
Abstract
Despite asthma has a considerable genetic component, an important proportion of genetic risks remain unknown, especially for non-European populations. Canary Islanders have the largest African genetic ancestry observed among Southwestern Europeans and the highest asthma prevalence in Spain. Here we examined broad chromosomal regions previously associated with an excess of African genetic ancestry in Canary Islanders, with the aim of identifying novel risk variants associated with asthma susceptibility. In a two-stage cases-control study, we revealed a variant within HLA-DQB1 significantly associated with asthma risk (rs1049213, meta-analysis p = 1.30 × 10–7, OR [95% CI] = 1.74 [1.41–2.13]) previously associated with asthma and broad allergic phenotype. Subsequent fine-mapping analyses of classical HLA alleles revealed a novel allele significantly associated with asthma protection (HLA-DQA1*01:02, meta-analysis p = 3.98 × 10–4, OR [95% CI] = 0.64 [0.50–0.82]) that had been linked to infectious and autoimmune diseases, and peanut allergy. HLA haplotype analyses revealed a novel haplotype DQA1*01:02-DQB1*06:04 conferring asthma protection (meta-analysis p = 4.71 × 10–4, OR [95% CI] = 0.47 [0.29– 0.73]).
Similar content being viewed by others


Introduction
Asthma is a complex respiratory disease characterized by reversible airflow obstruction and chronic inflammation of the lower respiratory tract, usually linked to allergic and atopic manifestations1. Asthma has an increasing prevalence, affecting more than 300 million people worldwide2,3, which represents a high health care cost. In Europe, the economic burden of asthma in adults is estimated to account for over €19 billion per year4. This global health problem is partly because of the heterogeneity of the disease that makes both its clinical management and research challenging. In this sense, asthma and the allergic traits are strongly conditioned by genetic factors, with heritability estimates ranging between 35 and 95%5. Several genome-wide association studies (GWAS) have revealed several asthma risk genes, including those encoding the ORMDL Sphingolipid Biosynthesis Regulator 3 (ORMDL3), Gasdermin B (GSDMB), and Cadherin Related Family Member 3 (CDHR3), as well as a number of genes related to innate immunity and immunoregulation6.
Despite the efforts made by large consortia, less than 5% of asthma variability can be explained by the genetic risk variants revealed to date7. Among other reasons, this could be because most of the previous genetic association studies have focused on patients of European ancestry8,9,10. Therefore, other genetic risks that are more frequent in other ethnicities could remain unknown. The key implications of ancestry in asthma risks have been evidenced in previous studies in non-European admixed populations that revealed novel gene risks for asthma in African American and Latino populations10,11,12,13,14. However, although in recent years the number of genetic studies in admixed populations has increased considerably, the statistical power in many of them has not been sufficient due to the large sample sizes required by GWAS to overcome the stringent significance penalties, making screening for asthma-related genes in admixed populations still a challenge10.
In this sense, studies leveraging local genetic ancestry analyses constitute an alternative to further disentangle the genetics underlying asthma in recently-admixed populations11,15,16,17,18,19, given that they attain better study power using more limited sample sizes20. Briefly, the genome of admixed individuals is constituted by chromosomal segments from their parental populations. The average proportion with which each parental group contributes to the genome of an admixed individual is known as global genetic ancestry, whereas the local genetic ancestry is defined as the ancestry proportion of each particular locus. Studies such as admixture mapping studies allow to reveal genomic regions where local ancestry correlates with disease risk, and the main challenge remains in the subsequent fine mapping studies of these regions to identify the causal variants17,21.
We previously assessed the local ancestry estimates in an admixed southwestern European population, the Canary Islands population (Spain), revealing the largest African genetic ancestry in Europe, with average proportions of 22.0% of North African (NAF) ancestry and 3.0% of Sub-Saharan African (SSA)22. Additionally, we identified five genomic regions showing an excess of African ancestry in this population within chromosomes 2, 3 (two regions), 6, and 13. These regions were enriched in genes linked to respiratory diseases, including asthma22, which is not surprising since the Canary Islanders have the highest prevalence of asthma in Spain23,24. As a matter of fact, we recently performed an admixture mapping of asthma in this population, revealing a novel locus associated with asthma risk16.
