
Abstract
Pectobacterium parmentieri (formerly Pectobacterium wasabiae), which causes soft rot disease in potatoes, is a newly established species of pectinolytic bacteria within the family Pectobacteriaceae. Despite serious damage caused to the potato industry worldwide, no field-deployable diagnostic tests are available to detect the pathogen in plant samples. In this study, we aimed to develop a reliable, rapid, field-deployable loop-mediated isothermal amplification (LAMP) assay for the specific detection of P. parmentieri. Specific LAMP primers targeting the petF1 gene region, found in P. parmentieri but no other Pectobacterium spp., were designed and validated in silico and in vitro using extensive inclusivity (15 strains of P. parmentieri) and exclusivity (94 strains including all other species in the genus Pectobacterium and host DNA) panels. No false positives or negatives were detected when the assay was tested directly with bacterial colonies, and with infected plant and soil samples. Sensitivity (analytical) assays using serially diluted bacterial cell lysate and purified genomic DNA established the detection limit at 10 CFU/mL and 100 fg (18–20 genome copies), respectively, even in the presence of host crude DNA. Consistent results obtained by multiple users/operators and field tests suggest the assay’s applicability to routine diagnostics, seed certification programs, biosecurity, and epidemiological studies.
Similar content being viewed by others


Introduction
Potato blackleg and soft rot, caused by bacterial species in the genera Pectobacterium and Dickeya, are among the most significant diseases with large economic impacts on potato crop production1,2,3,4. Soft rot causes one billion dollars’ loss to the potato industry annually5. Pectobacterium comprises eighteen species that utilize pectinolytic and cellulolytic enzymes to infect a broad range of crop and non-crop plants under wet and semi-anaerobic conditions6. Pectobacterium parmentieri, previously known as P. wasabiae, a gram-negative, rod-shaped virulent pectolytic pathogenic bacterium7, commonly isolated from potato plants and tubers displaying blackleg and soft rot symptoms, is able to survive under a range of environmental conditions. Latently infected potato seed-tuber and contaminated propagative plant materials contribute to the dispersal of P. parmentieri; pathogen has been identified in several regions of Europe8, Canada9, United States10, New Zealand, China11 and South Africa12.
Since soft rot diseases are caused by a complex of bacterial pathogens of different species and phenotypes, detection of an individual bacterial species requires a robust, accurate diagnostic tool13. At present, there are no validated methods reported for the specific detection of P. parmentieri. PCR-based methods are sensitive and specific, however, they can be time-consuming and are confined to laboratory settings with specialized equipment14. There are isothermal methods that can be used in field conditions and have advantages over the PCR-based methods, for example, recombinase polymerase amplification (RPA)—less sensitive to inhibitors and eliminates the need for DNA isolation15,16. Previously RPA was used to differentiate between Pectobacterium and Dickeya, but did not specifically identify the species P. parmentieri17. The cost per RPA reaction is higher than for other field-deployable techniques, such as loop-mediated isothermal amplification (LAMP). The LAMP assay has gained popularity for pathogen detection and point-of-need application18,19. This isothermal nucleic acid amplification technique is based on auto-cyclic amplification and a high DNA strand displacement activity facilitated by a Bacillus stearothermophilus (Bst) polymerase18,20. LAMP is typically performed at 65 °C, a temperature ideal for Bst polymerase activity21. LAMP provide a sensitive and straightforward detection suitable for field applications that doesn’t require expensive reagents or sophisticated equipment. Pathogen detection by LAMP can be achieved in 10 to 20 min, and the amplified products can be observed visually with SYBR Green dye22,23.
Several closely related Pectobacterium species cause soft rot and blackleg diseases in potatoes and shared high pairwise homology in their genomic regions. Therefore, it is crucial to identify the signature genomic region for designing taxon-specific primers24,25,26,27. The comparative genomic analysis allows identification of unique and conserved genomic regions suitable for a robust and highly specific diagnostic assay22,26. Primer specificity for P. parmentieri is important since it occurs in highly heterogeneous populations in different geographic locations28. This selectivity eliminates cross-reactivity with non-target pathogens..
This study described the development of a LAMP assay for specific detection of P. parmentieri in infected potato tissues and soil samples. Both field and laboratory assays confirmed the robustness of the diagnostic method. The resulting protocol is simple for field applications, routine diagnostics, surveillance, biosecurity, epidemiology, and disease management to mitigate the damaging effects and economic losses caused by P. parmentieri in agricultural production.
Results
Target selection and primer in-silico specificity
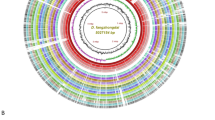
A signature region of the ferredoxin gene petF1, was selected to design and develop a highly specific and robust assay. The signature region within petF1 was not found in any other species of Pectobacterium but it was detected within all genomes of P. parmentieri. Six LAMP primers designed using petF1 gene matched 100% in query coverage and identity with all P. parmentieri genomes available publicly in the NCBI GenBank genome sequence database (Table 1). No crossmatch was observed with any other sequence present in the database. The petF1 gene region in P. parmentieri, is indicated in the BLAST Ring Image Generator (BRIG) image (Fig. 1), which includes genomes from P. parmentieri and other species of Pectobacterium.

