
Abstract
In this study, we analyzed the complete mitochondrial genome (mitogenome) of Speiredonia retorta, which is a pest and a member of the Lepidoptera order. In total, the S. retorta mitogenome was found to contain 15,652 base pairs encoding 13 protein-coding genes (PCGs), 22 tRNAs, 2 rRNAs, as well as an adenine (A) + thymine (T)-rich region. These findings were consistent with the mitogenome composition of other lepidopterans, as we identified all 13 PCGs beginning at ATN codons. We also found that 11 PCGs terminated with canonical stop codons, whereas cox2 and nad4 exhibited incomplete termination codons. By analyzing the mitogenome of S. retorta using Bayesian inference (BI) and maximum likelihood (ML) models, we were able to further confirm that this species is a member of the Erebidae family.
Similar content being viewed by others


Introduction
Speiredonia retorta (Lepidoptera: Erebidae) is a pest species that is widely distributed throughout Southeast Asia. S. retorta larvae can adhere to Acacia leaves and branches where they undergo pupation, while adults can feed on a range of fruits including apples, pears, and grapes, leading to their more rapid decay. These moths produce three generations per year, with a life cycle consisting of an egg stage (6–18 days), a larval stage with six instars (23–47 days), and a pupal stage (8–13 days). These insects overwinter in the pupal stage, which can last from 194–228 days1. The primary predators of this species include Ophion luteus (Hymenoptera: Ichneumonidae), Brachymeria obscurata (Hymenoptera: Chalalcididae), and Exorista sorbillans (Diptera: Tachinidae). Given that these moths represent a significant economic threat, their management is a key agricultural concern and necessitates an in-depth understanding of their biology. Further exploration of the phylogeny and genetic characteristics of S. retorta has the potential to offer novel insights into how to appropriately combat the spread of these moths. Understanding and analyzing the mitochondrial genome (mitogenome) of this species, in particular, may facilitate key comparative phylogenetic and evolutionary studies that can support such preventative efforts2,3.
The mitogenome of metazoan species generally ranges from 14,000–19,000 bp in size, with few or no intergenic spacer regions4, encoding 13 protein-coding genes (PCGs), 22 tRNAs, and 2 rRNAs5,6. In addition to these more broadly conserved features, members of the Lepidoptera order generally exhibit a conserved adenine and thymine (A + T)-rich region within their mitogenome. The mitogenome represents an ideal tool for the analysis of phylogenetic relationships, given that it has a simple structure, is maternally inherited, rarely undergoes recombination, and is conserved over the course of evolution7,8,9. Modern advances in sequencing techniques have led to the publication of mitogenomes from many different insects and other species, thereby supporting a wide range of evolutionary analyses6.
By conducting a comprehensive analysis of the mitogenome of a given insect species, we have the opportunity to perform intricate phylogenetic or population genetics studies and to identify potentially novel genes that may serve as valuable targets in future research efforts. The Lepidoptera order is the second-largest Insecta order and is composed of > 155,000 species of moths and butterflies10. Noctuoidea is the largest Lepidopteran sub-family, with > 42,400 species11. Several characteristic mitogenomic markers associated with these phylogenetic classes of insects have been identified to date, enabling us to reliably explore the phylogenetic relationship between S. retorta and other species through mitogenomic analyses. Six Noctuidae families were proposed in a phylogenetic framework constructed by Zahiri et al., including the Euteliidae, Erebidae, Nolidae, Notodontidae, Oenosandridae, and Noctuidae families12. Given the clear value of such analyses, in the present study we sequenced the full S. retorta mitogenome in an effort to more fully explore the evolutionary relationship between this agriculturally important insect and other Noctuidae species.
