
Abstract
The difference between the skeletal muscle growth rates of Western and domestic breeds is remarkable, but the potential regulatory mechanism involved is still unclear. Numerous studies have pointed out that long intergenic noncoding RNA (lincRNA) plays a key role in skeletal muscle development. This study used published Yorkshire (LW) and Tibetan pig (TP) transcriptome data to explore the possible role of lincRNA in the difference in skeletal muscle development between the two breeds. 138 differentially expressed lincRNAs (DELs) were obtained between the two breeds, and their potential target genes (PTGs) were predicted. The results of GO and KEGG analysis revealed that PTGs are involved in multiple biological processes and pathways related to muscle development. The quantitative trait loci (QTLs) of DELs were predicted, and the results showed that most QTLs are related to muscle development. Finally, we constructed a co-expression network between muscle development related PTGs (MDRPTGs) and their corresponding DELs on the basis of their expression levels. The expression of DELs was significantly correlated with the corresponding MDRPTGs. Also, multiple MDRPTGs are involved in the key regulatory pathway of muscle fiber hypertrophy, which is the IGF-1-AKT-mTOR pathway. In summary, multiple lincRNAs that may cause differences in skeletal muscle development between the two breeds were identified, and their possible regulatory roles were explored. The findings of this study may provide a valuable reference for further research on the role of lincRNA in skeletal muscle development.
Similar content being viewed by others


Introduction
Tibetan pigs, also known as ginseng pigs, have less subcutaneous fat, more lean meat, higher amounts of protein and amino acids, and better taste than Yorkshire pigs1. Tibetan pigs are particularly popular in the high-end market. The price of Tibetan pigs is at least five times the prices of other commercial pork varieties in the Chinese market. However, compared with traditional commercial pig breeds, such as Yorkshire, Tibetan pigs have slower growth rates and lower reproductive rates. At 12 months old, Tibetan pigs usually weigh approximately 25 kg2. As a typical Western breed, Yorkshire pigs have a high lean rate, rapid muscle growth, and heavy body weight3. Therefore, understanding the difference in skeletal muscle growth and development between the two breeds is beneficial to the genetic improvement of pigs in the future.
In pig breeding, the growth and development of skeletal muscles directly affect the quantity and quality of animal meat. Related studies have shown that skeletal muscle growth is affected by the number, size, and type of muscle fibers4. and the number and size of muscle fibers are closely related to the tenderness of pork5,6.
Long intergenic non-coding RNA (lincRNA) is defined as RNA transcripts longer than 200 nucleotides7. Many studies have revealed the potential effects of lincRNA between different pig species on skeletal muscle development. Zou et al8. found that multiple lincRNAs in Yorkshire pigs and Wunahua pigs may cause differences in growth and meat quality between the two breeds in unknown ways. Liang Zhou et al9. found that Linc-YY1 can promote myogenic differentiation and muscle regeneration by interacting with the transcription factor YY1. However, the function of most lincRNAs in muscle is still unclear. This study focuses on key lincRNAs and explores its possible effects on pig skeletal muscle development by analyzing the regulatory pathways it may participate in.
Earlier studies obtained transcriptome expression profiles of Tibetan pigs and Yorkshire through RNA-seq data. Among them, 209 genes were screened in TP, including multiple genes related to the formation of muscle fibers. The author speculates that it may play an important role in determining the growth rate and potential weight of pigs after birth10. In this study, we used this set of RNA-seq data to explore the potential role of lincRNA on porcine skeletal muscle development. DELs were obtained through pipeline analysis and differential expression analysis, and potential target genes in cis were predicted. A co-expression network of MDRPTG and its corresponding long intergenic non-coding RNA (lincRNA) was constructed and used to explore the potential role of DEL in the process of muscle fiber hypertrophy.
Result
Transcriptome assembly and lincRNA identification
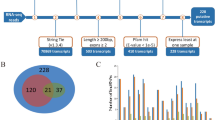
Six published RNA-seq data (Fig. S1,Table 1) sets of two pig breeds (Tibetan and Yorkshire)10 were used in identifying lincRNAs that may cause phenotypic differences between the breeds. Potential lincRNAs were obtained on the basis of this pipeline(Fig. 1A). Approximately 245.05 of 258.43 million reads were mapped to the pig reference genome (Sus scrofa 11.1) by Hisat2. The mapped reads of each data were assembled into one set of transcripts with StringTie, and all of the transcripts from six data were merged into a nonredundant transcript set. A total of 828 transcripts, which were > 200 bp intergenic transcripts with more than two exons, were obtained from this pipeline. Finally, three different methods namely, Cpc, Hmmer, and Blast were used for the assessment of the coding potential of the transcripts. A total of 361 potential lincRNAs were obtained. In addition, 53 of the potential lincRNAs had no overlaps with currently annotated coding or noncoding transcripts (Fig. 1B). These lincRNAs are distributed in all chromosomes except the Y chromosome (Fig. 1C).

