
Abstract
Hydrogen peroxide (H2O2) is ubiquitous in natural waters, and plays an important role in both biological and chemical processes. This study investigated the influence of dissolved organic matter (DOM) and its substituents on the accurate measurement of H2O2 by peroxidase-mediated depletion of scopoletin fluorescence method which is one of the most widely used methods for the determination of low concentration H2O2 in water. Six DOM and its 24 substituents interfered the determination of H2O2 at environmentally relevant concentration of 200 nM with different levels except 2,6-dimethoxy-1,4-benzoquinone and benzoic acid, which may be associated with origin and types of DOM, and numbers and position of active functional groups in DOM constituents. Each substance concentration and the corresponding decreasing ratio to the measured H2O2 concentration was fitted well to the linear model (R2 > 0.9), and the obtained interfering ratios (k, (mgC L−1)−1), expressing the degree of DOM or its substituents per unit concentration to the measurement of H2O2, were approximate for DOM, but the order of magnitude of k values of DOM constituents took on a large span from 10–3 to 10–7. When DOM levels exceed 0.1 mgC L−1 or its substituent concentration is at nM level (low to 20 nM), the H2O2 content will be underestimated substantially. A quantitative structure–activity relationship model with remarkable stability and strong predictability for the k of DOM substituents to H2O2 measurement was established, and the k was related to the electron transfer capacity, hydrophobicity and stability of these compounds.
Similar content being viewed by others


Introduction
As a relatively stable reactive oxygen species (ROS) with a long half-life (several to 100 h)1, hydrogen peroxide (H2O2) is ubiquitous in natural waters such as freshwater, rainwater, and seawater with a steady-state concentration ranging from nM to μM2,3. The source of H2O2 include the interactions of sunlight and light-absorbing substances (e.g., chromophoric dissolved organic matter (CDOM)), redox cycling of metals and biological processes, and atmospheric deposition4,5,6,7,8,9,10. The high redox-active transient plays an important role in the environmental fate of organic compounds and geochemical cycling of trace elements (e.g., iron, copper, chromium, manganese)11,12,13, thereby indirectly affecting their biological availability and/or toxicity to organisms14. Also, H2O2 can pose deleterious effects on biological systems directly. For instance, H2O2 at sufficiently high concentrations can pass through cell membranes and cause oxidative stress, mutagenesis and the bleaching of chlorophy II15,16,17. Therefore, it is not surprising that research on H2O2 has been carried out by numerous investigators, especially the development of its detection techniques which has profound implication on estimating net production of H2O2 in natural waters and our understanding of ecosystem biogeochemistry.
Up to now, more than 30 analytical methods based on absorbance, voltammetry, fluorescence or chemiluminescence have been developed to determine H2O2 distribution in various aquatic environment such as surface waters18. Each method attempts to accelerate the analysis process, reduce the detection limits and avoid potential interferences such as fluorescence quenching by DOM19. Among these methods, fluorescence (FL) method is by far the most popular one, where the probe compound is oxidized to yield products that either exhibit FL or whose FL is diminished in the presence of peroxidase and H2O218. The most commonly used and highly cited FL method for determination of H2O2 in natural waters in early studies was scopoletin-horseradish peroxidase (HRP) method18. Although no longer the most commonly used method for the quantitative determination of H2O2, it is the seminal method from which many current methods evolved and so a presentation of some methodological detail is appropriate for any review18. Currently, the method have been demonstrated to suffer from the potential interference from various factors including naturally occurring substances such as DOM and organic peroxide, pH and buffer2,18,20,21. For example, Cooper et al. (1988) determined H2O2 concentrations in natural water matrices including freshwater, agricultural water, sewage, seawater and estuarine exposed to sunlight using scopoletin method. The samples were prepared in phosphate buffer (PBS) adjusted to pH 7 and diluted if their total organic carbon (TOC) of was higher than 2 mgC L−1 prior to analysis. Miller and Kester (1988) discussed interferences of DOM and organic peroxide to H2O2 determination using another HRP-mediated (p-Hydroxyphenyl) acetic acid (POHPAA) method, and recommended using standard additions to determine H2O2 in natural water samples to overcome DOM interferences. Afterward, standard addition method has been widely applied in H2O2 determination in natural waters22. In fact, both the aqueous and soil environments contain large amounts of natural phenols (e.g., ferulic acid, syringaldehyde, pyrogallol, hydroxybenzoic acid, or catechol) that originate from lignin decomposition and are major substrates for oxidative coupling reaction leading to the formation of humus23. In addition, some xenobiotic compounds, e.g., chlorinated phenols and anilines, were introduced into the environment by accidental spills, illegal release of industrial and municipal wastewater, and excessive use of pesticides, and were regarded as analogues of DOM constituents24. As DOM constituents with phenolic, amino, and/or aromatic alcohols moiety as the core components in their chemical structures were also the excellent substrates of peroxidases, it is speculated that DOM constituents may interfere the measurement of H2O2 by peroxidase-mediated depletion of scopoletin fluorescence method. However, with the exception of DOM and organic peroxide, those heretofore neglected or overlooked other interferences in the determination of H2O2 in natural waters should be of concern. In view of this idea, knowledge related to the effect of DOM constituents on the determination of H2O2 in waters is of great environmental significance.