Results and discussion
Mitogenome structure, organization, and composition
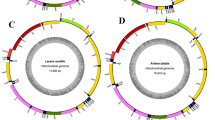
Our sequencing revealed the S. retorta mitogenome to be 15,652 bp in length (Fig. 1), consistent with reported mitogenomic lengths in other Lepidopteran species such as Thitarodes pui (Hepialidae; 15,064 bp) and Plutella xylostella (Plutellidae; 16,179 bp). We then aligned the S. retorta mitogenome sequences with those of other Lepidopteran species, enabling us to identify 13 PCGs (atp6, atp8, cox1, cox2, cox3, cytb, nad1, nad2, nad3, nad4, nad5, nad6, and nad4L), two rRNAs (large and small rRNA), 22 tRNAs, and a non-coding A + T-rich region that is conserved within the mitogenome of most known animal species5 (Table 1). The S. retorta mitogenome also harbored a trnM-trnI-trnQ gene arrangement that was distinct from the ancestral trnI-trnQ-trnM gene order6.

A map of the S. retorta mitogenome. Labeling of tRNA genes was conducted in accordance with IUPAC-IUB single-letter amino acid codes. The cytochrome c oxidase subunits (cox1-3), cytochrome b (cob), NADH dehydrogenase components (nad1-6), and rRNAs (rrnL and rrnS) are as indicated.
The composition and skewness of the S. retorta mitogenome were compared to those of other Noctuoidea species (Table 2). The major strand was composed of A, T, G, and C nucleotides at relative frequencies of 35.59%, 41.23%, 7.37%, and 11.80%, respectively (80.83% A + T), thus exhibiting negative AT and GC skewness (− 0.020 and − 0.231, respectively). The AT skewness of other Noctuoidea mitogenomes has been found to range between 0.016 (L. dispar) and − 0.027 (A. formosae), whereas GC skewness values range from − 0.266 (A. formosae) to − 0.178 (A. ipsilon). The negative AT skewness in S. retorta indicates that there are more T residues than A residues, as previously reported for many Lepidopteran species including A. formosae (− 0.027), and A. ipsilon (− 0.006) (Table 2).
PCGs and codon usage
We identified 13 total PCGs in the S. retorta mitogenome, spanning 71.1% of the mitogenome sequence (11,134 bp). These genes were between 159 bp (atp8) and 1713 bp (nad5) in length. This is consistent with findings from other Lepidopteran species in which these two mitogenes are often the shortest and longest, respectively11,13. All detected PCGs began with an ATN codon (2 ATA, 3 ATT, and 8 ATG). Atypical cox1 start codons (including TTAG, ACG, and TTG) have been reported in the mitogenomes of a range of insect species14,15,16. The TAA stop codon was the most common among these PCGs (nad2, cox1, atp8, atp6, cox3, nad3, nad5, nad4L, nad6, cytb), while nad1 utilized a TAG stop codon, and nad4 and cox2 harbored incomplete stop codons (T) (Table 1). This is a common conserved mitogenomic feature among invertebrates17,18,19, and this single T residue can be still be recognized by endonucleases during polycistronic pre-mRNA transcription, with polyadenylation from contiguous PCGs ultimately yielding a functional stop codon5,20,21.
Next, we assessed codon usage among a range of Lepidopteran species, including three Noctuoidae members as well as Bombycoidea, Tortricoidea, and Geometroidea members (Fig. 2). Through this approach, we found Asn, Ile, Leu2, Lys, Met, Phe, and Tyr to be the amino acids that were used most often. In these 6 species, we identified 4 codon families with at least 80 codons per thousand codons (Ile and Phe), and 3 with a minimum of 60 codons per thousand codons (Leu2, Met and Asn). The Arg and Cys codon families were the least represented. The codon distributions were consistent among the three Noctuoidae species analyzed herein, with the exception of Lys, which was rarely encoded in S. retorta (Fig. 3).

A comparative assessment of mitogenomic codon usage among Lepidopteran insects. Superfamily membership is indicated above species names using lowercase letters (a: Noctuoidea, b: Bombycoidea, c: Tortricoidea, d: Geometroidea).

The distribution of codons among Lepidopteran species. CDspT codons per thousand codons.
The RCSU was next assessed in the PCGs encoded by the mitogenomes of the four Lepidopteran superfamilies (Fig. 4). We found that the PCGs of S. retorta contained all possible combinations of codons other than GCG, CGC, GGC, AGG, CCG, ACG, and TGG. This lack of GC-rich codons is common to many other Lepidopterans including H. cunea (GCG and GTG), P. atrilineata (CGG), and C. pomonella (GCG). Utilized codons showed a bias towards A + T content, contributing to the overall mitogenomic AT-bias that was observed both in S. retorta and across other insect mitogenomes19,22.