LincRNA analysis pipeline, classification and chromosome distribution. (A) LincRNAs identification pipeline. (B) Venn diagram of the proportion of different lincRNAs. (C) LincRNAs chromosome distribution.
Characterization of pig lincRNAs
Previous studies have shown by comparing structural features that pig lincRNA is identical to the lincRNAs of other mammals (human and mouse)11,12. The pig lincRNA has fewer and longer exons than the coding gene; the lincRNA transcript, owing to their small number of exons, is shorter than the coding gene13. Thus, the present study compared the difference in exon number (Fig. 2A), length (Fig. 2B), and length of transcription (Fig. 2C) between lincRNA (known lincRNAs and novel lincRNAs) and protein coding genes in this data, consistent with previous reports14. The accuracy of lincRNAs obtained from the pipeline of this study was confirmed.

Characterization of identified lincRNAs. Characterization of identified lincRNAs. (A) The number of exons of different lincRNAs and protein-coding genes. (B) Probability density graph of the transcription length of different lincRNA and protein coding genes. (C) Probability density graph of the exon length of different lincRNAs and protein-encoding genes.
Expression analysis of lincRNAs
The mammalian genome is universally transcribes and encodes thousands of lincRNAs distributed throughout the genome, which are less conserved and have low expression levels15,16. The present study compared the average expression levels of 361 lincRNAs (known lincRNAs and novel lincRNAs) and protein coding genes from six samples to investigate whether this expression pattern is also present in pigs. The results showed that the average expression level of lincRNAs (known lincRNAs and novel lincRNAs) in pigs is generally lower than that of genes encoding proteins (Fig. 3A). In order to study the lincRNA that may cause phenotypic differences between the two breeds of pigs (Yorkshire pigs and Tibetan pigs). Deseq2 in the R was used to perform differential expression analysis on the two breeds of pig samples on the basis of expression levels. Between the two breeds, 66 of the 138 DELs of Yorkshire pigs were upregulated and 72 were downregulated (Fig. 3B). Between the two pig breeds, 326 of the 682 differentially expressed coding genes identified were downregulated and 356 were upregulated (Fig. S2).

The results of expression analysis. (A) Expression levels of different lincRNA and protein-encoding genes. (B) Differential expression analysis of differential lincRNAs, the bar code represents the color scale of the log10(FPKM) .
DELs target genes prediction and GO and KEGG enrichment analysis
Given that lncRNA can silence or activate cis-gene expression, it can act on neighboring genes at lncRNA sites17.Target genes (Table S1, the methods of target genes prediction refer to methods) in the range of 100 kb upstream and downstream of the DELs position were searched. Conducting the online GO and KEGG analysis18,19,20 (Table S2) through Metascape to explore the functions of target genes that may be regulated by DELs21. The results showed that 409 PTGs were significantly (P < 0.05) involved in 155 biological processes and 21 pathways. Many biological processes and pathways involved in muscle development (Fig. 4A). In addition, we found that the genes in the pathways related to muscle development differently expressed between the two species (Fig. 4B). It is speculated that the differential expression of target genes may be related to the differential expression of lincRNA between the two species.

The results of Gene ontology and pathway analysis. (A) Gene ontology and pathway related to muscle development. (B) Gene expression in Gene ontology and pathway related to muscle development, the bar code represents the color scale of the log10 (FC).
QTLs analysis of DELs and functional prediction
The functions of DELs were further explored by performing QTL mapping analysis (Table S3) after the prediction of the target genes of DELs. The results indicated that approximately 37% of QTLs are associated with muscle growth and development (Fig. 5A). We calculated the chromosome distribution of QTLs associated with muscle development were mainly distributed on chromosomes 4 and 6 (Fig. 5B). Interestingly, the top 10 QTLs were associated with muscle development, and were mainly concentrated on the average back fat thickness QTL, waist muscle area QTL and body weight QTL (Fig. 5C). This result speculates that the potential function of DELs may be related to muscle growth and development.

QTL analysis results. (A) Percentage of muscle development-related QTLs among all QTLs. (B) Distribution of QTL in each chromosome. (C) The Top 10 of QTLs.