The aims of the present study were (1) to investigate and compare the influence of 6 representing forms of DOM and its constituents on the determination of environmental level H2O2 using the HRP-mediated depletion of scopoletin fluorescence method. In light of the diversity of specific DOM constituents (Fig. S1), 24 compounds with phenolic, carbonyl, carboxyl, and amino groups attached to benzene skeleton were selected; (2) to establish a quantitative structure–activity relationship (QSAR) model based on the data of interfering effect of DOM constituents on H2O2 concentration determination with good stability and prediction ability, thereby trying to predict the effect of other DOM constituent analogues on the H2O2 measurement in water.
Results and discussion
Interference of DOM on H2O2 determination
The effects of 6 kinds of representative DOM on H2O2 determination were presented in Fig. 1. For each investigated DOM, it was apparent that DOM significantly interfere the accurate measurement of H2O2 content, and the measured concentrations were lower than the nominal concentration (200 nM). To be clear, here the decreasing ratio (C/C0) of measured H2O2 concentration caused by the presence of DOM was defined as the ratio of measured concentration of H2O2 (experimental values, C) and initial concentration of H2O2 (theoretical values, C0). Figure 2 displayed the relationship between each DOM concentration and the corresponding decreasing ratio to H2O2. As can be seen, the determinable H2O2 concentration became decreased with the increasing contents of DOM. There is a fine linear relationship between DOM concentrations and the natural logarithm of decreasing ratios (R2 > 0.9), thus the obtained slope k ((mgC L−1)−1) values were called as interfering ratios, expressing the interfering degree of DOM per unit concentration to the measurement of H2O2, and the greater the k values, the larger the interfering degree of DOM to H2O2 measurement. The interfering ratios of DOM from different sources and types were showed in Table 1. The interfering ratio of HA from Nordic lake (NLHA) was the highest, and significantly higher than SRFA, PLFA and SRNOM (p < 0.05). The interfering ratio of FA from Pony lake (PLFA) is significantly lower than NLFA, SRHA, NLHA and SRNOM (p < 0.05) (Table S1). The parameter EC10 (mgC L−1) in Table 1 expressed DOM concentrations added when the decreasing ratio of measured H2O2 concentration reach to 10%, which could be considered as not significant. As showed in Table 1, the EC10 values of most FAs were greater than 0.1 mgC L−1, and generally greater than that of HAs. On the whole, accurate determination of H2O2 using peroxidase-mediated depletion of scopoletin fluorescence method will be disturbed substantially when DOM levels exceed 0.1 mgC L−1 in natural waters. In fact, the level of DOM in actual water bodies usually ranged from a few to tens of mgC L−17,25. As a result, the water samples should be diluted by tens to hundreds of times to accurately determine H2O2 levels in natural waters.

The effect of DOM originated from different sources on H2O2 determination.

The relationship between SUVA254 and E2/E3 and the k of DOM.
The SUVA254 determined the absorbance at 254 nm divided by DOM concentration and E2/E3 determined the absorbance at 254 nm divided by the absorbance at 365 nm are important indicators that reflect the aromaticity and molecular weight of DOM, respectively26. Here the SUVA254 and E2/E3 of investigated six DOM at environmentally relevant concentration of 5 mgC L−1 were calculated in Table S2, and the relationship between them and the k of DOM was plotted in Fig. 2. As can be seen, the k of DOM displayed a negative correlation with E2/E3 values, and a good positive correlation with their SUVA254 values, which could be explained that DOM with the larger molecular weight and aromaticity may wrap the substrate, and thus prevent it from contacting HRP26. Based on our finding, the accurate determination of H2O2 in natural waters by a peroxidase-mediated depletion of scopoletin fluorescence method could be corrected according to the measurement of SUVA254 and E2/E3 of DOM. Our results would also provide information on the reliability evaluation of the interference of DOM on the determination of H2O2.
Influence of DOM constituents on H2O2 determination
Similarly, impacts of 24 DOM constituents on H2O2 at the initial concentration of 200 nM were explored, and the results were showed in Fig. 3. In general, most compounds interfered the measurement of H2O2 in different degree, and there is good linear relationship between compound concentrations and the natural logarithm of decreasing ratios (R2 > 0.9) with the exception of 2,6-dimethoxy-1,4-benzoquinone and benzoic acid. For 2,6-dimethoxy-1,4-benzoquinone, the fluorescence signal became weaker with its concentration increasing, which was probably explained by the quenching effect of the compound on the fluorescence intensity of scopoletin. As for benzoic acid, there was no obvious interference for H2O2 measurement regardless of its concentration.

The effects of natural DOM constituents on H2O2 determination.
