
Abstract
The present study aimed to improve the understanding of non-uterine leiomyosarcoma (NULMS) prognostic genes through system biology approaches. This cancer is heterogeneous and rare. Moreover, gene interaction networks have not been reported in NULMS yet. The datasets were obtained from the public gene expression databases. Seven co-expression modules were identified from 5000 most connected genes; using weighted gene co-expression network analysis. Using Cox regression, the modules showed favorable (HR = 0.6, 95% CI = 0.4–0.89, P = 0.0125), (HR = 0.65, 95% CI = 0.44–0.98, P = 0.04) and poor (HR = 1.55, 95% CI = 1.06–2.27, P = 0.025) prognosis to the overall survival (OS) (time = 3740 days). The first one was significant in multivariate HR estimates (HR = 0.4, 95% CI = 0.28–0.69, P = 0.0004). Enriched genes through the Database for Annotation, Visualization, and Integrated Discovery (DAVID) revealed significant immune-related pathways; suggesting immune cell infiltration as a favorable prognostic factor. The most significant protective genes were ICAM3, NCR3, KLRB1, and IL18RAP, which were in one of the significant modules. Moreover, genes related to angiogenesis, cell–cell adhesion, protein glycosylation, and protein transport such as PYCR1, SRM, and MDFI negatively affected the OS and were found in the other related module. In conclusion, our analysis suggests that NULMS might be a good candidate for immunotherapy. Moreover, the genes found in this study might be potential candidates for targeted therapy.
Similar content being viewed by others


Introduction
Sarcomas are heterogeneous and rare mesenchymal malignancies that are originated from different tissues. The sarcomas' biological characteristics are not understood well due to the high heterogeneity and uncommonness of this disease. Leiomyosarcoma (LMS), which are originated from smooth muscle cells, accounts for 14% of sarcomas and are the most popular soft tissue sarcomas1. Microarray analysis divides the LMSs into three subtypes. Subtype I expresses muscle associated genes, subtype II shows no significant differentiation from smooth muscle, and subtype III shows specific anatomic sites and is originated from the uterus2. In recent years, the efforts to explain the molecular heterogeneity of LMS have been increased. High throughput technologies generate opportunities to create new insight into different aspects of biological systems. This opportunity may compensate for the rare number of clinical trials in finding new LMS treatments in the future.
There are some studies on gene expression analysis of LMS3,4,5. Some discovered genes were differentially expressed in LMS in comparison with healthy tissues3. Moreover, higher expression of BCL2-associated agonist of cell death (BAD), SRC proto-oncogene, non-receptor tyrosine kinase (SRC), serum response factor (SRF), and myocardin (MYOCD) were confirmed in LMS in comparison with other subtypes of sarcomas6. Loss of fragments in chromosomes 1, 4, 16, and 18 were also reported in comparative genome hybridizations in LMS7,8. Despite many distinguishing efforts to find treatment options by identifying gene expression levels in LMS, surgery is still the main treatment. The currently available systematic therapies are not always effective in this cancer. Moreover, no targeted therapy exists, and personalized medicine approaches seem far away in LMS management. This situation is exacerbated in metastatic LMS. In other cancers, estimating the prognosis of the patients help to decide about the appropriate treatment9,10. But, the studies reporting the effect of gene expression in the survival of patients with LMS are rare11.
Most of the investigations on LMS gene expression has used differential expressed genes (DEGs). Although DEGs elicit vital information from high throughput data, it has some limitations. In fact, in DEG analyses, individual genes are identified, so the interactions between genes are ignored. In other words, DEGs fail to recognize the expression and organization of thousands of genes simultaneously. Gene expression is highly regulated, and it forms a pattern of co-expression networks in cells12. It is hypothesized that most of the time, carcinogenesis is not the result of several genes' deregulation. It is the consequence of complex mechanisms, such as subtle interconnection between genes in the regulatory networks13. Learning such patterns is crucial in cancer-associated studies that cannot be obtained with simple DEGs. To the best of our knowledge, no research has focused on non-uterine leiomyosarcoma (NULMS) based on gene interaction networks in recent years. However, a study was published that investigated all types of LMSs together14.
Weighted gene co-expression analysis (WGCNA) is a general framework that provides a system biology approach. By applying WGCNA, detailed characteristics have been investigated at the genetic network level15. This framework has been successfully utilized to study different cancers and non-cancer diseases16,17.Finding co-expression patterns can also associate the unknown function genes with biological processes due to the guilt-by-association (GBA) basis of WGCNA.