Based on the previous evidence, we hypothesized that the targeted screening of the five genomic regions from the current Canary Islander population that were found to be enriched in African alleles could reveal novel asthma risks. To test this possibility, we conducted a two-stage association study focused on the regions of interest in Canary Islanders, followed by a fine-mapping study of the highly variable human leukocyte antigen (HLA) region.
Results
After quality controls, 930 Canary Islanders from stage 1 (313 cases and 617 controls) and 557 from stage 2 (251 cases and 306 controls) remained in the study. Detailed information of these participants can be found in Table 1. Association testing focused on a total of 140,955 imputed genetic variants located on the five loci enriched in African ancestry in the Canary Islands population (2q21.2-q22.3, 3p25.3, 3q26.32, 6p22.3-p21.32, and 13q21.1-q21.33). The number of variants of each region can be found in Supplementary Table S1. After meta-analysis of results from the two stages, the variant rs1049213 from 6p22.3-p21.32 was significantly associated with asthma risk (OR [95%CI] = 1.74 [1.42–2.14]; p = 1.30 × 10–7) (Table 2). This variant is in the 3′untranslated region (3′ UTR) of the Major Histocompatibility Complex (MHC), Class II, DQ Beta 1 gene (HLA-DQB1) (Supplementary Figure S1). We accessed public GWAS data available at Open Target Genetics25 and found that this single nucleotide polymorphism (SNP) has been associated with asthma (p-value = 1.13 × 10–38, OR = 1.12) (http://www.nealelab.is/uk-biobank/) and broad allergic phenotype26 before. No significant variants were identified in the other regions of interest on chromosomes 2, 3, and 13.
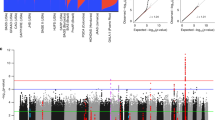
The HLA region is one of the most polymorphic regions of our genome, which makes it difficult to investigate its role in the disease. In this sense, new methods have emerged to impute alleles of classical HLA genes (“classical alleles” from now on) and amino acid polymorphisms from SNP genotyping data27,28,29, providing additional information that can be used to assess the role of this region in asthma physiopathology. We fine mapped this region using a specific method to impute a total of 172 common HLA classical alleles from 3 class I genes (-A, -B, -C) and four class II genes (-DPB1, -DQA1, -DQB1, -DRB1) and assessed their association with asthma susceptibility (Supplementary Table S2). A total of 10 classical alleles within six different HLA genes showed a nominal significance with asthma in stage 1 (p < 0.05) (Table 3). Two of them, HLA-DQA1*01:02 and HLA-DQB1*06:04, showed nominal significance and consistent direction of effects in stage 2 (p = 0.007 and p = 0.025, respectively). The association with asthma protection of HLA-DQA1*01:02 reached study-wise significance in the meta-analysis results from stage 1 and stage 2 (OR [95% CI] = 0.64 [0.50–0.82], p = 3.98 × 10–4) (Table 3, Fig. 1). This association was robust to the model adjustments for sex, age, body mass index, and local NAF or SSA ancestry (Supplementary Table S3) which suggests a correlation between both HLA-DQA1 and HLA-DQB1 variants. Accordingly, HLA haplotype analyses revealed that the haplotype DQA1*01:02-DQB1*06:04 was significantly associated with asthma protection (meta-analysis p = 4.71 × 10–4, OR[95% CI] = 0.47[0.29–0.73]) after setting a Bonferroni threshold at p = 2.17 × 10–3 (based on the 23 DQA1-DQB1 haplotypes tested, Table 4). Interestingly, HLA-DQB1*06:04 was the allele with the second most significant association in our study.

Manhattan plot of meta-analysis results for the association study of chromosome 6 region (grey) and the classical HLA alleles (blue). The y-axis displays transformed p-values (–log10) while the x-axis represents chromosome positions (GRCh37/hg19). The horizontal lines correspond to the significance thresholds of each study after Bonferroni correction: the upper for the targeted association (p = 1.20 × 10–6) and the lower for the classical HLA alleles mapping (p = 4.50 × 10–4). The significant variants are highlighted in orange (targeted association testing of SNPs) and red (fine mapping of classical HLA alleles). The code used to plot the data was obtained from HATK30.