Construction of co-expression networks
To further explore the potential role of DELs in muscle development. We collected PTGs from biological processes and pathways involved in muscle development (Table S4). Such as the second messenger adenosine 3′, 5′-adenosine monophosphate (cAMP) pathway, which can affect the size of muscle fibers for a long time. Continuous activation of the cAMP pathway can lead to a pronounced hypertrophic response in skeletal muscle fibers22,23. And can promote animal hypertrophy, including muscular dystrophy, age-related atrophy and other animal models of various diseases against atrophy24,25. cAMP is also involved in muscle development and regeneration mediated by muscle precursor cells26,27. On the other hand, our predicted pathway also includes the core pathway IGF1–Akt–mTOR pathway that affects skeletal muscle hypertrophy28. Muscle hypertrophy is caused by increased protein synthesis and decreased protein degradation29. This pathway plays an important role in protein accumulation30.Based on the expression levels of these PTGs and the corresponding lincRNA, we construction the DELs- MDRPTGs co-expression network by using Cytoscape_3.6.131 (Fig. 6). It was found that 34 of the 138 DELs may regulate PTGs which associated with muscle development, and we found that 25 of 34 DELs upregulate their target genes. In addition, there are eight DELs corresponding to more than two MDRPTGs, two MDRPTGs correspond to multiple DELs. Therefore, we speculate that there are some regulatory mechanisms related to muscle development between MDRPTG and DEL.

DELs-MDRPTGs co-expression network diagram. Green dots represent MDRPTG, red dots represent DEL; green lines indicate that DEL is negatively correlated with the corresponding MDRPTG, and red indicates that DEL is positively correlated with the corresponding MDRPTG.
Correlation verification between DEL genes and their PTGs
In the PTG prediction section, we predicted 409 PTGs corresponding to 138 DELs. To confirm this result, we randomly selected 5 lincRNA genes with significant positive correlation based on their expression levels. The correlation coefficients were all greater than 0.80, and the p values were less than 0.05. We performed RT-qPCR experiments on nine samples, and the results were analyzed using linear regression. The expression levels suggested that the five pairs of lincRNA genes and their PTGs are significantly positively correlated, with a correlation coefficient greater than 0.80 and p value of less than 0.05. The experimental results of RT-qPCR showed that the results of the two datasets are in good agreement, further improving the reliability of the present study (Fig. 7).

Linear regression of lincRNA and PTG expression. The r0 and p0 represent the Pearson correlation coefficient and p value of each pair of lincRNA and PTG in 6 samples, respectively; The r and p represent verification in 9 samples. (A) MSTRG.12010 vs IGF-2. (B) MSTRG.2530 vs WDR5. (C) MSTRG.24143 vs SESN3. (D) MSTRG.21771 vs SRF. (E) MSTRG.14875 vs UNKL.
Discussion
The skeletal muscle is the largest organ in mammalian animals. In pigs, skeletal muscle have important economic significance for production, and understanding the development of skeletal muscle is important for improving productivity and meat quality. In this study, we identified 361 potential lincRNAs based on the designed pipeline and found that 53 of them are novel lincRNAs. At the same time, we obtained 138 DELs. LincRNA generally indirectly exerts its potential regulatory effect by regulating the target protein-coding genes32. Therefore, we predicted the PTG of DEL and the potential function of these PTGs. We found that multiple biological processes of these PTGs are related to skeletal muscle development. Such as muscle structure development, muscle organ development, muscle cell differentiation and skeletal muscle tissue development. Further, We also found that there are multiple PTGs involved in cAMP signaling pathway, which can promote muscle growth and protein synthesis33, and play a key role in embryonic muscle growth and development34. In addition, in muscle tissue, there is a balance between muscle synthesis and degradation, and rapamycin (mTOR) signaling plays a key role in regulating protein synthesis35. We found that multiple PTGs are related to mTOR signaling pathway. On the other hand, QTL mapping analysis of DELs showed that 1137 of the 3018 QTLs were related to skeletal muscle development. The proportion of loin muscle area QTL and body weight QTL is the highest.
Finally, to further explore the potential regulatory role of DEL on muscle development. We generated the MDRPTG-DEL co-expression network and studied PTG related to muscle development. The growth of skeletal muscle depends on muscle fiber hypertrophy, and the size of muscle fibers is increased when the rate of protein synthesis is higher than the rate of degradation. Under normal physiological conditions, the IGF-1-Akt-mTOR pathway plays a key regulatory role in skeletal muscle protein synthesis36,37. After IGF-1 binds to the membrane receptor IGFR1, it promotes protein synthesis by activating Akt-mTOR38. The Akt–mTOR pathway is also a meeting point for other signaling pathways known to promote muscle growth. For example, Wnt7a can participates in this pathway to induce muscle fiber hypertrophy by activating its receptor Fzd739. Correspondingly, it has been reported that 5′-adenosine monophosphate-activated protein kinase (AMPK) can inhibit the activity of mTOR complex 1 (mTORC1), thereby inhibiting protein synthesis40. In addition, the WNK1-FOXO4 axis has been reported to affect muscle fiber hypertrophy. With-no-lysine (K) (WNK) kinases, which can mediate the nuclear localization and transcriptional activity of forkhead box protein O4 (FOXO4) to promote skeletal muscle cell hypertrophy41. But in general, the most well-studied pathway is still the IGF-1-Akt-mTOR pathway. A large number of studies have confirmed that it is a core pathway that affects muscle fiber hypertrophy and is essential for myotube formation and muscle hypertrophy28.