In this paper, the authors utilized the WGCNA algorithm as a system biology method to identify critical co-expressed genes and hub genes; affecting the NULMS survival. Eventually, the function, cellular compartment, and pathways related to patients' relapse were investigated through gene ontology. The study aimed to improve the understanding of NULMS prognostic genes through constructing a co-expression network with RNA sequencing data.
Results
Network construction reveals seven co-expression modules
We were interested in identifying clusters (modules) of co-expressed genes from transcriptomic data of NULMS. A network module is a subset of nodes that forms a sub-network inside a larger network. Soft and hard thresholding are two approaches to construct a co-expression network. WGCNA is a framework principally proposed for analyzing weighted networks. In this study, the soft-thresholding approach was selected to build the NULMS co-expression network.
The parameter β is essential for fulfilling the scale-free topology property of the co-expression network. Biological networks which are based on gene expression data are most likely to be scale-free18. Therefore, β = 17 was considered to obtain scale-free topology by the fit index greater than 0.8. Figure 1 shows the result of several powers for finding a network with scale-free topology properties.

Analysis of network topology for several soft-thresholding powers in WGCNA. (a) Scale-free fit index for different powers (β). (b) Mean connectivity analysis for various soft-thresholding powers (β). (c) Linear model fitting of R2 index showed quality of relationship between connectivity (k) and P(K).
The adjacency matrix was then produced through the adjacency function; using the β and gene expression matrix. The hierarchical clustering was built based on the TOM dissimilarity measure, as shown in Fig. 2. We identified seven co-expression modules. From large to small, these modules are turquoise, blue, brown, yellow, green, red, and black, respectively. In this study, each gene was assigned to separate modules.

Gene dendrogram and module colors for TCGA NULMS.
Module validation
Identified co-expressed modules in the reference dataset should be examined for validity with a standalone dataset. We used the module preservation statistics to achieve reliable and preserved modules. To this end, the co-expression network was constructed again by the NULMS Stanford dataset, and genes were assigned to modules based on the module assignment scheme in the reference dataset. Figure 3 shows that blue, brown, and green modules are strongly preserved (i.e., Z-summary more than 10); while the red and turquoise are moderately preserved (i.e., 5 < Z-summary < 10). The median rank of the green and black module is 2 and 7, respectively. Those values indicate that the green is more strongly preserved than the black module.

Median rank and Z-summary statistics in the module preservation process. (a) The plot shows the module position in the test dataset based on the Median rank. (b) The plot illustrates the analysis of the Z-summary between different modules in the test dataset.
Prognostic modules identification
Module–trait relationship
Finding the relationship between gene expression profiles and clinical traits is one of the WGCNA framework's advantages. The association between module eigengenes and clinical information such as age, different survival status, and time was computed through the Pearson’s correlation coefficient. Moreover, the P value was calculated for the given correlation. As shown in Fig. 4, the green module had a significant correlation with survival endpoint times including overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI) (P < 0.05).

The module–trait relationships were demonstrated by correlation values and P values (In parenthesis) with a range of colors; the degree of correlation between modules and clinical features is shown. Rows are module eigengene (ME) regards to each module, and the columns indicate traits.
Survival analysis
In this study, we were interested in finding the effect of significant modules on patients’ survival. For this purpose, we used module eigengene (ME) as a module representative for the survival analysis. As shown in Table 1, turquoise, green, and red modules had a significant association with OS endpoint in univariate analysis. Moreover, significant modules (MEturquoise, MEgreen, and MEred) were selected as the multivariate analysis covariates. We evaluated if the significant modules in combination had a significant effect on survival. As illustrated in Table 1, green and red were significant in multivariate analysis (Supplementary Table S1). That was statistically significant in the log-rank analysis (P value = 0.0003).