The HLA-DQA1*01:02 sequence translation to the amino acid sequence revealed a specific missense substitution within exon 2 at position 57 (Glu57Gln, E57Q) (Supplementary Figure S2). Mapping each nucleotide position within the altered codon indicated that E57Q corresponds to rs10093, which did not reach statistical significance in our study (meta-analysis OR [95%CI] = 1.13 [0.96–1.33], p = 0.139). However, a few of its linkage disequilibrium (LD) proxies (r2 > 0.77 in Europeans) were nominally significant in the meta-analysis (results for the leading variant rs9271588, OR [95% CI] = 1.20 [1.02–1.41], p = 0.027). Bioinformatic tools for variant prioritization (DSNetwork, VEP, and RegulomeDB) showed that the top-ranked proxy for rs10093 was rs9271588, an intergenic variant located between HLA-DQA1 and HLA-DRB1, which was predicted to have the higher confidence of regulatory impact (Supplementary Table S4). In this sense, rs9271588 was found to be linked to relevant functional consequences based on diverse in silico approaches (Supplementary Table S4). As a summary, this SNP is located within regulatory elements of genes in a subset of tissues and cell types, including enhancer and promoter histone marks and DNase I hypersensitive sites (Supplementary Table S4). Furthermore, rs9271588 is also implicated in disrupting regulatory motifs and protein binding (Supplementary Table S4). Hi-C experiment results supported physical chromatin interactions between the region harbouring rs9271588 and HLA-DQB1 and HLA-DRB1 promoter regions in diverse cell lines (Supplementary Table S4). Additionally, GTEx data supported that rs9271588 is an eQTL and sQTL for HLA-DQA1, HLA-DQB1, and HLA-DRB1 in different tissues, including lung, esophagus, and whole blood (Supplementary Table S4). This SNP also has high CellulAr dePendent dEactivating (CAPE) scores for eQTLs for HLA-DQA1, HLA-DQB1, and HLA-DRB1 in a lymphoblastoid cell line (Supplementary Table S4). Finally, transcriptomic data from bronchial brushing and bronchoalveolar lavage (BAL) samples revealed that HLA-DQA1 (only available for bronchial brushing), HLA-DQB1, and HLA-DRB1 were downregulated in patients with severe asthma compared to healthy controls (Supplementary Figure S3 and Supplementary Figure S4).
Discussion
Several studies support that non-European populations would carry the greatest burden of asthma10. However, the assessed genetic diversity of the published GWAS of asthma is strongly biased towards Central and Northern Europeans, and additional genetic studies in more diverse populations are required to identify novel genetic risk factors for asthma10. Our study provides significant insights of genetics underlying asthma susceptibility in a population with the largest recent African admixture recorded so far among southwestern Europeans31. Besides, the available estimates support that the Canary Islands population has the greatest prevalence of asthma in Spain23,24. Here we performed a targeted staged association study of five genomic regions that we previously described to be enriched in African ancestry among Canary Islanders, and revealed that genetic variants in 6p22.3–p21.32 and within HLA-DQA1 and HLA-DQB1 were significantly associated with asthma risk. HLA genes are located within the MHC and encode a group of proteins with important functions in cell–cell interactions and are critical regulators of the immune response32. In fact, a number of genetic variants from the MHC (some of them linked to HLA-DQA1 and HLA-DQB1) have been associated with asthma in large genetic association studies10,33,34. Interestingly, other studies in populations with African ancestry have allowed to link classical HLA class II gene alleles to the total serum IgE levels in patients with asthma35,36.
Our staged association study revealed the association of rs1049213 with asthma risk, which has been previously linked to asthma, supporting the robustness of our results. Accordingly, it is broadly known that interpreting SNP associations can be problematic, partly due to the difficulty in identifying the causal variant37, and this interpretation becomes even more complicated when the prioritized variant is in the highly polymorphic HLA region. Because of that, classical HLA alleles are more biologically informative and can be related to stronger effects than individual SNPs38,39. Our results revealed that the classical HLA allele HLA-DQA1*01:02 was significantly associated with asthma protection. HLA-DQA1*01:02 had never been linked to asthma risk before, although it has been related with protection from infectious40 and autoimmune diseases, including type-1 diabetes41, Crohn’s disease42, tubulointerstitial nephritis, and uveitis syndrome43, and risk for peanut allergy44. Additionally, HLA haplotype analyses revealed, for the first time, the association of DQA1*01:02-DQB1*06:04 with asthma protection, which supports that the combination of variants within both genes could influence the pathophysiology of the disease. This is in line with previous studies in asthma where HLA haplotypes were identified to be significantly associated with the disease45. The protein encoded by HLA-DQA1 binds to the protein encoded by HLA-DQB1, constituting a heterodimer that plays a central role in the immune system. Interestingly, our analyses prioritized an LD proxy of the missense change defining the HLA-DQA1*01:02 as a clear lung eQTL for HLA-DQA1 and HLA-DQB1. This evidence agrees with human transcriptomic observations revealing a reduction of the HLA-DQA1 and HLA-DQB1 lung gene expression in patients with severe asthma compared to healthy controls. Thus, a lower activity of the heterodimer among asthma patients would be expected in carriers of this classical HLA allele.