Mitogenome Relative Synonymous Codon Usage (RSCU) for the four Lepidopteran superfamilies, with codon families on the x-axis. Any codons above the bar were absent within the mitogenome.
Ribosomal and transfer RNA genes
Two rRNA genes were identified within the S. retorta mitogenome, consistent with findings in most other animal species. The 1413 bp large ribosomal RNA gene (rrnL) was located in between tRNA Leu (CUN) and tRNA Val, while the 781 bp small ribosomal RNA gene (rrnS) was between tRNA Val and the A + T-rich region (Table 1). These rRNA genes were A + T rich (84.40%), falling within the range observed for other Noctuoidae species including A. formosae (83.77%) and A. ipsilon (85.15%). These rRNA AT and GC skewness values have been found to be negative in the majority of analyzed Lepidopteran mitogenomes17, however, in S. retorta these values were positive (0.040 and 0.356, respectively), as has been reported in a subset of prior studies23,24.
We identified a full set of 22 tRNA genes (65–71 nucleotides long) in the S. retorta mitogenome, consistent with findings from other Lepidopterans. These tRNA regions were heavily A + T biased (81.39%), with positive AT and GC skewness (0.031 and 0.171, respectively; Table 2). All of these tRNAs exhibited expected cloverleaf-like secondary structures, although the DHU stem was lacking from trnS1 (Fig. 5), as has previously been observed in other Lepidopterans23. Furthermore, 8/22 tRNAs were encoded on the L-strand while 12/22 were encoded on the H-strand.

Predicted secondary structures for the 22 S. retorta mitogenome-encoded tRNAs.
Overlapping and intergenic spacer regions
There are 6 overlapping regions in the S. retorta mitogenome. These regions range from 1 to 8 bp in size (26 bp total), with the longest region of overlap being located between trnC and trnY (Table 1). When we aligned the region of overlap between atp6 and atp8, we found that S. retorta mitogenome contained an ATGATAA nucleotide sequence common to other Lepidopterans (Fig. 6). In addition, we identified the ‘ATACTAA’ motif within the 17 bp region between trnS2 (UCN) and nad1 (Fig. 7A), with this motif being highly conserved in insect mitogenomes in addition to being a potential mitochondrial transcription termination peptide-binding site (mtTERM protein)25.

Alignment of atp8 and atp6 overlap among Lepidopteran species and other insects, with the number of intergenic nucleotides shown on the right.

(A) The intergenic spacer region between trnS2 (UCN) and nad1 was aligned for multiple Lepidopteran species, with the conserved ‘ATACTAA’ motif being highlighted. (B) Features of the S. retorta A + T-rich region are shown in the reverse strand, with the ATATGA motif being highlighted. Double underlines were used to mark the poly-A region, while a single underline was used to mark the poly-T region. A dotted underline was used to mark single microsatellite T/A repeats.
A + T-rich region analysis
An A + T-rich region that was 417 bp long was detected between rrnS and trnM in the S. retorta mitogenome. This region was composed of 93.76% AT residues and exhibited negative AT and GC skewness (− 0.038 and − 0.462, respectively) (Table 2). This region contained a number of short repeated sequences, such as a 19 bp poly-T region that flanked an ‘ATAGA’ motif near rrnS, an ‘ATTTA’ roughly in the center of this A + T-rich region, as well as a poly-A element that was located upstream relative to trnM, consistent with mitogenomic findings from other Lepidopteran species (Fig. 7B). In addition, while the exact poly-T region length varies among Lepidopterans4,17,26, the ATAGA motif is highly conserved27.