Interestingly, we comprehensive analysis found that multiple DELs may participate in the IGF-1-Akt-mTOR signaling pathway by regulating their PTGs. Mitochondrial calcium unidirectional transporter (MCU) is a highly selective channel for Ca2+ transport into the mitochondria. Mammucari et al.42 reported that MCU participates in IGF-1-Akt-mTOR signaling by increasing Ca2+ level in the mitochondria, activating the PGC-1α4, which is a transcriptional coactivator; the IGF-1 gene is activated through the PGC-1α4, leading to muscle hypertrophy42,43. In the present study, our analysis showed that compared with the TP group, the LW group had higher MCU expression level (Fig. S3), which may be associated with the growth characteristics of Yorkshire pig breeds. More importantly, we found that DEL-MSTRG.8035 is positively related to the expression of MCU and highly expressed in the LW group. Insulin-like growth factor 2 (IGF-2) is a maternal blotting growth factor that regulates prenatal skeletal muscle development44. It can be involved in the IGF1-Akt-mTOR signaling pathway by activating the IGF1 receptor45. A significant positive correlation exists between the DEL-MSTRG.12010 and IGF-2, which were upregulated in the LW group. Interestingly, MSTRG.12010 was significantly negatively correlated with the troponin T-3 (TNNT3) gene. Wang et al. It is predicted that TNNT3 can regulate muscle growth and muscle fibers46. TNNT3 is an important part of pig skeletal muscle filaments, which can affect the taste and tenderness of pork47,48. Its expression level was low in the LW group. This may, on the other hand, find the reason for the decrease in meat quality as the growth rate increases. In addition, a potential target gene, serum response factor (SRF), plays an important role in controlling muscle fiber hypertrophy49,50. SRF can control the transcription of miR-486, which as a potential regulator of PI3K/Akt signal transduction in muscle cells, can phosphorylate Akt and activate the IGF-1-Akt-mTOR signaling pathway, leading to muscle fiber hypertrophy51. The co-expression network, suggests that DEL-MSTRG.21771 is significantly positively correlated with SRF expression and highly expressed in the LW group. It is speculated that MSTRG.21771 regulates the high expression of its potential target gene SRF, in the LW group, which possibly useful in maintaining the fast skeletal muscle rate in Yorkshire pig. PLD1 is an isoform of phospholipase D (PLD)52, which can stimulate phosphatidylcholine (PC) to produce phosphatidic acid (PA), which can bind to mTOR and participate in the IGF-1-Akt-mTOR signaling pathway53. The substrate S6K1 of mTORC1 is phosphorylated to enhance protein translation, resulting in muscle fiber hypertrophy54. Furthermore, DEL-MSTRG.6293 was positively correlated with its expression, and highly expressed in the LW group. QTL results indicated that MSTRG.8035, MSTRG12010, MSTRG21771, and MSTRG.6293 were all mapped to the QTL loci, such as Loin weight QTL, Loin muscle area QTL, and backfat above muscle dorsi QTL, which are related to muscle development. It is speculated that these DELs may be related to skeletal muscle development, and participate in the IGF-1-Akt-mTOR signaling pathway by regulating the expression of its potential target genes, thereby affecting the muscle fibrous hypertrophy process. However, the specific molecular regulation of this phenomenon remains unclear, and further studies are needed. We speculate that DEL may affect muscle protein synthesis by regulating its PTG to participate in the IGF-1-Akt-mTOR pathway (Fig. 8).

Schematic diagram of lincRNAs involvement in muscle fiber hypertrophy signaling by regulating its target genes. IGF-1, insulin-like growth factor-1; IRS, insulin receptor substrate; PI3K, phosphatidylinositol 3 kinase; mTORC1, mechanistic target of rapamycin in complex 1; IGF-2, Insulin-like growth factor 2; TNNT3, troponin T-3; SRF, Serum response factor; PLD, phospholipase D; PC, phosphatidylcholine; PA, phosphatidic acid; MCU, Mitochondrial calcium unidirectional transporter. Red indicates key genes in the IGF-1-Akt-mTOR signaling pathway; orange indicates lincRNA; and green indicates PTG.
In conclusion, based on our analysis of this data. We identified linRNAs that may cause differences in the growth of skeletal muscles of Tibetan and Yorkshire pigs, and discovered a number of novel linRNAs. In addition, we speculate that multiple DELs may participate in the IGF-1-Akt-mTOR pathway by regulating their potential target genes, ultimately affect muscle protein synthesis and the size of muscle fiber. Our findings provide valuable references and new ideas for lincRNA research.