We acknowledge some limitations of this study. First, all individuals were selected based on their self-declaration of having two generations of ancestors born in the Canary Islands. Nevertheless, this agrees with the National Institutes of Health (NIH) guidelines, and previous genetic studies in the same population have shown that this selection has no major effect on the findings of the study16,22. Additionally, the use of population controls could bias the results in the sense that it is possible that these individuals could develop asthma or related phenotypes during their lives. However, this type of controls has been widely used in genetic and genomic studies and is the most optimal control group to get the most representative subset of the population, reducing the selection bias10,46. We did not include other confounders such as environmental factors or responses to asthma treatment because data were lacking for most individuals. However, we validated our results in an independent case–control sample, supporting that our results may not be biased by these variables. Significant differences in sex and age were found among cases and controls from both stages, although results remained significant after including these variables in the models. Finally, in addition to the difficulties inherent to the highly polymorphic HLA region, because of technical limitations (e.g., array content and imputation algorithm), we did not assess all common classical HLA alleles of this population, which could have masked additional asthma associations.
Conclusions
A two-stage association study targeting the genomic regions with an excess of African ancestry in the Canary Islands population revealed a novel classical HLA allele (HLA-DQA1*01:02) associated with asthma protection. This suggests that not all shared genetic risks between asthma and autoimmune diseases have opposite directions of effect47. Further studies will be needed to validate these results in other populations, as well as to assess the potential of HLA-DQA1*01:02 as an asthma biomarker.
Methods
Study design and participants
The study was performed in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) and approved by the Research Ethics Committee from the Hospital Universitario Nuestra Señora de Candelaria. Written informed consents were collected from all subjects or their representatives.
We performed a two-stage case–control study of asthma susceptibility targeting the five genomic regions that showed an excess of African ancestry in the current population of the Canary Islands (chr2:133,952,040–144,266,489; chr3:10,539,482–11,710,471; chr3:177,443,968–178,679,751; chr6:24,703,442–36,288,651; and chr13:57,962,413–70,091,195)22. The stage 1 was performed to prioritize genetic variants within these regions, while the stage 2 allowed us to validate these associations in an independent data set. A meta-analysis combining stage 1 and stage 2 association results was performed to establish the genetic variants associated with asthma in this study.
All case and control individuals declared at least two generations of ancestors born in the Canary Islands (Spain). The stage 1 consisted of 314 cases of asthma and 674 controls, while the stage 2 comprised a total of 278 asthma patients and 349 controls. All asthma patients were part of the Genetics of Asthma study in the Spanish population (GOA) study48 and were diagnosed according to the Global Initiative for Asthma (GINA) guidelines49. Population controls were obtained from the Cardiovascular, Diabetes, and Cancer (CDC) cohort study from the Canary Islands50. DNA was extracted from peripheral blood following column-based methods. Further details of these studies can be found elsewhere16,22,51.
Genotyping and statistical analyses
All DNA samples were subject to genotyping of 587,352 SNPs using the Axiom Genome-Wide Human CEU 1 Array (Thermo Fisher Scientific, Waltham, MA, USA) by the Spanish Genotyping Center (CeGen). Variant calling was performed separately in cases and controls from the discovery study and low-quality SNPs and samples were excluded according to the manufacturer’s instructions. Stringent quality controls were subsequently conducted using R programming (v3.2.2)52 and PLINK v1.953. Genetic variants with genotype call rates (CR) < 95%, low minor allele frequency (MAF < 5%), or that deviated from Hardy Weinberg equilibrium (HWE, p < 1.0 × 10−6) were excluded. A total of 403,615 filtered high-quality SNPs were kept for downstream analyses. Additionally, we also excluded individuals with missing clinical information, sex mismatches between records and those inferred from the genotype data, CR < 95%, high degree of kinship with others included in the study (PIHAT > 0.2), or that constituted heterozygosity outliers. A principal component analysis (PCA) was performed using PLINK v1.953 to obtain the leading principal components (PCs) to correct for population stratification during association testing. The PCA was assessed for stages 1 and 2, separately, and individuals were projected on data from The 1000 Genomes Project54 (Fig. 2).