Phylogenetic analyses
A number of recent studies have explored phylogenetic relationships among Noctuoidea species. In one recent analysis, Zahiri et al. proposed the following relationship among these families: (Notodontidae + (Euteliidae + (Noctuidae + (Erebidae + Nolidae))))28. In contrast, Yang et al. published another study in which the following grouping scheme was proposed: (Notodontidae + (Erebidae + (Nolidae + (Euteliidae + Noctuidae))))29. In this study, we utilized BI and ML methods as well as the MAFFT alignment approach in order to explore the relationship between S. retorta and other Noctuidae insects according to its mitogenome sequence. We utilized the NT dataset to conduct phylogenetic analyses of 65 full mitogenomes which were representative of six Noctuoidea families (Erebidae, Lymantriidae, Euteliidae, Noctuidae, Notodontidae, and Nolidae). For outgrouping purposes, we additionally utilized the mitogenomes of Ahamus yunnanensis (NC_018095) and Thitarodes pui (NC_023530) in the present analysis (Fig. 8). According to Homziak’s study, our analyses revealed a topology within Erebinae that was as follows: ((Catocala sp + Speiredonia retorta) + (Grammodes geometrica + Parallelia stuposa) + Eudocima phalonia). The result indicated that Catocalini belongs to Erebinae subfamily30, as confirmed via BI (Fig. 8A) and ML (Fig. 8B) analyses. This approach further confirmed that S. retorta is a member of Erebidae: (Notodontidae + (Erebidae + Lymantriidae + (Nolidae + (Euteliidae + Noctuidae)))). This is distinct from a study conducted by Zahiri et al. that yielded different phylogenetic results: (Notodontidae + (Euteliidae + (Noctuidae + (Erebidae + Nolidae))))28.

Tree showing the phylogenetic relationships among Noctuoidea insects, constructed using (A) Bayesian inference (BI). (B) Maximum Likelihood method (ML). As outgroups for this analysis, we utilized Ahamus yunnanensis (NC_018095) and Thitarodes pui (NC_023530).
S. retorta has previously been identified as a member of the Noctuoidea superfamily within the Erebidae family and the Erebinae subfamily30. Our data were consistent with this hypothesis. Even so, our findings were distinct from those of the prior study with respect to some of the identified relationships, suggesting that the sequencing of the mitogenomes of additional Noctuoidea species will be required to more accurately resolve these phylogenetic relationships.
Materials and methods
Insects and DNA collection
Samples of S. retorta were obtained from Bengbu Medical College, Anhui Province, China. The collected specimens were identified as Speiredonia retorta based on their morphological characteristics by a taxonomist of the Department of Entomology, Anhui Agricultural University, Hefei, China (AHAU). After a careful examination of the morphological characteristics and the comparison of voucher specimens to the referenced publications regarding Healthy plantations from the Forest Science Institute of Vietnam1. A Genomic DNA Extraction Ki (Aidlab Co., Beijing, China) was used to extract DNA from these samples, after which 1% agarose gel electrophoresis (AGE) was used to evaluate DNA quality. Samples were then used for mitogenome isolation.
Mitogenome sequencing
The S. retorta mitogenome was amplified using 12 primer pairs designed based on known conserved mitogenome sequences in other Lepidopteran species (Table 3; BGI Group Co., Guangdong, China)2,31,32. An Eppendorf Mastercycler and Mastercycler gradient were used to amplify mitogenomic sequences in a 50 μL reaction volume containing 35 μL dH2O, 5 μL 10 × Taq buffer (Mg2+ plus), 4 μL dNTP (25 mM), 1.5 μL DNA, 2 μL of each primer (F + R; 10 μM) as well as 0.5 μL TaqDNA polymerase (Takara Co., Dalian, China). Thermocycler settings were: 94 °C for 4 min; 38 cycles of 94 °C for 30 s, 48–59 °C for 1–3 min (based on predicted fragment length); 72 °C for 10 min. Samples were then separated via 1% AGE and collected with a DNA gel extraction kit (Transgen Co., Beijing, China), after which direct sequencing was conducted with appropriate PCR primers.
Sequence assembly and annotation
NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast) and SeqMan II (DNASTAR Inc.; WI, USA) were used to annotate the sequenced mitogenome. The Invertebrate Mitochondrial Genetic Code was used to identify putative proteins encoded by specific PCGs. An approach previously detailed by Junqueira et al.33 was used to assess skewness, with base composition being assessed as follows: AT skew = [A − T]/[A + T], GC skew = [G − C]/[G + C].