Methods
Datasets used in this study
In this study, we obtained previously published RNA-seq data representing six transcriptomes from the GEO database (ID: GSE99749). Two sows (Tibetan pigs (TP) & Yorkshire pigs (LW)) used for RNA-seq were raised in the Tibet Agricultural and Animal Husbandry College Farm, under the same dietary and drinking water standards. Animal care and all experimentation were conducted in accordance with the guidelines pre-approved by the Tibet Agricultural and Animal Husbandry College Farm Institutional Animal Care and Use Committee. All samples were taken from longissimus dorsi muscle tissue of the embryo as described by Shang et al10. Divided into two groups (Tibetan pigs (TP) & Yorkshire pigs (LW)) according to breed. After 60 days of fertilization, each group randomly selected a pregnant sow and randomly took out nine embryo samples. The nine embryonic samples were randomly divided into three parts, each part as a biological replicate (each RNA library contains an equimolar ratio of RNA from the three samples.), and each group contains three biological replicates. The pig gene annotations were downloaded from ftp://ftp.ensembl.org/pub/release-91/gtf/sus_scrofa, and the non-redundant reference sequence database was downloaded from https://ftp.ncbi.nih. gov/blast/db/.
Ethics statement
Animals care and all the experimentation in this study were carried out in accordance with the pre-approved guidelines from Regulation Proclamation No. 5 of the Standing Committee of Hubei People’s Congress. All experimental protocols were approved by the Ethics Committee of Huazhong Agricultural University, Wuhan City, Hubei Province, P. R. China.
Animals and sample collection
The experimental Yorkshire pigs were provided by the National Livestock Engineering Research Center of Huazhong Agricultural University. All Yorkshire pigs were raised under the same temperature, humidity, ventilation conditions and feeding standards. After fasting for 12 h, three sows 55 days of gestation were randomly selected and euthanized by electric shock and rapid bleeding. Then, we collected 3 embryos from each sow for a total of 9 embryos. Collecting the longissimus dorsi muscle of the embryo and stored them in liquid nitrogen for later use.
RNA-Seq reads mapping and initial assembly
We use Fastqc to evaluate the quality of sequencing reads in the data. Low quality reads were removed using Trimmomatic (version 0.3.2)55 with the default parameters. The clean data obtained were aligned to the Sus scrofa genome (SusScrofa11.1) from University of California Santa Cruz (UCSC) using the Hisat2 (version 2.0.1) default parameters. Sorting mapped reads and remove duplicates via Samtools(version 0.1.19)56. In addition, we assembled the read map using the default parameters of Stringtie (version 1.2.2)57. At the same time, we set the Stringtie's "-G" option for the novel transcript assembly. Finally, we used the merge function of Stringtie software to combine the transcription files from six samples (GTF format) into a non-redundant transcriptome file.
Pipeline for lincRNA identification
361 lincRNAs were screened based on established pipelines in our laboratory8. The complete pipeline follows the one shown in Fig. 1A. Step 1, Transcripts representing intergenic transcripts classified as "U" were screened using the gffcompare program Stringtie (version1.2.2). Step 2, based on the transcript characteristics of lincRNA, transcripts with transcript lengths greater than 200 bp and exon numbers greater than 2 were screened. Step 3, the coding potential of lincRNAs were verified using a coding potential calculator (Cpc) tool58, and the lincRNAs with Cpc value < 0 was entered into the downstream screening program. Step 4, to improve the accuracy of lincRNA screening, we predicted the coding potential of transcripts from multiple perspectives. We translated the transcript into six possible protein sequences, which were then transcribed and compared to the Pfam database. Finally, no Pfam hit (E value < 1e−5) transcripts are retained. Step 5, Transcripts were aligned with NCBI NR and UniRef90 databases using the Blastx program, and transcripts of similar proteins in known proteins were filtered (E value < 1e-5). Step 6, to minimize the chance of false positives, transcripts that were not expressed (FPKM values) in all samples were removed.
Comparison of characteristics between protein- coding gene and lincRNA
We selected 45,788 "gene_biotype = protein_coding" transcripts from the pig's genome annotation file (SusScrofa 11.1) to define them as transcripts of protein coding genes. In addition, we use the "blastn" instruction to divide lincRNA into known lincRNA and novel lincRNA. We then identified and compared the transcript lengths, exon lengths, exon numbers, and FPKM averages for these three categories.
Differential expression analysis of lincRNA
We used the counting software Htseq59 to count the number of reads in the six samples, and then divided the six samples into two groups according to the variety, namely TP and LW, and compared them in R using the "Deseq2" package60. The gene of |log2FoldChange|> 1, padj < 0.05 is a differentially expressed gene. Then, it is calculated by taking the intersection of the potential lincRNA obtained from the pipeline to obtain the differentially expressed lincRNA (DELincRNA), at the same time, it is intersected with the protein coding gene expressed in the sample to obtain a differentially expressed protein encoding gene.
lincRNA target gene prediction
Consistent with previous research. Because lincRNA can regulate its potential target genes in cis. Based on this, we used Bedtools software (version 2.17.0) to search protein-coding gene in the 100 kb upstream and downstream of the lincRNA locus, and used R to calculate the Pearson correlation coefficient between DEL and protein-coding genes. Finally, protein-coding gene with a correlation coefficient greater than 0.9 were identified as potential target genes for DEL. At the same time, if the protein-coding gene is differentially expressed in the two groups, this protein-coding gene is a potential target gene for differential expression.