Locus representation of the target gene petF1 (ferredoxin) used for the specific detection of Pectobacterium parmentieri. The BLAST ring image was generated using the BRIG software29. The circular graphic shows the multiple alignment and genome comparison of six P. parmentieri strains and the other seventeen species that currently encompass the Pectobacterium genus. The three innermost layers in the graphic portray the genome coordinates (mega base pairs—mbp), GC content (zigzag black line) and GC skew (purple + /green − zigzag) of the P. parmentieri RNS 08-42-1A reference genome. The other colored rings, from the innermost to outermost, depict the nucleotide BLAST alignment of P. parmentieri RNS 08-42-1A (NZ_CP015749), P. parmentieri SCC3193 (NC_017845), P. parmentieri WPP163 (NC_013421), P. parmentieri IFB5619 (NZ_CP026985), P. parmentieri HC (NZ_CP046376), P. parmentieri IFB5486 (NZ_CP026982), locus of petF1 uniquely present in P. parmentieri (green line and label), P. actinidiae KKH3 (NZ_JRMH00000000), P. aquaticum A212-S19-A16 (NZ_QHJR00000000), P. aroidearum PC1 (NC_012917), P. atrosepticum JG10-08 (NZ_CP007744), P. betavasculorum NCPPB 2795 (NZ_JQHM00000000), P. brasiliense SX309 (NZ_CP020350), P. carotovorum WPP14 (NZ_CP051652), P. fontis M022T (JSXC00000000), P. odoriferum BC S7 (NZ_CP009678), P. parvum s0241 (OANP00000000), P. peruviense IFB5232 (NZ_LXFV00000000), P. polaris NIBIO 1006 (NZ_CP017481), P. polonicum DPMP315 (NZ_RJTN00000000), P. punjabense SS95 (NZ_CP038498), P. versatile 3–2 (NZ_CP024842), P. wasabiae CFBP 3304 (NZ_CP015750) and P. zantedeschiae 2 M (NZ_PESL00000000).
Specificity of LAMP assay
The effectiveness of the LAMP assay was demonstrated with extensive inclusivity and exclusivity panels (Tables 2, 3). The LAMP assay detected all 15 strains of P. parmentieri (mostly from North America) represented in the inclusivity panel (Fig. 2, Table 2). The exclusivity panel consisted of 94 bacteria, including strains from different but closely related genera, a complete representation of all other described species of Pectobacterium, and healthy potato DNA (Table 3). Sigmoid-shaped curves (Fig. 2A) and bell-shaped melt curves (Fig. 2B) were observed. Reaction tubes containing LAMP products from P. parmentieri strains changed color from orange to green after the addition of 3 ul of SYBR Green I (Fig. 2C). Ten strains of P. parmentieri, which included LMG29774, PL67, PL72, PL71, PL74, PL75, PL124, PL123, PL30, and PL183, were used to represent the inclusivity panel. The samples representing the exclusivity panel (Fig. 2) included P. carotovorum (PL 73), P. versatile (ICMP 9168), P. polaris (ICMP 9180), P. punjabense (LMG 30,622), P. actinidae (LMG 26,003), P. polonicum (LMG31077), P. fontis (LMG30744), Ralstonia solanacearum (A6117), Xanthomonas phaseoli pv. dieffenbachiae (PL37), and negative template control (NTC, water). No amplification was observed from bacterial strains of other Pectobacterium sp., Dickeya sp., other gram-positive bacteria, endophytes/saprophytes isolated from potato or healthy potato plants (Fig. 2, Table 2). These data indicate that no cross-reactivity occurred with non-target bacterial species.

Specificity assay of loop-mediated isothermal amplification (LAMP) for specific detection of Pectobacterium parmentieri. Ten representative strains of P. parmentieri and 10 representative strains from the exclusivity panels are shown. (A) Real-time amplification plot with strains from both inclusivity and exclusivity panels; (B) melt-curve of 10 strains of P. parmentieri, no melt curve was observed with the strains from exclusivity panel and negative controls; (C) visualization of LAMP products after adding 3 μL of SYBR Green I stain. Tube 1, positive control P. parmentieri (LMG29774), tubes 2–10 P. parmentieri (PL67, PL72, PL71, PL74, PL75, PL124, PL123, PL30, and PL183), tubes 11–19 P. carotovorum (PL 73), P. versatile (ICMP 9168), P. polaris (ICMP 9180), P. punjabense (LMG 30,622), P. actinidae (LMG 26,003), P. polonicum (LMG31077), P. fontis (LMG30744), Ralstonia solanacearum (A6117), Xanthomonas phaseoli pv. dieffenbachiae (PL37), and N, negative template control (NTC, water).