Plot of the first two principal components (explaining 78.95% of variability) of the individuals analysed in stage 1 and 2, projected on data of African (AFR), East Asian (EAS), European (EUR), and South Asian (SAS) populations from The 1000 Genomes Project.
Variant imputation of chromosomes 2, 3, 6, and 13 was then conducted on filtered data using the Michigan Imputation Server55 selecting SHAPE-IT v2.r790 for chromosome phasing56 and European population data from the Haplotype Reference Consortium release 1.1 as reference panel57. Logistic regressions were performed with EPACTS v3.2.658 using a binary Wald test and assuming an additive inheritance model. We included the first four PCs as covariates. Variants with MAF < 1% and a poor imputation quality (Rsq < 0.3) were excluded from the analysis. The genomic inflation factor (λ) of the results was calculated with the R package “qqman”59 (Supplementary Figure S5).
Genotyping and statistical analyses of the data from the two stages followed the same procedures. To assess the overall effect size of associated SNPs across the two stages, a meta-analysis was conducted using METASOFT v2.0.1, where effect heterogeneity was assessed with the Cochran's Q test significance60. Only variants showing the same direction of effects on both studies were considered. We estimated the effective number of independent tests with the Genetic type 1 error calculator (GEC)61 and established the meta-analysed significance at p = 1.20 × 10–6 based on Bonferroni correction.
Fine mapping of the HLA region
A more detailed assessment of the HLA region, residing in chromosome 6, was performed on both stages separately. Classical HLA alleles from three class I genes (-A, -B, -C) and four class II genes (-DPB1, -DQA1, -DQB1, -DRB1) were imputed with HLA Genotype Imputation with Attribute Bagging (HIBAG) v1.4 using imputation models adapted to the Axiom Genome-Wide Human CEU 1 Array and a European reference panel27. The association analysis with asthma was also performed with HIBAG using an additive model on those individuals with a high confidence score (probability threshold > 0.5). The four first PCs were included in the models. A meta-analysis was conducted with METASOFT v2.0.160 for the common alleles (MAF ≥ 1%) that showed a nominal association (p < 0.05) and the same direction of effects on both stages. For this study, a meta-analysed significance threshold was declared at p = 4.50 × 10–4 after Bonferroni correction based on the number of alleles tested. A post-hoc sensitivity analysis was also performed to address potential biases of the results including sex, age, and local ancestry estimates as covariables in the HIBAG association models. The estimation of local ancestry was carried out using ELAI62 assuming a three-way admixture as detailed elsewhere (European, NAF, and SSA)16,22. HLA haplotype analyses were performed using BIGDAWG63, focusing on those genes that were in the vicinity of the variants that showed significant association. Statistical testing of the haplotype difference was based on a Chi-squared test, using BIGDAWG’s default parameters.
Functional annotation of variants and gene expression
We used HIBAG to convert the associated P-coded HLA alleles to amino acid sequences, in order to reveal amino acid residues and, hence, those SNPs that may explain the classical HLA allele associations. We explored the potential biological consequences of the SNP predicting the altered codon in the amino acid in the classical HLA allele and its best proxies (i.e., in strong LD in Europeans, r2 > 0.7) by using different in silico tools. Variant prioritisation was based on results obtained with DSNetwork64, RegulomeDB65, and VEP66. Additionally, we assessed the potential regulatory role of the variants using HaploReg v4.167 and RegulomeDB, as well as the existence of long-distance physical interactions with Capture Hi-C Plotter68. We also accessed GTEx69, ExSNP70, and SNPdelScore71 to evaluate tissue-specific local expression quantitative trait loci (eQTLs) and splicing quantitative trait loci (sQTLs). Further details are provided in the Supplement.