The tRNAscan-SE program (http://lowelab.ucsc.edu/tRNAscan-SE/) was used to identify tRNA genes34, with additional predictions being made based upon sequences capable of adopting a tRNA secondary structure and containing an anticodon. Tandem repeats within the A + T-rich region were identified with a tandem repeat finder application (http://tandem.bu.edu/trf/trf.html)35.
Codon usage and RCSU
We assessed codon usage across a range of Lepidopteran species, including three Noctuoidae members as well as Bombycoidea and Geometroidea members that were closely related to Noctuoidae species and Tortricoidea species that were far more distantly related to these species24,36 (Fig. 2). MEGA7.0 was used to calculate relative synonymous codon usage (RSCU) values37.
Phylogenetic analysis
Lepidopteran phylogenetic relationships were evaluated by utilizing all 64 Noctuoidea mitogenomes available from Genbank (Table 3), with the Ahamus yunnanensis (NC_018095) and Thitarodes pui (NC_023530) mitogenomes serving as an outgroup for this analysis. MAFFT was then used to conduct multiple alignments of concatenated nucleotide sequences for the 13 PCGs using default settings38, and the resultant concatenated sequences were used for phylogenetic analyses, which were conducted via the Maximum Likelihood (ML) method using MEGA7.037 and via a Bayesian Inference (BI) approach using MrBayes v3.239. A total of 1000 bootstrap replicates were used for ML analysis in order to develop phylogenetic trees. For the BI analysis, the GTR + I + G model was used for the analyzed nucleotide sequences, with MrModeltest 2.3 being used in accordance with Akaike's information criterion (AIC). In the BI analysis, four simultaneous MCMC chains were run for 10,000,000 generations, with sampling being conducted in 1000 generation intervals with a 2500 generation burn-in. FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) was then used to visualize the resultant phylogenetic trees.
References
Thu, P. Q. G. et al. Healthy Plantations. A Field Guide to Pests and Pathogens of Acacia, Eucalyptus and Pinus in Vietnam 7–8 (Agri-Science Queensland in the Department of Empolyment, Economic Development and Innovation, DEEDI, Queensland, 2010).
Sun, Y. et al. Comparative mitochondrial genome analysis of Daphnisnerii and other lepidopteran insects reveals conserved mitochondrial genome organization and phylogenetic relationships. PLoS ONE 12, e0178773. https://doi.org/10.1371/journal.pone.0178773 (2017).
Xin, Z. Z. et al. Mitochondrial genome of Argopectenirradians reveals higher-level phylogenetic relationships in Anisomyaria. Int. J. Biol. Macromol. 117, 1089–1092. https://doi.org/10.1016/j.ijbiomac.2018.06.008 (2018).
Jiang, S. T. et al. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogynapyretorum (Lepidoptera: Saturniidae). Int. J. Biol. Sci. 5, 351–365 (2009).
Cameron, S. L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 39, 400–411 (2014).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Lin, C. P. & Danforth, B. N. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogenet. Evol. 30, 686–702. https://doi.org/10.1016/S1055-7903(03)00241-0 (2004).
Cameron, S. L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. https://doi.org/10.1146/annurev-ento-011613-162007 (2014).
Wolstenholme, D. R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Sun, Y. et al. Characterization of the complete mitochondrial genome of Spilarctiarobusta (Lepidoptera: Noctuoidea: Erebidae) and its phylogenetic implications. Eur. J. Entomol. 113, 558–570. https://doi.org/10.14411/eje.2016.076 (2016).
Huang, Y. et al. Comparative mitochondrial genome analysis of Grammodesgeometrica and other noctuid insects reveals conserved mitochondrial genome organization and phylogeny. Int. J. Biol. Macromol. 125, 1257–1265. https://doi.org/10.1016/j.ijbiomac.2018.09.104 (2019).
Zahiri, R. et al. Molecular phylogenetics of Erebidae (Lepidoptera, Noctuoidea). Syst. Entomol. 37, 102–124. https://doi.org/10.1111/j.1365-3113.2011.00607.x (2012).