Bacterial colony detection using LAMP assay
The LAMP detection was performed with pure colonies of P. polaris (ICMP 9180), P. versatile (ICMP 9168), D. dianthicola (A6058), Pantoea sp. (A1865), P. odoriferum (A1089), D. dadantii (A 5419), P. odoriferum (A2686), P. atrosepticum (A6163), Klebsiella aerogenes (A3131) and P. parmentieri (LMG29774). The DNA template of P. parmentieri (LMG29774) and nuclease free water were used as positive and negative controls, respectively. Amplifications were observed with both P. parmentieri heat-killed cells and the DNA template (Fig. 3A). Results were validated by adding 3 μL of SYBR Green I (Fig. 3B). No sigmoid curves (Fig. 3A) and subsequent color changes (Fig. 3B) were observed with other non-target strains from exclusivity panel and NTC. Therefore, indicating no cross-reactivity with non-target species. The developed LAMP assay accurately detected the target directly from heat-killed bacterial cells without prior DNA isolation or purification.

Specific detection of Pectobacterium parmentieri using loop-mediated isothermal amplification (LAMP) assay from heat-killed bacterial cells. (A) Real-time amplification plot, no sigmoidal curve was observed with strains of exclusivity panel and non-template control; (B) visualization of LAMP products after adding 3 μl of SYBR Green I stain. 1- Positive control DNA (P. parmentieri LMG29774), 2–11 heat-killed colonies: P. parmentieri (LMG29774), P. polaris (ICMP 9180), P. versatile (ICMP 9168), D. dianthicola (A6058), Pantoea sp. (A1865), P. odoriferum (A1089), D. dadantii (A5419), P. odoriferum (A2686), P. atrosepticum (A6163), Klebsiella aerogene (A3131), and negative template control (NTC, water).
Limit of detection
The limit of detection was determined using four independent assays with tenfold serially diluted purified genomic DNA and heat-killed bacterial cells. The LAMP assay detected purified P. parmentieri genomic DNA down to 100 fg per reaction (Fig. 4A–D). The detection limit with heat-killed cells was 10 CFU/mL (Fig. 5A–D). No adverse effect on the sensitivity was observed when 5 µl of crude host DNA was added in each reaction containing 1 µl of serially diluted genomic DNA (Fig. 4E–H) or lysate of heat-killed cells (Fig. 5E–H). No discrepancies were observed among the results of the different chemistries (fluorescence, SYBR Green I, UV, and gel electrophoresis) used for the cross-validation of the assay (Figs. 4, 5).

Detection limit of Pectobacterium parmentieri specific loop mediated isothermal amplification (LAMP) assay using pure genomic DNA. Ten-fold serially diluted genomic DNA (LMG29774) was used from 10 ng to 1 fg per reaction, indicated with lane 1 to 8. (A, E) Sigmoidal curves; (B, F) by adding SYBR Green I dye, color change from orange to bright green observed with naked eyes; (C, G) tubes observed under the UV light; (D, H) electrophoresis of amplified products on 2% agarose gel stained with ethidium bromide. Positive amplification was observed up to 100 fg (lane 6). Lane 9 is negative control (non-template control—water) and L is a 100 bp ladder. In the spiked assay, a 5 µl of crude host DNA was added to each reaction containing tenfold serially diluted genomic DNA.

Detection limit of Pectobacterium parmentieri specific loop-mediated isothermal amplification (LAMP) assay using heat-killed bacterial cells. Ten-fold serially diluted bacterial cells (LMG 29,774) starting from 109 CFU/mL to 1 CFU/mL per reaction, indicated with lane 1 to 10. (A, E) Sigmoidal curves; (B, F) by adding SYBR Green I dye, color change from orange to bright green observed with naked eyes; (C, G) tubes observed under the UV light; (D, H) electrophoresis of amplified products on 2% agarose gel stained with ethidium bromide. Positive amplification was observed up to 10 CFU (lane 9). Lane N is negative control (non-template control—water) and L is a 100 bp ladder. In the spiked assay, a 5 µl of crude host DNA was added to each reaction containing tenfold serially diluted heat killed bacterial cells.
LAMP assay validation with naturally and artificially infected plant and soil samples
The developed assay’s diagnostic capabilities were tested on 14 naturally infected plant samples, 10 artificially inoculated plant samples and 7 artificially infested soil samples. The LAMP assay accurately detected P. parmentieri in the DNA isolated from all of the above samples and did not cross-react with samples infected/infested with other Pectobacterium or Dickeya species (Table 4).
LAMP validation with artificially inoculated potato tubers to assess the applicability for field applications
Potato slices were inoculated with different Pectobacterium species (P. parmentieri, P. punjabense, P. fontis, P. polonicum, P. carotovorum, and P. wasabiae) and D. dianthocola. The LAMP assay accurately detected the target pathogen in P. parmentieri-inoculated potato tubers. After the addition of SYBR Green, the samples, containing LAMP products, changed from orange to green, indicating a positive amplification. Additionally, no color change was observed for the non-target species, non-template control, or healthy potato slices (Fig. 6).

