurs at about 191 °C; this was reasonable due to the loss of the adsorbed water. The second occurred between 191 and 850 °C with a mass loss of 5.6% and is related to the removal of the crystal water. The endothermic peaks revealed by the DTA curve at 122 °C reflects the dehydration process and is in agreement with findings by Zubowa et al. and Huo et al.8, 17. At about 970 °C there is an exothermic peak which testifies the transformation into a new phase.

DTA-TG analysis of the sample at 91 h (100 °C).
The high temperature X ray diffraction on sample at 91 h (65 °C) (Fig. 7) reveals the lower of simmetry to tetragonal NaP structure, just visible at 100 °C; then the tetragonal phase transforms into phillipsite in the temperature interval 400–800 °C, and finally the passage to the stable phase of nepheline is testified at 1000 °C. The phase transitions observed are in agreement with the findings of Huo et al.8. The exothermic peak revealed by the DTA curve is indicative of the crystallisation of nepheline. Rietvield refinement of sample at 1000 °C provides crystallographic values of a = b = 10.15 Å, c = 8,44 Å, α = β = 90°, γ = 120°.

High temperature XRD pattern of sample at 91 h (100 °C). Full squares: Na-P1; empty squares: tetragonal NaP; empty stars : phillipsite; full stars: nepheline.
Figure 8a shows the N2 adsorption–desorption plots at 77 K for zeolite Na-P1. There is evidence of a hysteresis loop indicating the presence of mesopores; the vertical hysteresis loop indicates cylindrical mesopores. There is a steep increase in the adsorbed amount of nitrogen when the relative pressure (P/P0) is higher than 0.8. Micropore-size distribution was calculated according to the method proposed by Horvath and Kawazoe (Fig. 8b). The absence of an inflection point demonstrates the absence of micropores in the sample analyzed.

(a) Nitrogen adsorption–desorption isotherms of the Na-P1 zeolite and (b) pore size distribution pattern.
Conclusion
This work describes the synthesis of zeolite Na-P1 using a kaolinitic rock. Appearance of Na-P1 phase begins at about 24 h at 65 °C and at about 8 h at 100 °C. The existence field of the Li-A(BW) zeolite is very large, in fact no phase replaces the zeolite in the time interval 1–91 h at 65 °C and 1–54 h at 100 °C. The chemical-physical, morphological and spectroscopic characterization of experimental products proved the efficacy of the experimental procedure proposed here.
When our results are compared with those of other authors who have synthesized the same zeolite starting from a natural precursor, a reduction of the calcination temperature of kaolinite, of synthesis temperature, and crystallization time is evident. Lovat et al.23 and Li et al.15 in fact, operate a calcination temperature of kaolinite of 800 °C, while we reduced it to 650 °C. In addition, the same authors investigated the reaction of metakaolinite in presence of fluoride ions; here the synthesis protocol does not include the use of these additives. Another important observation is the reduction of synthesis times, which in our case makes the experimental synthesis protocol very attractive from an economic, and therefore industrial, point of view. In fact, the previous authors synthesize the Na-P1 zeolite at 85 °C in 60 days23 and at 180 °C in 48 h15. In the present research the zeolite instead appears at 65 °C already at 24 h and at 100 °C at 8 h. Another substantial difference between our work and that of previous authors lies in the effective assessment of the degree of success of the experiment from calculation by QPA of the percentage of crystallization vs. amorphous material and other impurities. The industry requires at least 90% pure products. And our powders reach 91.98% purity at 65 °C (91 h) and 92.7% at 100 °C (54 h). The results of the QPA analyses and the wide temporal range of stability of this zeolite suggests that transfer to an industrial production scale would be possible.
References
Nery, J. G., Mascarenhas, Y. P. & Cheetham, A. K. A study of the highly crystalline, low-silica, fully hydrated zeolite P ion exchanged with (Mn2+, Cd2+, Pb2+, Sr2+, Ba2+) cations. Micropor. Mesopor. Mat. 57, 229–248 (2003).
Adams, C. J., Araya, A., Cunningham, K. J., Franklin, K. R. & White, I. F. Measurement and prediction of Ca–Na ion-exchange equilibrium in maximum aluminium P (MAP), a zeolite with the GIS framework topology. J. Chem. Soc. Faraday Trans. 93, 499–503 (1997).
Novembre, D., Di Sabatino, B., Gimeno, D. & Pace, C. Synthesis and characterization of Na-X, Na-A and Na-P zeolites and hydroxysodalite from metakaolinite. Clay Miner. 46, 336–354 (2011).
Brown, G. T., Osinga, T. J., Parkington, M. J. & Steel, A. T. Europatent No. 89311284, 7 (1989).
Hakansson, U., Falth, L. & Hansen, S. Structure of a high-silica variety of zeolite Na-P. Acta Crystallogr. C 46, 1363–1364 (1990).