In parallel, we accessed the results of public gene expression studies of asthma available in Gene Expression Omnibus (GEO). Differential gene expression between cases with asthma and healthy controls was examined for those genes in the vicinity of the SNPs and classical HLA alleles significantly associated with asthma susceptibility in our study, to provide additional information on the role of these genes in asthma physiopathology. First, we accessed transcriptomic data of bronchial brushing samples from 27 healthy controls and 128 individuals with asthma (72 non-severe asthma, 56 severe asthma) (GSE63142)72. Then, we accessed gene expression results of BAL samples from 12 healthy controls and 74 asthmatic individuals (28 non-severe asthma, 46 severe asthma) (GSE74986)73. The differential expression was assessed with two-sample t-tests and one-way analyses of variance (ANOVA). Further details are provided in the Supplement.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The genotype and sequence data are not publicly available due to privacy or ethical restrictions.
References
National Asthma Education and Prevention Program. Expert panel report 3 (EPR-3): Guidelines for the diagnosis and management of asthma. J. Allergy Clin. Immunol. 120, S94-138 (2007).
To, T. et al. Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Public Health 12, 204 (2012).
Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention 2014. Available from: www.ginasthma.org (2014).
Accordini, S. et al. The cost of persistent asthma in Europe: An international population-based study in adults. Int. Arch. Allergy Immunol. 160, 93–101 (2013).
Ober, C. & Yao, T.-C. The genetics of asthma and allergic disease: A 21st century perspective. Immunol. Rev. 242, 10–30 (2011).
Mathias, R. A. Introduction to genetics and genomics in asthma: genetics of asthma. Adv. Exp. Med. Biol. 795, 125–155 (2014).
Demenais, F. et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat. Genet. 50, 42–50 (2018).
Lockett, G. A. & Holloway, J. W. Genome-wide association studies in asthma; perhaps, the end of the beginning. Curr. Opin. Allergy Clin. Immunol. 13, 463–469 (2013).
Mills, M. C. & Rahal, C. A scientometric review of genome-wide association studies. Commun. Biol. 2, 9 (2019).
Schoettler, N., Rodríguez, E., Weidinger, S. & Ober, C. Advances in asthma and allergic disease genetics: Is bigger always better?. J. Allergy Clin. Immunol. 144, 1495–1506 (2019).
Roth, L. A. et al. GWAS and admixture mapping identify different asthma-associated loci in Latinos: The GALA II Study. J. Allergy Clin. Immunol. 134, 295–305 (2014).
Mathias, R. A. et al. A genome-wide association study on African-ancestry populations for asthma. J. Allergy Clin. Immunol. 125, 336–346 (2010).
Yan, Q. et al. A genome-wide association study of severe asthma exacerbations in Latino children and adolescents. Eur. Respir. J. 57, 2002693 (2021).
Herrera-Luis, E. et al. Genome-wide association study reveals a novel locus for asthma with severe exacerbations in diverse populations. Pediatr. Allergy Immunol. 32, 106–115 (2021).
Gignoux, C. R. et al. An admixture mapping meta-analysis implicates genetic variation at 18q21 with asthma susceptibility in Latinos. J. Allergy Clin. Immunol. 143, 957–969 (2019).
Guillen-Guio, B. et al. Admixture mapping of asthma in southwestern Europeans with North African ancestry influences. Am. J. Physiol. Cell. Mol. Physiol. 318, 965–975 (2020).
Hernandez-Pacheco, N., Flores, C., Oh, S. S., Burchard, E. G. & Pino-Yanes, M. What ancestry can tell us about the genetic origins of inter-ethnic differences in asthma expression. Curr. Allergy Asthma Rep. 16, 53 (2016).
Goddard, P. C. et al. Integrative genomic analysis in African American children with asthma finds three novel loci associated with lung function. Genet. Epidemiol. 45, 190–208 (2021).
Daya, M. et al. Association study in African-admixed populations across the Americas recapitulates asthma risk loci in non-African populations. Nat. Commun. 10, 880 (2019).
Shriner, D., Adeyemo, A., Ramos, E., Chen, G. & Rotimi, C. N. Mapping of disease-associated variants in admixed populations. Genome Biol. 12, 1–8 (2011).
Suarez-Pajes, E., Díaz-de Usera, A., Marcelino-Rodríguez, I., Guillen-Guio, B. & Flores, C. Genetic ancestry inference and its application for the genetic mapping of human diseases. Int. J. Mol. Sci. 22, 6962 (2021).