Increased expression of genes in green modules indicates a good prognosis related to OS in NULMS (HR = 0.6, 95% CI = 0.4–0.89, P = 0.0125); while red module genes shows poor prognosis (HR = 1.55, 95% CI = 1.06–2.27, P = 0.025; Table1). For seven modules, survival curves were plotted through Kaplan–Meier. Plots for green and red modules were illustrated (Supplementary Figure S3). Likewise, univariate analysis revealed that 39% and 20% of genes were significant in green and red modules, respectively (P value ≤ 0.01) (Supplementary Table S2 and S3). To validate the result of survival analysis, the GSE7111919 was used as an independent cohort. Regarding univariate analysis in green and red modules, fifteen genes with the lowest P.Cox value were selected. We ran multivariate Cox regression on selected genes. In the green module, ICAM3, IL18RAP, LCK, CTSW, and GRAP2 were significant. Also, PYCR1, B3GALT6, GALNT1, UNC5B, MEX3A, and DCN were significant in the red module (Supplementary Table S4).
Identification of hub genes for prognostic modules
We ranked and picked the top 20 genes based on module membership (MM) and intramodular connectivity separately in each module. In the green and red module, 19 out of 20 and 16 out of 20 genes were common in both lists, respectively (Table 2)20. Our findings clearly showed that there is a strong positive correlation between MM and intramodular connectivity. Although all the hub genes were significant with OS (P < 0.05) in the green module, they were not the most significant genes or one with the least HR related to the OS. The most important hub gene in the green module was in rank 14 in the list (Table 2).
In the red module, 13 out of 16 hub genes (81%) had a significant relationship with OS (P < 0.05). Except for PYCR1 in the first rank, the hub genes were not the most significant genes or one with the high HR related to the OS.
Functional enrichment analysis of prognostic module genes
Functional analysis was performed through the DAVID bioinformatics tool for all genes with a P value smaller than 0.01 in the green and red modules (Supplementary Table S2 and S3). As shown in Table 3, the green module genes were significantly enriched for immune response, inflammatory response, positive regulation of natural killer cell-mediated cytotoxicity, T cell activation, and B cell activation. Cytokine–cytokine receptor interaction and immunoregulatory interactions between a lymphoid and a non-lymphoid cell were significant pathways.
Discussion
In this study, we used the WGCNA framework to analyze the mRNA expression data to find essential modules and genes related to clinical information, especially the survival of the NULMS. The studies on this type of cancer are limited, mainly based on network analysis. WGCNA, as an unsupervised algorithm, can establish and detect the relationship between gene expression and clinical traits. In the present study, seven distinct co-expression modules were identified from 5000 most connected genes; two of them were significantly related to OS status in multivariate Cox regression analysis. For more insight and finding biological mechanisms, hub genes were explored. Increased expression of genes in the green module indicated favorable prognosis related to OS in NULMS; while the red module showed poor prognosis associated with OS. Based on univariate Cox regression, the green module's top five most significant genes were ICAM3, NCR3, KLRB1, IL18RAP, and CECR1. In order to GO analysis, most of the genes of the green module were in the plasma membrane (GO:0005886), integral component of membrane (GO:0016021), T cell receptor complex (GO:0042101), immunological synapse (GO:0001772), and alpha–beta T cell receptor complex (GO:0042105). Based on GO biological function, there were enriched in regulation of the immune response (GO:0050776, GO:0006955), T cell activation (GO:0042110), adaptive immune response (GO:0002250), T cell costimulation (GO:0031295), chemokine-mediated signaling pathway (GO:0070098), inflammatory response (GO:0006954), positive regulation of natural killer cell-mediated cytotoxicity (GO:0045954), B cell activation (GO:0042113), and many other critical biological responses which are listed in supplementary files.
The green module deduced that our WGCNA model successfully separated gene expression of immune cells in the tumor microenvironment from cancer cells and other cancer tissues' cellular components. Numerous studies showed the link between immune cell infiltration in the tumor site and better response to therapy and prognosis in carcinomas21. For example, infiltration of CD8 + and CD57 + cells (as markers of CD8 + T-cells and NK-cells) in tumors was shown as an independent prognostic factor for a more prolonged disease-free survival22. Several studies23,24 in cancer favor immune cell infiltration and better survival even in different sarcoma24 and Ewing sarcoma25. But, there are still some controversies in carcinomas and sarcomas23,26.