Atkins, M., Glasser, F. P. & Jack, J. J. Zeolite P in cements: Its potential for immobilizing toxic and radioactive waste species. Waste Manag. 15(2), 127–135 (1995).
Moirou, A., Vaxevanidou, A., Christidis, G. E. & Paspaliaris, I. Ion exchange of zeolite Na-Pc with Pb2+, Zn2+, and Ni2+ ions. Clays Clay Min. 48, 563–571 (2000).
Huo, Z. et al. Thermal study of NaP zeolite with different morphologies. J. Therm. Anal. Calorim. 111, 365–369 (2013).
Szala, B., Bajda, T. & Jelen, A. Removal of Chromium (VI) from aqueous solutions using zeolites modified with HDTMA and ODTMA surfactants. Clay Miner 50(1), 103–115 (2015).
Meier, W.M. & Olson, D.H. Atlas of Zeolite Structure Types, 4th ed. (ed. Elsevier Science) 405pp (London, 1996).
Baerlocher, Ch. & Meier, W. M. The crystal structure of synthetic zeolite Na-P1, an isotope of gismondine. Z. Kristallogr. 135, 339–354 (1971).
Hansen, S., Hakansson, U., Landa-Canovas, A. R. & Falth, L. On the crystal chemistry of NaP zeolites. Zeolites 13, 276–280 (1993).
Albert, B. R., Cheetham, A. K., Stuart, J. A. & Adams, C. J. Investigations on P zeolites: synthesis, characterization, and structure of highly crystalline low-silica NaP. Micropor. Mesopor. Mat. 21, 133–142 (1998).
Ali, I.O., El-Sheikh, S.M., Salama, T.M., Bakr, M.F. & Fodial, M.H. Controllable synthesis of NaP zeolite and its application in calcium adsorption. Sci. China Mater. 1–13 (2015).
Li, J.T., Zeng, X, Chen, R.Y., Yang, X.B. & Luo, X.T. Synthesis of pure zeolite Na-P from kaolin and enhancement of crystallization rate by sodium floride in Advanced Materials, Structure and Mechanical Engineering- (ed. Kaloop, Taylor and Francis Group) (London, 2016).
Meftha, M., Oueslati, W., Chorfi, N. & Ben Haj Amara, A. Effect of the raw material type and the reaction time on the synthesis of halloysite based Zeolite Na-P1. Results Phys., 7, 1475–1484 (2017).
Zubowa, H. L. et al. Crystallization of phase-pure zeolite NaP from MCM-22-type gel compositions under microwave radiation. Micropor. Mesopor. Mat. 108, 542–548 (2008).
Sathupunya, M., Gulari, E. & Wongkasemjit, S. ANA and GIS zeolite synthesis directly from alumatrane and silatrane by sol-gel process and microwave technique. J. Eur. Ceram. Soc. 22, 2305–2314 (2002).
Pal, P., Das, J. K., Das, N. & Bandyopadhyay, S. Synthesis of NaP zeolite at room temperature and short crystallization time by sonochemical method. Ultrason. Sonochem. 20, 314–321 (2013).
Srinivasan, A. & Grutzeck, M. W. The adsorption of SO2 by zeolites synthesized from Fly Ash. Environ. Sci. Technol. 33, 1464–1469 (1999).
Murayama, N., Yamamoto, H. & Shibata, J. Mechanism of zeolite synthesis from coal fly ash by alkali hydrothermal reaction. Int. J. Miner. Process. 64, 1–17 (2002).
Kazemian, H., Naghdali, Z., Kashani, T. G. & Farhadi, F. Conversion of high silicon fly ash to Na-P1 zeolite: Alkaline fusion followed by hydrothermal crystallization. Adv. Powder Technol. 21, 279–283 (2010).
Lovat, V. C. & Chandrasekhar, S. Hydrothermal reaction of kaolinite in presence of fluoride ions at pH<10. Zeolites 13, 534–541 (1993).
Baccouche, A., Srasra, E. & El Maaoui, M. Preparation of Na-P1 and sodalite octahydrate zeolites from interstratified illite-smectiote. Appl. Clay Sci. 13, 255–273 (1998).
Khabuanchalead, S., Khemthong, P., Prayoonpokarack, S. & Wittayakun, J. Transformation of zeolite NaY synthesized from rice husk silica to NaP during hydrothermal synthesis. J. Sci. Technol. 15(3), 225–231 (2008).
Kats, A., Brough, A. R., Kirkpatrick, R. J., Struble, L. & Young, F. Effect of solution concentration on the properties of a cementitious GRout wasteform for low-level nuclear waste. Nucl. Technol. 129(2), 236–245 (2017).
Novembre, D., Di Sabatino, B. & Gimeno, D. Synthesis of Na-A zeolite from 10 Å halloysite and a new crystallization kinetic model for the transformation of Na-A into HS zeolite. Clay. Clay Miner. 53(1), 28–36 (2005).