Guillen-Guio, B. et al. Genomic analyses of human European diversity at the southwestern edge: Isolation, African influence and disease associations in the Canary Islands. Mol. Biol. Evol. 35, 3010–3026 (2018).
Sánchez-Lerma, B. et al. High prevalence of asthma and allergic diseases in children aged 6 and 7 years from the canary islands: The international study of asthma and allergies in childhood. J. Investig. Allergol. Clin. Immunol. 19, 383–390 (2009).
Juliá-Serdá, G. et al. High prevalence of asthma and atopy in the Canary Islands, Spain. Int. J. Tuberc. Lung Dis. 15, 536–541 (2011).
Ghoussaini, M. et al. Open targets genetics: Systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021).
Ferreira, M. A. et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 49, 1752–1757 (2017).
Zheng, X. et al. HIBAG–HLA genotype imputation with attribute bagging. Pharmacogenom. J. 14, 192–200 (2014).
Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 8, e64683 (2013).
Dilthey, A. T., Moutsianas, L., Leslie, S. & McVean, G. HLA*IMP—an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics 27, 968–972 (2011).
Choi, W., Luo, Y., Raychaudhuri, S. & Han, B. H. A. T. K. HLA analysis toolkit. Bioinformatics 37, 416–418 (2021).
Botigue, L. R. et al. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe. Proc. Natl. Acad. Sci. 110, 11791–11796 (2013).
Charles A. Janeway, Jr, Paul Travers, Mark Walport, and M. J. S. Immunobiology, 5th edition The Immune System in Health and Disease. (Garland Science, 2001).
Dessaint, J. P. Genetics of asthma and allergic diseases. Rev. Fr. d’Allergologie d’Immunologie Clin. 45, 200–207 (2005).
Moffatt, M. F. et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 363, 1211–1221 (2010).
Vince, N. et al. Association of HLA-DRB1∗09:01 with tIgE levels among African-ancestry individuals with asthma. J. Allergy Clin. Immunol. 146, 147–155 (2020).
Akenroye, A. T. et al. Genome-wide association study of asthma, total IgE, and lung function in a cohort of Peruvian children. J. Allergy Clin. Immunol. https://doi.org/10.1016/j.jaci.2021.02.035 (2021).
Scheinfeldt, L. B., Schmidlen, T. J., Gerry, N. P. & Christman, M. F. Challenges in translating GWAS results to clinical care. Int. J. Mol. Sci. 17, 6–8 (2016).
Nishida, N. et al. Understanding of HLA-conferred susceptibility to chronic hepatitis B infection requires HLA genotyping-based association analysis. Sci. Rep. 6, 1–7 (2016).
Meyer, D. & Nunes, K. HLA imputation, what is it good for?. Hum. Immunol. 78, 239–241 (2017).
Lu, L. et al. Association of polymorphisms of human leucocyte antigen -DRB1 and -DQA1 allele with outcomes of hepatitis B virus infection in Han population of north China. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 28, 134–142 (2006).
Khalil, I. et al. Dose effect of cis- and trans-encoded HLA-DQ heterodimers in IDDM susceptibility. Diabetes 41, 378–384 (1992).
Nakajima, A. et al. HLA-linked susceptibility and resistance genes in Crohn’s disease. Gastroenterology 109, 1462–1467 (1995).
Santoro, D. et al. Drug-induced TINU syndrome and genetic characterization. Clin. Nephrol. 78, 230–236 (2012).
Hong, X. et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat. Commun. 6, 6304 (2015).
Takejima, P. et al. Allergic and nonallergic asthma have distinct phenotypic and genotypic features. Int. Arch. Allergy Immunol. 172, 150–160 (2017).
Weiss, S. T. Association studies in asthma genetics. Am. J. Respir. Crit. Care Med. 164, 2014–2015 (2001).
Li, X. et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J. Allergy Clin. Immunol. 130, 861–868 (2012).
Barreto-Luis, A. et al. Genome-wide association study in Spanish identifies ADAM metallopeptidase with thrombospondin type 1 motif, 9 (ADAMTS9), as a novel asthma susceptibility gene. J. Allergy Clin. Immunol. 137, 964–966 (2016).
Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention 2015. Available from: www.ginasthma.org (2015).