Function enrichment analysis
Due to the limitations of pig genome annotation, this study included background human orthologous genes61. After transforming the pig gene into a human gene in the Ensembl website, gene ontology (GO) and the Kyoto Gene and Genomic Encyclopedia (KEGG)18,19,20 pathway enrichment analysis were performed in Metascape21. Subsequent selection of p value less than 0.05 is a valid result.
Prediction of DELs function by QTL
In this study, the pig QTL annotation file was downloaded from the animal QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/SS/index), and the location information of DEL was proposed in the non-redundant transcription file according to the ID of DEL. After that, we performed QTL mapping on DEL using the BedTools (version 2.17.0).
Correlation verification between DEL and its PTG
We used the longissimus dorsi muscle from nine 55-day-old embryos of Yorkshire pigs and performed RT-qPCR to verify the expression correlation between DELs and PTGs. For quantitative verification, in the first step, total RNA was extracted using Trizol reagent (Invitrogen, Life Technologies, CA, USA) and performed according to the manufacturer's instructions. To prevent the degradation of RNA, before cDNA synthesis, we measured the purity and concentration of total RNA at 260 and 280 nm with a microphotometer (Thermo, NanoDrop 2000, United States). At the same time, we conduct a gel electrophoresis test to detect whether the RNA is degraded. There are usually three frequency bands, of which 28S and 18S are clear, and the brightness ratio is about 2:1, indicating that there is no degradation. Next, cDNA synthesis was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo, Wuhan, Cat#k1622).According to the manufacturer's instructions, qPCR for DELs and PTGs detection in Roche LightCyler 480 system (Roche, Mannheinm, Germany) was performed using SYBR Green (CWBIO, Beijing, China, CW0957). Ten pairs of RT-qPCR primers were designed using the Primer 5 program (Tables S5, S6). 18S rRNA is used as an endogenous control gene. The RT-qPCR data were analyzed using the 2−△△CT method and R scripts were used to perform related linear regression analysis.
Data availability
The sequence datasets of this paper from the GEO database. The GEO data set ID is GSE65983 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP108727&o=acc_s%3Aa).
References
Li, M. et al. An atlas of DNA methylomes in porcine adipose and muscle tissues. Nat. Commun. 3, 850. https://doi.org/10.1038/ncomms1854 (2012).
Mi, S., Li, X., Zhang, C. H., Liu, J. Q. & Huang, D. Q. Characterization and discrimination of Tibetan and Duroc x (Landrace x Yorkshire) pork using label-free quantitative proteomics analysis. Food Res. Int. (Ottawa, Ont.) 119, 426–435. https://doi.org/10.1016/j.foodres.2019.02.016 (2019).
Liu, H. et al. Comparative transcriptomic analysis of skeletal muscle tissue during prenatal stages in Tongcheng and Yorkshire pig using RNA-seq. Funct. Integr. Genom. 18, 195–209. https://doi.org/10.1007/s10142-017-0584-6 (2018).
Xu, X. et al. Differential transcriptome analysis of early postnatal developing longissimus dorsi muscle from two pig breeds characterized in divergent myofiber traits and fatness. Anim. Biotechnol. 30, 63–74. https://doi.org/10.1080/10495398.2018.1437045 (2019).
Ryu, Y. C. & Kim, B. C. The relationship between muscle fiber characteristics, postmortem metabolic rate, and meat quality of pig longissimus dorsi muscle. Meat Sci. 71, 351–357. https://doi.org/10.1016/j.meatsci.2005.04.015 (2005).
Elminowska-Wenda, G. Structure traits of Longissimus lumborum muscle in wild boar/domestic pig hybrids. Folia Biol. 54, 133–137 (2006).
Deniz, E. & Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 17, 135–143. https://doi.org/10.1007/s10142-016-0524-x (2017).
Zou, C. et al. Transcriptome analysis reveals long intergenic noncoding RNAs contributed to growth and meat quality differences between Yorkshire and Wannanhua pig. Genes https://doi.org/10.3390/genes8080203 (2017).
Zhou, L. et al. Linc-YY1 promotes myogenic differentiation and muscle regeneration through an interaction with the transcription factor YY1. Nat. Commun. 6, 10026. https://doi.org/10.1038/ncomms10026 (2015).