Dai, L. S., Kausar, S., Abbas, M. N. & Wang, T. T. Complete sequence and characterization of the Ectropisoblique mitochondrial genome and its phylogenetic implications. Int. J. Biol. Macromol. 107, 1142–1150. https://doi.org/10.1016/j.ijbiomac.2017.09.093 (2018).
Lee, E. S. et al. The mitochondrial genome of the smaller tea tortrix Adoxophyeshonmai (Lepidoptera: Tortricidae). Gene 373, 52–57. https://doi.org/10.1016/j.gene.2006.01.003 (2006).
Ogoh, K. & Ohmiya, Y. Complete mitochondrial DNA sequence of the sea-firefly, Vargulahilgendorfii (Crustacea, Ostracoda) with duplicate control regions. Gene 327, 131–139. https://doi.org/10.1016/j.gene.2003.11.011 (2004).
Lutz-Bonengel, S., Sanger, T., Pollak, S. & Szibor, R. Different methods to determine length heteroplasmy within the mitochondrial control region. Int. J. Legal Med. 118, 274–281. https://doi.org/10.1007/s00414-004-0457-0 (2004).
Liu, Q. N., Chai, X. Y., Bian, D. D., Zhou, C. L. & Tang, B. P. The complete mitochondrial genome of Plodiainterpunctella (Lepidoptera: Pyralidae) and comparison with other Pyraloidea insects. Genome 59, 37–49. https://doi.org/10.1139/gen-2015-0079 (2016).
Yang, L. et al. Characterization of the complete mitochondrial genome of Orthagaolivacea Warre (Lepidoptera Pyralidae) and comparison with other Lepidopteran insects. PLoS ONE 15, e0227831. https://doi.org/10.1371/journal.pone.0227831 (2020).
Li, J. et al. Mitochondrial genome characteristics of Somenascintillans (Lepidoptera: Erebidae) and comparation with other Noctuoidea insects. Genomics 111, 1239–1248. https://doi.org/10.1016/j.ygeno.2018.08.003 (2019).
Lu, C. et al. Complete sequence determination and analysis of Bombyxmori mitochondrial genome. J. Agric. Biotechnol. 10, 163–170 (2002).
Li, Q., Wang, X., Chen, X. & Han, B. Complete mitochondrial genome of the tea looper caterpillar, Ectropisobliqua (Lepidoptera: Geometridae) with a phylogenetic analysis of Geometridae. Int. J. Biol. Macromol. 114, 491–496. https://doi.org/10.1016/j.ijbiomac.2018.02.038 (2018).
Zhu, X. Y. et al. The complete mitochondrial genome of Closteraanastomosis (Lepidoptera: Notodontidae) and implication for the phylogenetic relationships of Noctuoidea species. Int. J. Biol. Macromol. 118, 1574–1583. https://doi.org/10.1016/j.ijbiomac.2018.06.188 (2018).
Xin, Z. Z. et al. Comparative mitochondrial genome analysis of Spilarctiasubcarnea and other noctuid insects. Int. J. Biol. Macromol. 107, 121–128. https://doi.org/10.1016/j.ijbiomac.2017.08.153 (2018).
Ramirez-Rios, V., Franco-Sierra, N. D., Alvarez, J. C., Saldamando-Benjumea, C. I. & Villanueva-Mejia, D. F. Mitochondrial genome characterization of Teciasolanivora (Lepidoptera: Gelechiidae) and its phylogenetic relationship with other lepidopteran insects. Gene 581, 107–116. https://doi.org/10.1016/j.gene.2016.01.031 (2016).
Taanman, J. W. The mitochondrial genome: Structure, transcription, translation and replication. Biochem. Biophys. Acta 1410, 103–123 (1999).
Sun, Y. U. et al. Mitochondrial genome of Abraxassuspecta (Lepidoptera: Geometridae) and comparative analysis with other Lepidopterans. Zootaxa 4254, 501–519. https://doi.org/10.11646/zootaxa.4254.5.1 (2017).
Cameron, S. L. & Whiting, M. F. The complete mitochondrial genome of the tobacco hornworm, Manducasexta (Insecta : Lepidoptera : Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408, 112–123. https://doi.org/10.1016/j.gene.2007.10.023 (2008).