Cabrera de León, A. et al. Presentación de la cohorte ‘CDC de Canarias’: objetivos, diseño y resultados preliminares. Rev. Esp. Salud Publica 82, 519–534 (2008).
Pino-Yanes, M. et al. HLA-DRB1∗15:01 allele protects from asthma susceptibility. J. Allergy Clin. Immunol. 134, 1201–1203 (2014).
Bunn, A. & Korpela, M. R. A language and environment for statistical computing. 2, 1–12 (2016).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 1–16 (2015).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Delaneau, O., Howie, B., Cox, A. J., Zagury, J. F. & Marchini, J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 93, 687–696 (2013).
McCarthy, S. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
H., K. Efficient and parallelisable association container toolbox (EPACTS). http://genome.sph.umich.edu/wiki/EPACTS (2014).
Turner, S. D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv Prepr. doi:https://doi.org/10.1101/005165.
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Li, M. X., Yeung, J. M. Y., Cherny, S. S. & Sham, P. C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756 (2012).
Guan, Y. Detecting structure of haplotypes and local ancestry. Genetics 196, 625–642 (2014).
Pappas, D. J., Marin, W., Hollenbach, J. A. & Mack, S. J. Bridging immunogenomic data analysis workflow gaps (BIGDAWG): An integrated case-control analysis pipeline. Hum. Immunol. 77, 283–287 (2016).
Lemaçon, A. et al. DSNetwork: An integrative approach to visualize predictions of variants’ deleteriousness. Front. Genet. 10, 1–9 (2020).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 1–14 (2016).
Ward, L. D. & Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, 930–934 (2012).
Schofield, E. C. et al. CHiCP: A web-based tool for the integrative and interactive visualization of promoter capture Hi-C datasets. Bioinformatics 32, 2511–2513 (2016).
Lonsdale, J. et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Yu, C. H., Pal, L. R. & Moult, J. Consensus genome-wide expression quantitative trait loci and their relationship with human complex trait disease. Omi. A J. Integr. Biol. 20, 400–414 (2016).
Alvarez, R. V., Li, S., Landsman, D. & Ovcharenko, I. SNPDelScore: Combining multiple methods to score deleterious effects of noncoding mutations in the human genome. Bioinformatics 34, 289–291 (2018).
Modena, B. D. et al. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am. J. Respir. Crit. Care Med. 190, 1363–1372 (2014).
Sun, Y. et al. Inhibition of the kinase ITK in a mouse model of asthma reduces cell death and fails to inhibit the inflammatory response. Sci. Signal. 8, 1–14 (2015).
Funding
This work was supported by Instituto de Salud Carlos III (ISCIII; PI14/00844, PI17/00610, FI18/00230, and CD19/00231), cofinanced by the European Regional Development Fund (ERDF) “A Way of Making Europe” from the European Union (EU); by the Wellcome Trust (221680/Z/20/Z); by Fundación CajaCanarias and Fundación Bancaria “La Caixa” (2018PATRI20); and by Agreement OA17/008 with Instituto Tecnológico y de Energías Renovables to strengthen scientific and technological education, training, research, development, and innovation in genomics, personalized medicine, and biotechnology. The genotyping service was performed at Centro Nacional de Genotipado-Plataforma de Recursos Biomoleculares PRB3-ISCIII; it is supported by Grant PT17/0019, of the Programa Estatal de Investigación, Desarrollo e Innovación 2013–2016, funded by ISCIII and ERDF-EU.
Author information
Authors and Affiliations
Contributions
C.F. and B.G.G. designed and supervised the study. E.S.P., C.D.G., H.R.P., J.M.LS., I.M.R,. A.Co., X.Z., A.Ca., E.P.R., J.C.G.R., R.G.M., B.G.G. did the analyses and experiments. E.S.P., C.D.G., C.F., and B.G.G. interpreted results of experiments. E.S.P., C.F., and B.G.G. wrote the draft of the manuscript. C.F. obtained the funding. All authors participated in the critical revision and final approval of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suarez-Pajes, E., Díaz-García, C., Rodríguez-Pérez, H. et al. Targeted analysis of genomic regions enriched in African ancestry reveals novel classical HLA alleles associated with asthma in Southwestern Europeans. Sci Rep 11, 23686 (2021). https://doi.org/10.1038/s41598-021-02893-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02893-w
- Springer Nature Limited