Novembre, D., Gimeno, D., Pasculli, A. & Di Sabatino, B. Synthesis and characterization of sodalite using natural kaolinite: an analytical and mathematical approach to simulate the loss in weight of chlorine during the synthesis process. Fresen. Environ. Bull. 19(6), 1109–1117 (2010).
Novembre, D., Pasculli, A., Pace, C., Gimeno, D. & Di Sabatino, B. Synthesis of sodalite from natural kaolinite. A way to simulate the loss in weight of chlorine during synthesis process by an analytical and mathematical modelling. Rend. Online Soc. Geol. It. 11(2), 548–549 (2010).
Novembre, D., Gimeno, D., d’Alessandro, N. & Tonucci, L. Hydrothermal synthesis and characterization of kalsilite by using a kaolinitic rock from Sardinia, Italy, and its application in the production of biodiesel. Mineral. Mag. 82(4), 961–973 (2018).
Novembre, D., Gimeno, D. & Del Vecchio, A. Improvement in the synthesis conditions and studying the physicochemical properties of the zeolite Li-A(BW) obtained from a kaolinitic rock. Sci. Rep. 10(1), 5715–5723 (2020).
Novembre, D., Pace, C. & Gimeno, D. Synthesis and characterization of wollastonite-2M by using a diatomite precursor. Mineral. Mag. 82(1), 95–110 (2018).
Larson A.C. & Von Dreele R.B. GSAS: general structure analysis system. Document Laur 86–748, Los Alamos National Laboratory (1997).
Toby, B. H. EXPGUI, a graphical user interface for GSAS. J. Appl. Crystallogr. 34, 210–213 (2001).
Gatta, G. D. & Angel, R. J. Elastic behavior and pressure-induced structural evolution of nepheline: Implications for the nature of the modulated superstructure. Am. Mineral. 92, 1446–1455 (2007).
Novembre, D., Di Sabatino, B., Gimeno, D., Garcia Valles, M. & Martinez-Manent, S. Synthesis of Na-X zeolites from tripolaceous deposits (Crotone, Italy) and volcanic zeolitized rocks (Vico Volcano, Italy). Micropor. Mesopor. Mat., 75, 1–11 (2004).
Fernandez-Turiel, J. L., Gimeno, D., Valero, F., Carnicero, M. & Rodríguez, J. J. Spatial and seasonal water quality in a mediterranean catchment: the Llobregat river (NE Spain). Environ. Geochem. Health 25, 253–474 (2003).
Ruggieri, F., Fernandez-Turiel, J. L., Saavedra, J., Gimeno, D. & Garcia-Valles, M. Environmental geochemistry of ancient volcanic ashes. J. Hazard. Mater. 183, 353–365 (2010).
Novembre, D. & Gimeno, D. The solid-state conversion of kaolin to KAlSiO4 minerals: The effects of time and temperature. Clays Clay Min. 65(5), 355–366 (2017).
Novembre, D., Gimeno, D. & Poe, B. Synthesis and characterization of leucite using a diatomite precursor. Sci. Rep. 9, 10051–10061 (2019).
Novembre, D., Pace, C. & Gimeno, D. Syntheses and characterization of zeolites K-F and W type using a diatomite precursor. Mineral. Mag. 78, 1209–1225 (2014).
Breck, D.W. Zeolite Molecular Sieves. Structure, Chemistry and Use. Robert E. Krieger Publishing Company, Malabar, Florida, 771 pp. (1984).
Flanigen, E.M., Khatami, H. & Szymanski, H.A. Infrared structural studies of zeolite frameworks. In: Flanigen, E.M., and Sand, L.B., Molecular Sieve Zeolites, Advances in Chemistry 101, American Chemical Society, Washington DC, 201–229 (1971).
Horwath, G. & Kawazoe, K. Method for the calculation of effective pore size distribution in molecular sieve carbon. J. Chem. Eng. Jpn. 16, 470–475 (1983).
Acknowledgements
The authors greatly acknowledge the technical staff at CCiT (UB) for their help during the development of the work.
Author information
Authors and Affiliations
Contributions
D.N., D. G. and A.D.V. wrote the main manuscript text and D.N. prepared figures and tables. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Novembre, D., Gimeno, D. & Del Vecchio, A. Synthesis and characterization of Na-P1 (GIS) zeolite using a kaolinitic rock. Sci Rep 11, 4872 (2021). https://doi.org/10.1038/s41598-021-84383-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84383-7
- Springer Nature Limited
This article is cited by
-
Optimized removal of chloroform and DMDS using synthetic zeolite (Na-P1) and activated carbon composite
Modeling Earth Systems and Environment (2024)
-
A Review on Zeolite: Application, Synthesis and Effect of Synthesis Parameters on Product Properties
Chemistry Africa (2022)
-
Synthesis and characterization of analcime (ANA) zeolite using a kaolinitic rock
Scientific Reports (2021)