Detection of Pectobacterium parmentieri from infected potato samples. (A) Infected potato slices infected with different Pectobacterium species and Dickeya dianthicola; LMG29774 P. parmentieri, PL30 P. parmentieri, LMG30622 P. punjabense, LMG30744 P. fontis, LMG31077 P. polonicum, PL182 P. carotovorum, PL188 P. wasabiae, PL191 D. dianthicola, NC (negative control) healthy potato. (B) Standard curve diagram–only two P. parmentieri infected potato slices and positive control LMG29774 P. parmentieri were positive, no curve was observed with NC (negative control) healthy potato and NTC, non-template control (water). (C) Visualization of LAMP products after addition of SYBR Green I dye—green color represents positive amplification. (D) Visualization of SYBR Green I results under UV light—fluorescence indicative of positive amplification. 1, LMG29774 P. parmentieri; 2, PL30 P. parmentieri; 3, LMG30622 P. punjabense; 4, LMG30744 P. fontis; 5, LMG31077 P. polonicum; 6, PL182 P. carotovorum; 7, PL188 P. wasabiae; 8, PL191 D. dianthicola; 9, NC healthy potato.
Multi-operator validation
Three independent operators performed the LAMP assay with blind samples, including P. parmentieri and closely related species, plant material infected with P. parmentieri, and a non-template control (Table 5). As indicated, all three operators correctly identified P. parmentieri and there was no cross-reactivity with any other non-target samples.
Discussion
To effectively contain the pathogen and manage the disease, an effective diagnostic test is an essential requirement. Here, we designed and developed a LAMP assay to specifically detect P. parmentieri and optimized the test for both field and laboratory diagnostics. LAMP is a popular and well-established rapid and cost-effective diagnostic technique with high specificity and sensitivity which is easily applied at point-of-need. We performed various validation tests to ensure the quality of the assay31.
Precision, dependability, and accuracy are important components of a robust and specific detection assay to be utilized in monitoring and surveillance programs. The foundation of a robust and specific assay depends on target selection32. The low cost of genome sequencing and availability of whole genomic data in public databases increases the use of comparative genomic approaches for identifying for signature genomic regions exclusively present in target species32. In this study, we designed P. parmentieri primers to amplify a unique petF1 gene region, a genomic region highly conserved in all P. parmentieri strains tested, but not in other closely related bacterial strains, pathogenic or non-pathogenic (Fig. 1). Comparing the whole genomes of bacteria of different origin and host ranges for target-specific primer development ensures assay’s specificity greatly minimizing inadvertent cross-reactivity with non-target microorganisms and hosts33, this virtually eliminating false-positives in the identification of P. parmentieri. The designed primers, validated in silico by against the NCBI GenBank database, showed high specificity to P. parmentieri (Table 1).
The in-silico validation of primers is required to initially eliminate non-specific targets but does not guarantee the target’s in-vitro specificity. Therefore, extensive inclusivity and exclusivity panels were composed of multiple strains of P. parmentieri and other closely-related species, respectively (Tables 2, 3). All 15 strains in the inclusivity panel were positive with the LAMP assay (Table 2), while 94 bacterial strains of 18 closely-related species of the genus Pectobacterium, including closely-related species that cause similar potato blackleg and soft rot symptoms, were negative (Table 3). Additionally, the assay was tested and validated with endophytic and saprophytic bacteria and DNA from healthy host plants (Table 3). The detection capability and accuracy of an assay can be adversely affected by inclusion of infected plant and soil materials that may contain inhibitors34. Similarly, the soil contains compounds that inhibit enzymes involved in DNA manipulation35. The developed LAMP assay was not inhibited by naturally or artificially infected plant materials, infested soil samples or plant samples infected with closely-related species. The assay’s accuracy was evaluated by infecting the potato slices with several closely-related species—no false positives or negatives were detected (Fig. 6). The diagnostic assay’s speed and simplicity was achieved by incorporating a Plant Material Lysis Kit (less than 5 min preparation time, as described by Ocenar et al.22) with the LAMP assay (10–15 min), which reduced the total assay time to 15–25 min. During validation, neither cross-reactivity nor false positives were observed. Each run included a positive and a negative control.
The high sensitivity of a detection assay reduced the possibilities of false-negative results15. Host plant constituents can impact the detection limit of an assay; thus, it is imperative that the detection limits are assessed in the presence of crude plant DNA22. The developed LAMP assay’s sensitivity was confirmed by evaluating its performance at low concentrations of genomic DNA and heat-killed bacterial cells. The assay detected purified genomic DNA as low as 100 fg (18–20 genome copies) and a minimum of 10 CFU from bacterial lysate (Fig. 4). The detection limit was not affected by spiking the diluents with host crude plant DNA prepared using Plant Material Lysis Kit (Fig. 5), indicating that our assay will be highly effective in early diagnosis, and identify the pathogen at low concentrations in the plant sap. The dead bacterial cells present in bacterial cell lysate might have contributed to obtain higher sensitivity, and therefore, we believe assessing the assay’s limit of detection using CFU method is not appropriate. The Ocenar and Colleagues22 also reported a detection limit of 10 CFU, but reported lower assay sensitivity (1 pg) than was achieved in this study (100 fg) when performed with purified genomic DNA22. However, this difference may be due to quantification methods, since NanoDrop quantification is less accurate than the newer Qubit method used in the work reported here. The assay performance can also be affected by the operators, but we have confirmed that the developed assay is repeatable by obtaining concordant results when blind tests were performed by three independent operators (Table 5).