Shang, P. et al. A comparison of prenatal muscle transcriptome and proteome profiles between pigs with divergent growth phenotypes. J. Cell. Biochem. 120, 5277–5286. https://doi.org/10.1002/jcb.27802 (2019).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. https://doi.org/10.1101/gad.17446611 (2011).
Pauli, A. et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 22, 577–591. https://doi.org/10.1101/gr.133009.111 (2012).
Li, J. et al. Identification and functional analysis of long intergenic noncoding RNA genes in porcine pre-implantation embryonic development. Sci. Rep. 6, 38333. https://doi.org/10.1038/srep38333 (2016).
Zou, C. et al. Identification and functional analysis of long intergenic non-coding RNAs underlying intramuscular fat content in pigs. Front. Genet. 9, 102. https://doi.org/10.3389/fgene.2018.00102 (2018).
Hezroni, H. et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 11, 1110–1122. https://doi.org/10.1016/j.celrep.2015.04.023 (2015).
Liu, S. et al. Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain. Genome Res. 27, 1608–1620. https://doi.org/10.1101/gr.217463.116 (2017).
Batista, P. J. & Chang, H. Y. Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307. https://doi.org/10.1016/j.cell.2013.02.012 (2013).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: integrating viruses and cellular organisms. Nucl. Acids Res. https://doi.org/10.1093/nar/gkaa970 (2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019).
Kim, Y. S. & Sainz, R. D. Beta-adrenergic agonists and hypertrophy of skeletal muscles. Life Sci. 50, 397–407. https://doi.org/10.1016/0024-3205(92)90374-x (1992).
Minetti, G. C. et al. Gαi2 signaling promotes skeletal muscle hypertrophy, myoblast differentiation, and muscle regeneration. Sci. Signal 4, ra80. https://doi.org/10.1126/scisignal.2002038 (2011).
Griffin, C. A., Kafadar, K. A. & Pavlath, G. K. MOR23 promotes muscle regeneration and regulates cell adhesion and migration. Dev. Cell 17, 649–661. https://doi.org/10.1016/j.devcel.2009.09.004 (2009).
Hall, J. E., Kaczor, J. J., Hettinga, B. P., Isfort, R. J. & Tarnopolsky, M. A. Effects of a CRF2R agonist and exercise on mdx and wildtype skeletal muscle. Muscle Nerve 36, 336–341. https://doi.org/10.1002/mus.20820 (2007).
Zalin, R. J. & Leaver, R. The effect of a transient increase in intracellular cyclic AMP upon muscle cell fusion. FEBS Lett. 53, 33–36. https://doi.org/10.1016/0014-5793(75)80675-2 (1975).
Chen, A. E., Ginty, D. D. & Fan, C. M. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 433, 317–322. https://doi.org/10.1038/nature03126 (2005).
Philippou, A., Maridaki, M., Halapas, A. & Koutsilieris, M. The role of the insulin-like growth factor 1 (IGF-1) in skeletal muscle physiology. Vivo 21, 45–54 (2007).
Goldberg, A. L. Protein turnover in skeletal muscle. I. Protein catabolism during work-induced hypertrophy and growth induced with growth hormone. J. Biol. Chem. 244, 3217–3222 (1969).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293. https://doi.org/10.1016/j.cell.2012.03.017 (2012).
Su, G., Morris, J. H., Demchak, B. & Bader, G. D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 47, 8–13. https://doi.org/10.1002/0471250953.bi0813s47 (2014).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. https://doi.org/10.1101/gr.132159.111 (2012).
Berdeaux, R. & Stewart, R. cAMP signaling in skeletal muscle adaptation: hypertrophy, metabolism, and regeneration. Am. J. Physiol. Endocrinol. Metab. 303, E1–E17. https://doi.org/10.1152/ajpendo.00555.2011 (2012).
Zalin, R. J. & Montague, W. Changes in cyclic AMP, adrenylate cyclase and protein kinase levels during the development of embryonic chick skeletal muscle. Exp. Cell Res. 93, 55–62. https://doi.org/10.1016/0014-4827(75)90422-x (1975).
Ilha, J., do Espírito-Santo, C. C. & de Freitas, G. R. mTOR signaling pathway and protein synthesis: from training to aging and muscle autophagy. Adv. Exp. Med. Biol. 1088, 139–151. https://doi.org/10.1007/978-981-13-1435-3_7 (2018).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. https://doi.org/10.1016/j.cell.2017.02.004 (2017).
Erbay, E., Park, I. H., Nuzzi, P. D., Schoenherr, C. J. & Chen, J. IGF-II transcription in skeletal myogenesis is controlled by mTOR and nutrients. J. Cell Biol. 163, 931–936. https://doi.org/10.1083/jcb.200307158 (2003).