The immune infiltration may be prominent in response to immunotherapy drugs. Recently, a clinical trial in undifferentiated pleomorphic sarcoma (UPS) revealed a positive correlation between immune infiltration and response to pembrolizumab. Increased percentage of tumor-associated macrophages (TAM); expressing PD-L1 and higher accumulation of activated T cells (CD8 + CD3 + PD-1 +) were associated with better response to Pembrolizumab27. Immunotherapy is rapidly developing, and predicting the response to it is an enormous prerequisite. Moreover, finding a suitable target for immunotherapy is of utmost importance28. For patients with proper expression of immune markers, available drugs may be applied or a new medication might be designed. For patients or cancer types with lower expression of immune genes, an alternate therapy except for immunotherapy may be useful. This manuscript suggests that NULMS might be a good candidate for immunotherapy.
Based on GO analysis, most of the genes were in extracellular space and the extracellular exosome in the red module. Based on GO biological function, there were enriched for an extracellular matrix, collagen fibril, collagen catabolic process, etc. Genes in this module were enriched in biological processes, including angiogenesis, cell–cell adhesion, protein glycosylation, and protein transport functions (Supplementary Table S3).
The most significant gene in the red module was Pyrroline-5-carboxylate reductase 1 (PYCR1). It is a crucial proline biosynthesis enzyme. Most of the studies showed that this gene is an unfavorable prognostic marker in cancers29,30,31. It is also essential in cell proliferation in NSCLC32.
Spermidine synthase (SRM) is an unfavorable tumor marker that is expressed in renal and liver cancer based on Human Protein Atlas. Its function is a polyamine metabolic process based on GO cellular function. It was also showed that inhibition of SRM could slow B cell lymphoma onset in transgenic mice33. Studies on this protein are limited, and it is recommended to perform similar investigations for NULMS. Moreover, the SRM inhibitors would be a research line for therapy in this subtype of cancer.
Beta-1, 3-Galactosyltransferase 6 (B3GALT6) was another gene in the red module with a hazard ratio of 2. Based on GO biological function, this protein is vital in glycosaminoglycan synthesis and protein glycosylation. Mutation of this gene was also reported in connective tissue disorder34. Few studies have been performed on this protein in the cancer area. But, protein glycosylation was studied well in cancer formation, microenvironment, and metastasis35. Studies on this protein may contribute to our better understanding of NULMS.
According to Human Protein Atlas, high expression of NECAP2 is a favorable and unfavorable prognostic factor in colorectal and liver cancer, respectively. It was shown that NECAP2 is a crucial factor for recruiting AP-1 to early endosomes and the efficient recycling of EGFR. It controls the clathrin coat recruitment of endosomes for the recycling of EGFR36. EGFR signaling is one of the important pathways in cancer. Dysregulated intracellular trafficking of the EGFR family of receptor tyrosine kinases plays a critical role in oncogenesis37.
Moreover, metastasis is caused by increased cancer cell migration and invasion and is the leading cause of cancer-related mortality. NECAP2 function is also essential for the fast recycling of integrin αvβ3 and integrin αvβ3-dependent migration and cancer cell invasion38. The result of the present study showed that NECAP2 is a marker for poor prognosis. Thus, therapies on controlling endosome trafficking may be useful in NULMS.
Myod inhibitor (MDFI) is a tumor suppressor gene and can inhibit proliferation in breast cancer 4T1 cell line39. Down-regulation of MDFI through hyper-methylation may be a risk of NSCLC in young, smoker women40. The study of this protein in cancer is also limited and could be continued in LMS. It is noteworthy that validation of significant genes in both modules confirmed that these genes were important in an independent dataset and could be proper candidates for further experimental and clinical analysis.
Hub gene analysis showed that although all hub genes were significant with OS in the green and red modules, they were not the most significant genes or one with the least HR related to the OS, except for PYCR1. The effect of hub genes in survival was investigated in many studies and hub genes were introduced as important prognostic markers14,41. It is important to note that we should look at relapse as a consequence of complex mechanisms, and nodes, hub genes, are not the best options for predicting them. Every single gene in a significant module may have a cumulative effect on survival, and pinpointing nodes can not be the whole story.
Materials and methods
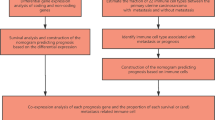
The research design and all steps of this study are presented in the flowchart of Fig. 5. Data collection, preprocessing, and filtering were executed in three steps that were performed before constructing the co-expression network. NULMS co-expression network was constructed on preprocessed data, and the validation process was achieved through the module preservation. Moreover, survival analysis was performed on preserved modules. The identification of prognostic modules was the next step of this study. Subsequently, hub genes in prognostic modules were investigated. Finally, the biological process and different pathways; related to identified modules were analyzed. R platform (version 3.6.1) was used for the computational analysis.