In conclusion, we demonstrated a simplified field-deployable LAMP assay for specific detection of P. parmentieri. The assay is sensitive and rapid, and has applications in pathogen detection, quarantine, eradication, border protection, seed certification, disease management, and epidemiology.
Materials and methods
Any plant and plant materials used in this research compliance with international, national and institutional guidelines.
Target selection and primer design
A total of 50 complete and draft genomes sequences were included in the analysis; 19 of these genomes came from various P. parmentieri strains isolated in different years from distinct geographic locations. All genomes sequences were re-annotated using Prokka36. The pan and core genomes among all Pectobacterium species were analyzed using the ROARY pipeline37. After conducting the pan-core analysis, ROARY output displayed the presence and absence of genes among the 50 genomes, allowing identification of unique gene regions present exclusively in all P. parmentieri strains. Candidate genes found exclusively in P. parmentieri were analyzed in silico using the nucleotide BLAST algorithm. The PetF1 gene was identified and used as the specific target for designing the LAMP primers. The criterion for gene selection was to display 100% identity with 100% query coverage of all P. parmentieri strains. Additionally, the selected gene had to be absent in the other Pectobacterium species and other closely related bacteria that share the same ecological niche of the target pathogen, P. parmentieri. After identifying the target gene petF1, a nucleotide comparison ring image (Fig. 1) was created to portray the gene’s location and unique presence across different P. parmentieri strains. The image was generated using BRIG (BLAST Ring Image Generator)29. The genome comparison was performed based upon the NCBI-BLAST version 2.10.0 + database; P. parmentieri RNS 08–42-1A served as a reference genome for nucleotide alignment. The complete genomes of six P. parmentieri strains and 17 complete/draft genomes other Pectobacterium species were included in the analysis. The locus of petF1 is highlighted in Fig. 1. The genomes included in the circular graphic were downloaded from the NCBI GenBank database with these accession numbers: P. parmentieri RNS 08-42-1A (NZ_CP015749), P. parmentieri SCC3193 (NC_017845), P. parmentieri WPP163 (NC_013421), P. parmentieri IFB5619 (NZ_CP026985), P. parmentieri HC (NZ_CP046376), P. parmentieri IFB5486 (NZ_CP026982), P. actinidiae KKH3 (NZ_JRMH00000000), P. aquaticum A212-S19-A16T (NZ_QHJR00000000), P. aroidearum PC1 (NC_012917), P. atrosepticum JG10-08 (NZ_CP007744), P. betavasculorum NCPPB 2795 (NZ_JQHM00000000), P. brasiliense SX309 (NZ_CP020350), P. carotovorum WPP14 (NZ_CP051652), P. fontis M022T (JSXC00000000), P. odoriferum BC S7 (NZ_CP009678), P. parvum s0241T (OANP00000000), P. peruviense IFB5232T (NZ_LXFV00000000), P. polaris NIBIO 1006T (NZ_CP017481), P. polonicum DPMP315T (NZ_RJTN00000000), P. punjabense SS95T (NZ_CP038498), P. versatile 3–2 (NZ_CP024842), P. wasabiae CFBP 3304 (NZ_CP015750) and P. zantedeschiae 2 M (NZ_PESL00000000).
Six LAMP primers, forward inner primer (Pp-FIP), forward outer primer (Pp-F3), backward inner primer (Pp-BIP), backward outer primer (Pp-B3), forward loop primer (Pp-LF) and backward loop primer (Pp-LB), were designed using PrimerExplorer V5 (https://primerexplorer.jp/e/) and are listed in Table 1. The NCBI GenBank BLASTn tool was used to confirm each primer’s specificity against the available genome database.
Source of bacterial strains and DNA isolation
A total of 110 bacterial strains from different hosts and geographic locations, including strains obtained from international culture collections, were used in this study (Tables 2, 3). Fifteen strains of P. parmentieri and 95 strains belonging to closely-related genera and species were chosen for inclusivity and exclusivity panels including 8 endophytic bacteria, respectively (Tables 2, 3). Bacterial strains listed with “A”, “PL”, and culture collection IDs were stored at − 80 °C, and revived by streaking onto 2,3,5-triphenyltetrazolium chloride (TZC) medium (peptone 10 g 1−1, dextrose 5 g l−1, 0.001% TZC and agar 17 g l−1) and TZC-sucrose medium (TZC-S: peptone 10 g l−1, sucrose 5 g l−1, 0.001% TZC and agar 17 g l−1), respectively (Norman and Alvarez 1989). The plates were incubated at 26 °C (± 2 °C) for 12–24 h. Single colonies were re-streaked onto a new TZC medium plate and later used to harvest pure bacterial growth for DNA isolation32.
For bacterial genomic DNA extraction from pure cultures, loopful of bacterial cells from TZC plates was suspended into phosphate‐buffered saline (PBS) or directly into 1.5 mL tubes containing 200 µl alkaline lysis buffer provided and proceed with DNA isolation using the DNeasy Blood and Tissue Kit following the manufacturer’s instruction (Qiagen, Germantown, MD).