Bodine, S. C. et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 3, 1014–1019. https://doi.org/10.1038/ncb1101-1014 (2001).
von Maltzahn, J., Bentzinger, C. F. & Rudnicki, M. A. Wnt7a-Fzd7 signalling directly activates the Akt/mTOR anabolic growth pathway in skeletal muscle. Nat. Cell Biol. 14, 186–191. https://doi.org/10.1038/ncb2404 (2011).
Williamson, D. L., Bolster, D. R., Kimball, S. R. & Jefferson, L. S. Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am. J. Physiol. Endocrinol. Metab. 291, E80–E89. https://doi.org/10.1152/ajpendo.00566.2005 (2006).
Mandai, S. et al. WNK1 regulates skeletal muscle cell hypertrophy by modulating the nuclear localization and transcriptional activity of FOXO4. Sci. Rep. 8, 9101. https://doi.org/10.1038/s41598-018-27414-0 (2018).
Mammucari, C. et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 10, 1269–1279. https://doi.org/10.1016/j.celrep.2015.01.056 (2015).
Ruas, J. L. et al. A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331. https://doi.org/10.1016/j.cell.2012.10.050 (2012).
Clark, D. L., Clark, D. I., Beever, J. E. & Dilger, A. C. Increased prenatal IGF2 expression due to the porcine intron3-G3072A mutation may be responsible for increased muscle mass. J. Anim. Sci. 93, 2546–2558. https://doi.org/10.2527/jas.2014-8389 (2015).
Jaafar, R. et al. Phospholipase D regulates myogenic differentiation through the activation of both mTORC1 and mTORC2 complexes. J. Biol. Chem. 286, 22609–22621. https://doi.org/10.1074/jbc.M110.203885 (2011).
Wang, Z. et al. iTRAQ-based proteomic analysis reveals key proteins affecting muscle growth and lipid deposition in pigs. Sci. Rep. 7, 46717. https://doi.org/10.1038/srep46717 (2017).
Chen, H., Zhang, J., Yu, B., Li, L. & Shang, Y. Molecular cloning, structural analysis, and tissue expression of the TNNT3 gene in Guizhou black goat. Gene 573, 123–128. https://doi.org/10.1016/j.gene.2015.07.038 (2015).
Ji, J. et al. A whole-genome sequence based association study on pork eating quality traits and cooking loss in a specially designed heterogeneous F6 pig population. Meat Sci. 146, 160–167. https://doi.org/10.1016/j.meatsci.2018.08.013 (2018).
Li, S. et al. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. USA 102, 1082–1087. https://doi.org/10.1073/pnas.0409103102 (2005).
Guerci, A. et al. Srf-dependent paracrine signals produced by myofibers control satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 15, 25–37. https://doi.org/10.1016/j.cmet.2011.12.001 (2012).
Small, E. M. et al. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. USA 107, 4218–4223. https://doi.org/10.1073/pnas.1000300107 (2010).
Jaafar, R. et al. Phospholipase D regulates the size of skeletal muscle cells through the activation of mTOR signaling. Cell Commun. Signal.: CCS 11, 55. https://doi.org/10.1186/1478-811x-11-55 (2013).
Toschi, A. et al. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 29, 1411–1420. https://doi.org/10.1128/mcb.00782-08 (2009).
Schiaffino, S., Dyar, K. A., Ciciliot, S., Blaauw, B. & Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294–4314. https://doi.org/10.1111/febs.12253 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. https://doi.org/10.1038/nbt.3122 (2015).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucl. Acids Res. 35, W345-349. https://doi.org/10.1093/nar/gkm391 (2007).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 31, 166–169. https://doi.org/10.1093/bioinformatics/btu638 (2015).
Varet, H., Brillet-Gueguen, L., Coppee, J. Y. & Dillies, M. A. SARTools: a DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS ONE 11, e0157022. https://doi.org/10.1371/journal.pone.0157022 (2016).
Yang, Y. et al. Dynamic transcriptome analysis reveals potential long non-coding RNAs governing postnatal pineal development in pig. Front. Genet. 10, 409. https://doi.org/10.3389/fgene.2019.00409 (2019).
Acknowledgements
The work was supported by National Natural Science Foundation of China (NSFC, 31872322), the Fundamental Research Funds for the Central Universities (2662017PY030), National R&D Project of Transgenic Animals of Ministry of Science and Technology of China (2016ZX08006-003) and National high technology research and development plan (863, 2011AA100302).
Author information
Authors and Affiliations
Contributions
C.C.L. conceived and designed the experiments and explained the data. Z.Y.H. analyzed main content of the data with the help of Q.Q.L. M.X.L. performed the experiment. Z.Y.H. wrote the paper with the help of C.C.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, Z., Li, Q., Li, M. et al. Transcriptome analysis reveals the long intergenic noncoding RNAs contributed to skeletal muscle differences between Yorkshire and Tibetan pig. Sci Rep 11, 2622 (2021). https://doi.org/10.1038/s41598-021-82126-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82126-2
- Springer Nature Limited