Flow chart of sequential steps for data preprocessing and analysis. Data Prep-aration includes data collection, preprocessing, and filtering. Three data sets were downloaded; the first one was TCGA NULMS. The GSE45510 was used for module preservation, and the GSE71119 was applied for validation of sur-vival analysis. The next step is NULMS co–expression network construction was based on the prepared data. Then, constructed modules were validated through the module preservation process and survival analysis was performed on validated modules. Identification of prognostic modules was performed in the next step. Then, hub genes in prognostic modules were investigated. Final-ly, the biological processes and different pathways; related to identified mod-ules were analyzed.
Dataset and preprocessing
Gene expression and clinical data of two datasets were used. The first one, as the main dataset, was from TCGA and included 74 NULMS cases. Those cases' clinical information was also obtained from the supplementary section of a TCGA paper42 on the integrated TCGA pan-cancer clinical data resource. In connection with biological data, The Cancer Genome Atlas (TCGA) was utilized as the primary source of RNA-seq. TCGA is a project, including different omics data related to various cancers. Through that project, more than 20,000 cancer and normal samples were collected (https://www.cancer.gov/tcga).
The second dataset (GSE45510) was downloaded from NCBI Gene Expression Omnibus, which included 50 NULMS cases2, and it is used as the validation dataset.
HTSeq–Count files of TCGA NULMS were downloaded through the "TCGAbiolinks" package43. TCGA HTseq-counts were normalized based on the Transcripts Per Million (TPM) method. These data were transferred to a new space; using the log2 function. "BatchQC" package44 was used for finding batch effects in the TCGA dataset. Batch effects correction was done; using the "sva"package45. For this purpose, the ComBat function was used with the parametric adjustment (Supplementary Figure S1). Among NULMS cases in TCGA, two patients "TCGA_IE_A3OV", "TCGA_K1_A6RT" were removed because they belonged to batches with just one patient. Hierarchical clustering was performed through samples for finding outliers. Also, the Adaptive branch pruning of hierarchical clustering (dendrogram) was applied by the "dynamicTreeCut" package (Supplementary Figure S2). Through that process, "TCGA_DX_A3UB" was detected as an outlier. Eventually, in the last step of preprocessing, both datasets were checked for missing entries and zero-variance genes; using the goodSamplesGenes function in the WGCNA package.
In this study, the expression matrix was constructed; using protein-coding genes. The analysis was restricted to the most connected genes with non-zero variance. At last, 5000, most connected genes were chosen by applying the softConnectivity function from the WGCNA package15, and connectivity was calculated between genes.
Weighted gene co-expression network construction
The co-expression network was constructed based on WGCNA functions. In this study, biweight midcorrelation (bicor) was used to compute the correlation between each pair of genes because of its robustness to noise in comparison to the Pearson correlation coefficient. Between 3 types of co-expression networks, we used the signed network. In this network, zero correlation gives rise to a non-zero adjacency, and the similarity is defined as (1 + cor)/2.
The correlation matrix was transferred to the adjacency matrix through the adjacency function from the WGCNA package with power β.
That power was calculated; using the pickSoftThreshold function. So, we used the arguments (corFnc = "bicor", corOptions = list (maxPOutliers = 0.1), network type = “signed”, power = “β”) to meet the need of scale-free topology property of the co-expression network. A generalized version of Topological Overlap Measure (TOM) was utilized to find clusters of highly co-expressed genes (modules).TOM in the TOMsimilarity function was applied, converting similarity values for each pair of genes to the new matrix, which was non-negative and symmetric. TOM calculates the similarities based on the number of shared neighbors between gene pairs in the resulting co-expression network46. Since TOM-based dissimilarity has better performance for the distinction gene module, in WGCNA, 1-TOM was used instead of TOM47. Hierarchical clustering was built by the average linkage hierarchical clustering algorithm implemented in the hClust function. Gene modules, groups of genes with a similar expression, were identified with the cutreeDynamic function48. In this function, the “deepSplit” argument value was 2, and a minimum cluster size was 50.
The module eigengene (ME) is a robust and proper representative for each module. It is the first principle component in each module that covers the highest percentage of variance for expression values of all genes in a module15. The moduleEigengenes function calculated the MEs. The close modules were merged through the mergeCloseModules function and determining of MEs threshold was applied on hierarchical clustering of computed modules eigengene.