Specificity determination
Specificity of the developed LAMP assay was determined using different bacterial strains included in the inclusivity and exclusivity panels listed in Tables 2 and 3, respectively. The inclusivity panel included 15 strains of P. parmentieri isolated from potato (Solanum tuberosum) from three different geographical locations (Table 2). Ninety-five samples consisting of all known Pectobacterium species, excluding P. parmentieri, isolated from different hosts and locations, plant pathogenic Gram-positive and Gram-negative bacteria, potato endophytic bacterial strains and healthy potato plant DNA were included in the exclusivity panel (Table 3). The LAMP reaction of 25 µl consisted of 15 µl Optigene Master Mix (Optigene, West Sussex, UK), 2 µl primer mix (1.6 µM each of Pp-FIP and Pp-BIP, 0.2 µM each of Pp-F3 and Pp-B3, 0.4 µM each of Pp-LF and Dd-LB), 7 µl of water (Invitrogen), and 1 µl DNA template. DNA templates from P. parmentieri strains was used as a positive control; DNA from healthy plants and no template DNA (nuclease-free water) were used as the negative control. The LAMP reaction mixture was incubated and amplified in the Rotor-Gene Q (Qiagen, Germantown, MD) at 65 °C for 20 min. The melt curves were analyzed using Rotor-Gene Q series software 2.3.1 (Built 49) at 80–99 °C with an increment of 0.2 °C/s. Positive target amplification was determined by melt curves above a designated threshold. Melt curves below the threshold were deemed as no amplification or negative. The results were also validated using colorimetric-based detection, by adding 3 µl of SYBR Green dye I (Life Technologies Corporation, Eugene, OR) in each amplified reaction. A positive LAMP reaction was indicated by a change in product color from orange to bright green, while negative reactions remained orange. Results obtained using SYBR Green I dye were observed directly either by the naked eye or by placing the reaction tubes under UV light in a Gel Doc XR + Gel Documentation System (Bio-Rad, Hercules, CA).
LAMP detection directly from heat-killed bacterial colonies
The LAMP detection was performed using colonies of 10 bacterial strains: P. parmentieri (LMG29774), P. polaris (ICMP 9180), P. versatile (ICMP 9168), D. dianthicola (A6058), Pantoea sp. (A1865), P. odoriferum (A1089), D. dadantii (A5419), P. odoriferum (A2686), P. atrosepticum (A6163), and Klebsiella aerogene (A3131). Pure DNA template (LMG29774) and nuclease-free water were used as a positive control and non-template control, respectively. Pure colonies from each strain were collected from TZC plates and added to a PCR tube containing 25 μl of nuclease-free water and heated at 95 °C for 10 min in a T-100 thermocycler (Bio-Rad). One microliter of colony lysate was used as a template for LAMP reactions. LAMP assays were performed following the protocol described above. Real-time amplification plots were obtained, and the results validated by adding 3 µl of SYBR Green I dye in the amplified reaction tubes. The tubes were observed directly by the naked eye for color change.
Limit of detection determination
The detection limit was determined by performing four independent assays—two with genomic DNA and two with heat-killed cells. To determine the limit of detection using genomic DNA, P. parmentieri (LMG 29774) purified genomic DNA was quantified using a Qubit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA). Ten-fold serial dilutions were prepared from 10 ng to 1 fg of genomic DNA in nuclease-free water. One microliter of DNA from each dilution was added into the individual LAMP reaction mixture. The LAMP assay was performed following the same conditions and components described above. A spiked assay was performed by adding 5 µl of crude host (potato stem tissues) DNA, prepared using Plant Material Lysis Kit (Optigene, Sussex, UK), in each LAMP reaction while containing 1 µl serially diluted P. parmentieri genomic DNA. To determine the limit of detection utilizing heat-killed bacterial cells, an overnight grown culture of P. parmentieri (LMG 29774) was tenfold serially diluted and enumerated by spread plating 100 µl of the 10−6, 10−7 and 10−8 cells onto nutrient agar medium (BD, Becton Dickinson) plates in triplicate. Plates were incubated at 28 °C for 18–24 h prior to counting. Bacterial colonies were counted, averaged, and calculated as log10 CFU/mL. The count was 1.1 × 109 CFU/mL. For the LAMP assay, bacterial cultures were serially diluted from 109 to 1 CFU in peptone water and heat-killed at 95 °C for 10 min in a T-100 thermocycler, centrifuged at maximum speed for 2 min. One microliter of supernatant from each dilution was used in individual LAMP reactions. LAMP assay conditions and components, except template, were as described above. Spiked assays were performed by adding 5 µl of crude host DNA, as described above, in each LAMP reaction while containing 1 µl of serially diluted heat-killed cells supernatant. A negative control was included in each run.