Zahiri, R. et al. A new molecular phylogeny offers hope for a stable family level classification of the Noctuoidea (Lepidoptera). Zool. Scr. 40, 158–173. https://doi.org/10.1111/j.1463-6409.2010.00459.x (2011).
Yang, X., Cameron, S. L., Lees, D. C., Xue, D. & Han, H. A mitochondrial genome phylogeny of owlet moths (Lepidoptera: Noctuoidea), and examination of the utility of mitochondrial genomes for lepidopteran phylogenetics. Mol. Phylogenet. Evol. 85, 230–237. https://doi.org/10.1016/j.ympev.2015.02.005 (2015).
Homziak, N. T., Breinholt, J. W., Branham, M. A., Storer, C. G. & Kawahara, A. Y. Anchored hybrid enrichment phylogenomics resolves the backbone of erebine moths. Mol. Phylogenet. Evol. 131, 99–105. https://doi.org/10.1016/j.ympev.2018.10.038 (2019).
Liu, Q. N. et al. The complete mitochondrial genome of the common cutworm, Spodopteralitura (Lepidoptera: Noctuidade). Mitochondrial DNA 27, 122–123. https://doi.org/10.3109/19401736.2013.873934 (2016).
Dai, L. S. et al. Mitochondrial genome of the sweet potato hornworm, Agriusconvolvuli (Lepidoptera: Sphingidae), and comparison with other Lepidoptera species. Genome 60, 128–138. https://doi.org/10.1139/gen-2016-0058 (2017).
Junqueira, A. C. et al. The mitochondrial genome of the blowfly Chrysomyachloropyga (Diptera: Calliphoridae). Gene 339, 7–15. https://doi.org/10.1016/j.gene.2004.06.031 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Sun, Y. et al. The complete mitochondrial genome of the Papilioparis (Lepidoptera: Papilionidae). Mitochondrial DNA B 5, 733–735 (2020).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Ronquist, F. et al. MrBayes 32: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Acknowledgements
We would like to thank Mr. Junjie Li for his help in taxonomy. This work was supported by the Natural Science Foundation of Bengbu Medical College (Grant number: BYKF1730), the Key Program of Natural Science of Education Department of Anhui Province (Grant number: KJ2019A0310), the Key Program of Natural Science of Education Department of Anhui Province (Grant number: KJ2019A0323), Anhui Students’ Platform for Innovation and Entrepreneurship Training Program (Grant number: S201910367024), the Department of Science and Technology of Anhui Province (Grant number: 1804h08020290) and the Natural Science Foundation of China (Grant number: 21806002).
Author information
Authors and Affiliations
Contributions
Y.S. and J.G. designed the research. Y.S. and H.H. performed the research. Y.S., H.H., S.L., Y.L. and J.X., analyzed the data. J.G. and K.Z. contributed reagents/materials/analysis tools. Y.S. and J.G. wrote the paper with other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, Y., Huang, H., Liu, Y. et al. Organization and phylogenetic relationships of the mitochondrial genomes of Speiredonia retorta and other lepidopteran insects. Sci Rep 11, 2957 (2021). https://doi.org/10.1038/s41598-021-82561-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82561-1
- Springer Nature Limited
This article is cited by
-
Novel insight into lepidopteran phylogenetics from the mitochondrial genome of the apple fruit moth of the family Argyresthiidae
BMC Genomics (2024)
-
Characterization and Molecular Phylogenetic Analysis of Subfamily Erebinae (Lepidoptera: Noctuoidea: Erebidae) Using Five Complete Mitochondrial Genomes
Biochemical Genetics (2024)
-
Characterization of complete mitochondrial genomes of five noctuoid moths (Lepidoptera: Noctuoidea) and their phylogenetic implications
Journal of Biosciences (2023)
-
Phylogenomics including the newly sequenced mitogenomes of two moths (Noctuoidea, Erebidae) reveals Ischyja manlia (incertae sedis) as a member of subfamily Erebinae
Genetica (2023)
-
Morphological and molecular identification of the entomopathogenic fungus Purpureocillium lilacinum and its virulence against Tuta absoluta (Meyrick) (Lepidoptera: Gelechiidae) larvae and pupae
Egyptian Journal of Biological Pest Control (2022)