Validation of identified co-expressed modules
If a module in the reference dataset is not determined randomly, it will be reproduced in other independent datasets across different conditions. In this study, validation of co-expressed modules in the TCGA NULMS dataset was done by an independent dataset (GSE45510); explained in the dataset and preprocessing part. Module preservation statistics was used to validate whether a defined module in one data set could also be found in another data set. The WGCNA used two composite preservation statistics for module preservation: First, Z-summary distinguished preserved modules from non-preserved ones through the permutation test (nPermutations = 200). Median rank is another statistic to compare the amount of preservation among modules. Compare the two modules, the one with a higher median rank was considered to have a lower preservation tendency49.
Finding modules of interest
Module-trait relationship
The relationship between modules and traits was calculated by ME. In other words, we applied ME for calculating the Pearson correlation coefficient between each module and traits through cor() function. Clinical traits included age, OS, DSS, DFI, and PFI status/time. Among the different survival endpoints, OS and PFI were selected for survival analysis due to complete available clinical data and no missing values.
Survival analysis
Survival50, Survminer51, and RegParallel52 packages were used to identify the module-survival relationship. The ME, as the representative of each module, was selected to define the association of each module with OS and PFI. Therefore, for multigene associations, each ME was dichotomized into positive and negative values53. Then, univariate Cox regression, the hazard ratio (HR), and K-M plot were applied for each module; using log-rank tests. In the next step, modules with P value ≤ 0.05 in univariate were selected for multivariate Cox regression. Finally, single-gene survival analysis was done on genes in significant prognostic modules.
Identification of hub genes
A hub gene is a highly-interconnected node in a module with the highest intra-modular connectivity that defines as module membership (MM)20. Hub genes were identified by calculating gene connectivity; using the intramodularConnectivity function from the WGCNA in the whole network (kTotal) and each module (kWithin). The MM, which is also a measure in WGCNA, assesses the correlation between a gene and the ME in a module. In this study, two lists of genes with the highest connectivity and MM were selected. In the end, hub genes were chosen through the intersection of these two lists.
Functional annotation
Gene enrichment analysis was performed for the genes within the significant modules; using the Database for Annotation, Visualization, and Integrated Discovery (DAVID). Depending on DAVID outcome, gene ontology and various pathways for selected genes were investigated. In the pathway analysis, we investigated the Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, and Biological Biochemical Image Database (BBID). P value ≤ 0.05 and false discovery rate (FDR) smaller than 0.05 were considered as the cut-off for determining significant terms.
Conclusion
In summary, WGCNA was used to construct a gene co-expression network. ICAM3, NCR3, KLRB1, IL18RAP, and CECR1 were identified as good prognosis genes, most of them related to immune cells. Our results revealed the immune cell infiltration as a favorable prognostic factor. Moreover, PYCR1, SRM, and MDFI negatively affected the OS. These genes are related to angiogenesis, cell–cell adhesion, protein glycosylation, and protein transport functions. We also found hub genes the most significant of which were LCK, FSCN1, CD3G, PGD, CD2, and TPM3. Our findings confirmed prior investigations that the hub genes were not necessarily the most effective genes related to the OS. The genes found in this study were validated in an independent cohort and provided a virtuous gene list for further experimental analysis. Experiments investigating the mechanism of function of these genes and multi-omics data integration in NULMS are further warranted.
References
Gage, M. M. et al. Sarcomas in the United States: Recent trends and a call for improved staging. Oncotarget 10, 2462 (2019).
Guo, X. et al. Clinically relevant molecular subtypes in leiomyosarcoma. Clin. Cancer Res. 21, 3501–3511 (2015).
Skubitz, K. M. & Skubitz, A. P. Differential gene expression in leiomyosarcoma. Cancer 98, 1029–1038 (2003).
Mas, A. et al. The differential diagnoses of uterine leiomyomas and leiomyosarcomas using DNA and RNA sequencing. Am. J. Obstet. Gynecol. 221, 320. e321–320. e323 (2019).
Michal, M. et al. Inflammatory leiomyosarcoma shows frequent co-expression of smooth and skeletal muscle markers supporting a primitive myogenic phenotype: a report of 9 cases with a proposal for reclassification as low-grade inflammatory myogenic tumor. Virchows Arch. 477, 219–230 (2020).