Detection from infected plant and soil samples
Naturally and artificially infected plant tissues were used to validate the assay. A total of 14 naturally infected potato plant samples containing Pectobacterium and/or Dickeya species were screened. A total of 10 infected plants (infected with strains PL67, PL70-75, PL123-124, and PL128) and 7 infected soil samples (infected with strains PL71-75, PL123 and PL128) were included in this study to validate the LAMP protocol. These DNA samples from artificially infected plant and infested soil samples were used from a previous study in our laboratory30. These samples were inoculated/infested with known cultures (Table 4) and DNA was isolated using DNeasy PowerSoil Kit (Qiagen) or a DNeasy Plant Mini Kit (Qiagen).
Potato tubers were cleaned using tap water and dipped into a 0.6% hypochlorite solution for 3 min followed by rinsing three times with sterile water then cut into slices. A loopful (~ 10 µl) of overnight grown bacterial culture was inoculated into each potato slice, placed into petri dishes and incubated for 12–18 h. A total of 100 mg macerated tissue was taken and used for crude DNA isolation using a Plant Material Lysis Kit (Optigene, West Sussex, UK). Five μl of crude DNA was used in each LAMP reaction following the above protocol.
Multi-operator validation
Multi-operator validation was included to confirm the repeatability of the developed assay. Three independent operators performed blind assays with total of 7 samples and one NTC. The samples included genomic DNA from two P. parmentieri strains, two DNA samples from P. parmentieri-infected plant samples, and DNA from P. polaris, P. zantedeschiae, and D. fangzhongdai (Table 5). Each operator performed the assay following the LAMP protocol mentioned above. The results were compared with initial diagnostic data.
References
Czajkowski, R., Grabe, G. J. & van der Wolf, J. M. Distribution of Dickeya spp. and Pectobacterium carotovorum subsp. carotovorum in naturally infected seed potatoes. Eur. J. Plant Pathol. 125, 263–275 (2009).
Ma, X., Schloop, A., Swingle, B. & Perry, K. L. Pectobacterium and Dickeya responsible for potato blackleg disease in New York State in 2016. Plant Dis. 102, 1834–1840 (2018).
Skelsey, P., Humphris, S. N., Campbell, E. J. & Toth, I. K. Threat of establishment of non-indigenous potato blackleg and tuber soft rot pathogens in Great Britain under climate change. PLoS ONE 13, e0205711 (2018).
Toth, I. et al. Dickeya species: An emerging problem for potato production in Europe. Plant. Pathol. 60, 385–399 (2011).
Chung, Y. S., Goeser, N. J., Cai, X. & Jansky, S. The effect of long term storage on bacterial soft rot resistance in potato. Am. J. Potato Res. 90, 351–356 (2013).
Ma, B. et al. Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163 (2007).
Khayi, S. et al. Transfer of the potato plant isolates of Pectobacterium wasabiae to Pectobacterium parmentieri sp. nov.. Int. J. Syst. Evol. Microbiol. 66, 5379–5383 (2016).
Ngoc Ha, V. et al. First report of Pectobacterium parmentieri causing stem rot disease of potato in Russia. Plant Dis. 103, 144–144 (2019).
De Boer, S., Li, X. & Ward, L. Pectobacterium spp. associated with bacterial stem rot syndrome of potato in Canada. Phytopathology 102, 937–947 (2012).
Dung, J., Johnson, D. & Schroeder, B. First report of Pectobacterium wasabiae causing aerial stem rot of potato in Washington State. Plant Dis. 96, 1819–1819 (2012).
Li-ping, W., Min, Z., Min-hua, R., Xing-wei, L. & Qiong-guang, L. Pectobacterium carotovorum subsp. brasiliensis and Pectobacterium parmentieri as causal agents of potato blackleg and soft rot in China. J. Plant Pathol. 102, 871–879 (2020).
Moleleki, L. N., Onkendi, E. M., Mongae, A. & Kubheka, G. C. Characterisation of Pectobacterium wasabiae causing blackleg and soft rot diseases in South Africa. Eur. J. Plant Pathol. 135, 279–288 (2013).
Charkowski, A. O. The changing face of bacterial soft-rot diseases. Annu. Rev. Phytopathol. 56, 269–288 (2018).
Arizala, D. et al. First report of bacterial soft rot and blackleg on potato caused by Pectobacterium parmentieri in Hawaii. Plant Dis. 104, 970 (2020).
Boluk, G. et al. Genome-informed recombinase polymerase amplification assay coupled with a lateral flow device for in-field detection of Dickeya species. Plant Dis. 104, 2217–2224 (2020).
Daher, R. K., Stewart, G., Boissinot, M. & Bergeron, M. G. Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 62, 947–958 (2016).
Ahmed, F. A., Larrea-Sarmiento, A., Alvarez, A. M. & Arif, M. Genome-informed diagnostics for specific and rapid detection of Pectobacterium species using recombinase polymerase amplification coupled with a lateral flow device. Sci. Rep. 8, 1–11 (2018).
Panno, S. et al. Loop mediated isothermal amplification: principles and applications in plant virology. Plants 9, 461 (2020).
Yasuhara-Bell, J., Marrero, G., De Silva, A. & Alvarez, A. M. Specific detection of Pectobacterium carotovorum by loop-mediated isothermal amplification. Mol. Plant Pathol. 17, 1499–1505 (2016).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63–e63 (2000).