Villacis, R. A. et al. Gene expression profiling in leiomyosarcomas and undifferentiated pleomorphic sarcomas: SRC as a new diagnostic marker. PLoS ONE 9(7), e102281 (2014).
Carneiro, A. et al. Indistinguishable genomic profiles and shared prognostic markers in undifferentiated pleomorphic sarcoma and leiomyosarcoma: Different sides of a single coin?. Lab. Investig. 89, 668–675 (2009).
Beck, A. H. et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene 29, 845–854 (2010).
Paik, S. et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 351, 2817–2826 (2004).
Gray, R. G. et al. Validation study of a quantitative multigene reverse transcriptase–polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J. Clin. Oncol. 29, 4611–4619 (2011).
Wang, Q. et al. OSlms: a web server to evaluate the prognostic value of genes in leiomyosarcoma. Front. Oncol. 9, 190 (2019).
Joehanes, R. Gene Expression Analysis ch. 16 325–341 (Humana Press, Totowa, 2018).
Bizzarri, M., Cucina, A., Conti, F. & D’Anselmi, F. Beyond the oncogene paradigm: understanding complexity in cancerogenesis. Acta. Biotheor. 56, 173–196 (2008).
Yang, J., Li, C., Zhou, J., Liu, X. & Wang, S. Identification of prognostic genes in leiomyosarcoma by gene co-expression network analysis. Front. Genet. 10, 1408 (2019).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 9, 559 (2008).
Wang, H. et al. Identification of gene modules and hub genes in colon adenocarcinoma associated with pathological stage based on WGCNA analysis. Cancer Genet. 242, 1–7 (2020).
Zhou, X. et al. Identification of key modules, hub genes, and noncoding rnas in chronic rhinosinusitis with nasal polyps by weighted gene coexpression network analysis. BioMed Res. Int. 2020, 6140728 (2020).
Barabasi, A.-L. & Oltvai, Z. N. Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113 (2004).
Lesluyes, T. et al. Genomic and transcriptomic comparison of post-radiation versus sporadic sarcomas. Mod. Pathol. 32, 1786–1794 (2019).
Tan, N. et al. Weighted gene coexpression network analysis of human left atrial tissue identifies gene modules associated with atrial fibrillation. Circ. Cardiovasc. Genet. 6, 362–371 (2013).
Jochems, C. & Schlom, J. Tumor-infiltrating immune cells and prognosis: The potential link between conventional cancer therapy and immunity. Exp. Biol. Med. 236, 567–579 (2011).
Menon, A. G. et al. Immune system and prognosis in colorectal cancer: A detailed immunohistochemical analysis. Lab. Investig. 84, 493–501 (2004).
Barnes, T. A. & Amir, E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 117, 451–460. https://doi.org/10.1038/bjc.2017.220 (2017).
Raj, S., Miller, L. D. & Triozzi, P. L. Addressing the adult soft tissue sarcoma microenvironment with intratumoral immunotherapy. Sarcoma 2018, 9305294. https://doi.org/10.1155/2018/9305294 (2018).
Stahl, D., Gentles, A. J., Thiele, R. & Gutgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology 8, e1674113. https://doi.org/10.1080/2162402X.2019.1674113 (2019).
Oike, N. et al. Prognostic impact of the tumor immune microenvironment in synovial sarcoma. Cancer Sci 109, 3043–3054. https://doi.org/10.1111/cas.13769 (2018).
Keung, E. Z. et al. Correlative analyses of the SARC028 trial reveal an association between sarcoma-associated immune infiltrate and response to pembrolizumab. Clin. Cancer Res. 26, 1258–1266. https://doi.org/10.1158/1078-0432.CCR-19-1824 (2020).
Liu, Y. A global immune gene expression signature for human cancers. Oncotarget 10, 1993–2005. https://doi.org/10.18632/oncotarget.26773 (2019).
Ding, J. et al. Human mitochondrial pyrroline-5-carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis 38, 519–531. https://doi.org/10.1093/carcin/bgx022 (2017).
Zhuang, J. et al. PYCR1 interference inhibits cell growth and survival via c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathway in hepatocellular cancer. J. Transl. Med. 17, 343. https://doi.org/10.1186/s12967-019-2091-0 (2019).
Chen, S. et al. SIRT3 regulates cancer cell proliferation through deacetylation of PYCR1 in proline metabolism. Neoplasia 21, 665–675. https://doi.org/10.1016/j.neo.2019.04.008 (2019).
Cai, F. et al. Pyrroline-5-carboxylate reductase 1 promotes proliferation and inhibits apoptosis in non-small cell lung cancer. Oncol. Lett. 15, 731–740. https://doi.org/10.3892/ol.2017.7400 (2018).
Forshell, T. P., Rimpi, S. & Nilsson, J. A. Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev. Res. (Phila.) 3, 140–147. https://doi.org/10.1158/1940-6207.CAPR-09-0166 (2010).
Malfait, F. et al. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 92, 935–945. https://doi.org/10.1016/j.ajhg.2013.04.016 (2013).
Peixoto, A., Relvas-Santos, M., Azevedo, R., Santos, L. L. & Ferreira, J. A. Protein glycosylation and tumour microenvironment alterations driving cancer hallmarks. Front. Oncol. 9, 380 (2019).
Chamberland, J. P., Antonow, L. T., Dias Santos, M. & Ritter, B. NECAP2 controls clathrin coat recruitment to early endosomes for fast endocytic recycling. J. Cell Sci. 129, 2625–2637. https://doi.org/10.1242/jcs.173708 (2016).
Tomas, A., Futter, C. E. & Eden, E. R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 (2014).
Chamberland, J. NECAP2-driven fast recycling controls cell migration and cancer cell invasion. Doctoral dissertation, Boston University (2018).
Cai, C. et al. Inhibitory effect of MyoD on the proliferation of breast cancer cells. Oncol. Lett. 11, 3589–3596. https://doi.org/10.3892/ol.2016.4448 (2016).
Goh, K.-I. et al. The human disease network. Proc. Natl. Acad. Sci. 104, 8685–8690 (2007).
Zhou, Z. et al. Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis. Int. J. Biol. Sci. 14, 124 (2018).
Liu, J. et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416. e411 (2018).
Mounir, M. et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 15, e1006701 (2019).
Manimaran, S. et al. BatchQC: interactive software for evaluating sample and batch effects in genomic data. Bioinformatics 32, 3836–3838 (2016).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Zhao, W. et al. Weighted gene coexpression network analysis: state of the art. J. Biopharm. Stat. 20, 281–300 (2010).
Spellman, P. T. et al. Comprehensive identification of cell cycle–regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9, 3273–3297 (1998).
Langfelder, P., Zhang, B. & Horvath, S. Defining clusters from a hierarchical cluster tree: The dynamic tree cut package for R. Bioinformatics 24, 719–720 (2007).
Langfelder, P., Luo, R., Oldham, M. C. & Horvath, S. Is my network module preserved and reproducible?. PLoS Comput. Biol. 7, e1001057 (2011).
Therneau, T. M. & Grambsch, P. M. in Modeling Survival Data: Extending the Cox Model 87–152 (Springer, Berlin, 2000).
51Kassambara, A., Kosinski, M., Biecek, P. & Fabian, S. Drawing Survival Curves using “ggplot2”[R package survminer version 0.4. 2]. Comprehensive R Archive Network (CRAN) (2018).
RegParallel: Standard regression functions in R enabled for parallel processing over large data-frames (bioconductor, 2019).
Zhang, C. & Sun, Q. Weighted gene co-expression network analysis of gene modules for the prognosis of esophageal cancer. J. Huazhong Univ. Sci. Technol. Med. Sci. 37, 319–325 (2017).
Acknowledgements
We thank Dr. Sadegh Azimzadeh for technical help and Dr. Habibollah Asghari for general support.
Author information
Authors and Affiliations
Contributions
Conceptualization, R.E. ; methodology, M.D., R.E.; software, M.D.; validation, M.D.; formal analysis, M.D.; data curation, M.D. ; Data interpretation, R.E.; writing original draft preparation, M.D.,R.E.; writing, review, and editing, S.G., R.E., K.M.A; supervision, S.G., R.E.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Darzi, M., Gorgin, S., Majidzadeh-A, K. et al. Gene co-expression network analysis reveals immune cell infiltration as a favorable prognostic marker in non-uterine leiomyosarcoma. Sci Rep 11, 2339 (2021). https://doi.org/10.1038/s41598-021-81952-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81952-8
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.