Chander, Y. et al. A novel thermostable polymerase for RNA and DNA loop-mediated isothermal amplification (LAMP). Front. Microbiol. 5, 395 (2014).
Ocenar, J. et al. Development of a robust, field-deployable loop-mediated isothermal amplification (LAMP) assay for specific detection of potato pathogen Dickeya dianthicola targeting a unique genomic region. PLoS ONE 14, e0218868 (2019).
Dobhal, S., Larrea-Sarmiento, A., Alvarez, A. M. & Arif, M. Development of a loop-mediated isothermal amplification assay for specific detection of all known subspecies of Clavibacter michiganensis. J. Appl. Microbiol. 126, 388–401 (2019).
Suárez, M. B., Feria, F., Martín-Robles, M., del Rey, F. & Palomo, J. Pectobacterium parmentieri causing soft rot on potato tubers in southern Europe. Plant Dis. 101, 1029–1029 (2017).
Arizala, D. & Arif, M. Genome-wide analyses revealed remarkable heterogeneity in pathogenicity determinants, antimicrobial compounds, and CRISPR-cas systems of complex phytopathogenic genus Pectobacterium. Pathogens 8, 247 (2019).
Boluk, G. et al. First report of Pectobacterium brasiliense causing soft rot on Brassica oleracea var. sabellica in Hawaii. U. S. Plant Dis. 104, 2721 (2020).
Nabhan, S., De Boer, S., Maiss, E. & Wydra, K. Taxonomic relatedness between Pectobacterium carotovorum subsp. carotovorum, Pectobacterium carotovorum subsp. odoriferum and Pectobacterium carotovorum subsp. brasiliense subsp. nov. J. Appl. Microbiol. 113, 904–913 (2012).
Zoledowska, S., Motyka, A., Zukowska, D., Sledz, W. & Lojkowska, E. Population structure and biodiversity of Pectobacterium parmentieri isolated from potato fields in temperate climate. Plant Dis. 102, 154–164 (2018).
Alikhan, N.-F., Petty, N. K., Zakour, N. L. B. & Beatson, S. A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 12, 1–10 (2011).
Arizala, D. et al. Multiplex TaqMan qPCR targeting unique genomic regions for specific, sensitive and robust detection of Pectobacterium species and P. parmentieri. Plant Health 2019. https://www.apsnet.org/meetings/annual/meetingarchives/planthealth2019/Documents/Abstracts/aps2019ab727.htm (2019).
Wong, Y. P., Othman, S., Lau, Y. L., Radu, S. & Chee, H. Y. Loop-mediated isothermal amplification (LAMP): A versatile technique for detection of micro-organisms. J. Appl. Microbiol. 124, 626–643 (2018).
Dobhal, S. et al. Comparative genomics reveals signature regions used to develop a robust and sensitive multiplex TaqMan real-time qPCR assay to detect the genus Dickeya and Dickeya dianthicola. J. Appl. Microbiol. 128, 1703–1719 (2020).
Lang, J. M. et al. Genomics-based diagnostic marker development for Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola. Plant Dis. 94, 311–319 (2010).
Dobhal, S., Olson, J. D., Arif, M., Suarez, J. A. G. & Ochoa-Corona, F. M. A simplified strategy for sensitive detection of Rose rosette virus compatible with three RT-PCR chemistries. J. Virol. Methods 232, 47–56 (2016).
Braid, M. D., Daniels, L. M. & Kitts, C. L. Removal of PCR inhibitors from soil DNA by chemical flocculation. J. Microbiol. Methods 52, 389–393 (2003).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Page, A. J. et al. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Acknowledgements
This work was supported by the USDA-ARS Agreement No. 58-2040-9-011, Systems Approaches to Improve Production and Quality of Specialty Crops Grown in the U.S. Pacific Basin; sub-project: Genome Informed Next Generation Detection Protocols for Pests and Pathogens of Specialty Crops in Hawaii. The strains were stored and maintained by the National Science Foundation funded project (NSF-CSBR Grant No. DBI-1561663). Barry and Barbara Brennan Endowment also supported this work. The mention of trade names or commercial products in this publication does not imply recommendation or endorsement by the University of Hawaii. This research paper is the outcome of the course “PEPS/MBBE 627 Molecular Diagnostics: Principles and Practices”.
Author information
Authors and Affiliations
Contributions
M.A. conceived and designed the study; R.D., C.P., D.K., H.V., A.C.T., X.W., A.C., J.N.U., M.S., D.A., and S.D. performed the experiments and wrote the manuscript; D.A., S.D., and G.B. collected and initially purified the strains used in this study; J.P.B., F.M.O.C., E.A., J.P.S., J.F., J.O., D.J., A.M.A. and M.A. revised the manuscript and provided ideas and support for the final submission; all authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Domingo, R., Perez, C., Klair, D. et al. Genome-informed loop-mediated isothermal amplification assay for specific detection of Pectobacterium parmentieri in infected potato tissues and soil. Sci Rep 11, 21948 (2021). https://doi.org/10.1038/s41598-021-01196-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-01196-4
- Springer Nature Limited