Table 2 listed the decreasing ratios of the DOM constituents on H2O2 determination. Except for 2,6-dimethoxy-1,4-benzoquinone and benzoic acid, the order of magnitude of interfering ratios of all other compounds take on a large span from 10–3 to 10–7 μM-1. In a word, catechol gave rise to the maximum interfering on H2O2 measurement and was significantly higher than that of all compounds with the interference for H2O2 measurement except for gallic acid (p < 0.05, Table S3). There was significant effect on measurement of H2O2 at 200 nM level when the concentrations of those 5 compounds reached to 20 nM in waters, and measurable H2O2 concentration can be completely suppressed when their concentrations increased to 200 nM. H2O2 determination will be disturbed by guaiacol, p-anisidine, 3,4-dimethoxyphenol, p-aminophenol and caffeic acid at 30 nM, and may be submerged absolutely on the condition of 700 nM of the same compounds. As for resorcinol and o-anisidine, when their concentration came up to 200 and 400 nM respectively, H2O2 measurement would be affected, and be completely suppressed at 5 and 7 μM of the 2 compounds respectively. The ones induced the weakest interfering effect on H2O2 were p-hydroxybenzoic acid, 2,5-dihydroxy-1,4-benzoquinone, aniline, p-aminobenzoic acid, salicylic acid and veratryl alcohol. The significant impact on H2O2 measurement required the levels of these compound reaching up to several μM, and even hundreds of μM.
Different inhibition ratios of DOM constituents may be connected with their functional group type, number and position of these compounds. For instance, in terms of isomers, the interfering effect of catechol to H2O2 measurement was close to that of hydroquinone, while was higher than that of resorcinol. Moreover, guaiacol versus p-methoxyphenol, and salicylic acid versus p-hydroxybenzonic acid have presented a parallel effect. For non-isomers with the same substituent position, type and number, a good agreement on the interfering impact existed between catechol and hydroquinone with -OH versus guaiacol and p-methoxyphenol with -OCH3, respectively, which was similar to hydroquinone versus p-aminophenol and p-methoxyphenol versus p-anisidine. A comparison for the interfering effect of hydroquinone versus p-hydroxybenzoic acid, catechol versus salicylic acid, and p-aminobenzoic acid, p-anisidine and p-aminophenol might indicate that -COOH caused a slight action on H2O2 determination.
These results were significant to determine precisely the levels of H2O2 in natural waters. Montero et al. detected phenol and several chlorophenols (e.g., 2-chlorophenol and 2,4-dimethylphenol) concentration ranged from ng L−1 to μg L−1 in lakes and ground waters, and the total amount of analyzed phenols ranged from 43 to 138 μg L−127. Davi et al. have found phenolic compounds (e.g., nonylphenol and alkylphenol) with the concentration of up to tens of μg L−1 in river Po water which is utilized to produce drinking water28. In coast of Thermaikos Gulf, Northern Greece, various nitro- and chlorophenols were monitored from 2003 to 2004, and the maximum concentration of chlorophenols were observed for pentachlorophenol (8.04 μg L−1) followed by 2,4-dichlorophenol (6.11 μg L−1)29. The levels of the mentioned phenolic compounds are in the range of nM to μM when their mass concentration was converted to molar concentration. In context, the current levels of these compounds in natural waters did interfere with the determination of H2O2 on the basis of given 200 nM H2O2 in our experiment, resulting in dramatically underestimating the actual concentration of H2O2 in the aquatic environment.
Relationship between the log k of DOM constituents and their molecular descriptors
We selected 22 compounds having significant interfering effect on H2O2 measurement as target, and tried to build a model between the obtained k and the calculated 14 physicochemical and quantum-chemical descriptors of the corresponding compound by stepwise multiple linear regression method embodied in SPSS 17.0. The resulting four-parameter optimal equation was obtained as following (Eq. 1):
where n, R, SE, q and F represent the number of compounds, the correlation coefficient, the standard error, the leave-one-out cross validation coefficient, and the Fisher test value of the equation, respectively. The robustness and internal predictive power of the model (Eq. 1) were assessed according to q2. Here, q2 (q2 = 0.638) is greater than 0.5, indicating that the model has good robustness and predictive power. The F value of Eq. (1) (F = 15.54) is greater than the critical value at the confidence levels of 95% (F0.05 = 2.965), indicating that the model has statistical significance. Equation (1) has high R and low SE values. Thus, all of the correlative relationships are significant, suggesting that using molecular descriptors to fit the log k of natural DOM constituents is successful. The four parameters Ehomo, qH+, H and V were taken into Eq. (1). It can be seen from Eq. (1) that log k value was positively correlated with Ehomo and qH+, and negatively with H and V. The reasons may be that (1) the compounds with higher Ehomo have the stronger electron donor capacity, and more easily replace scopoletin to react with phenolic radical, (2) the compounds with the larger qH+ have the stronger electron-withdrawing capacity, resulting in readily forming hydrogen bond with water and going into the water phase to react, (3) H is related to the stability of compound. A compound with larger H is more stable, and is hard to take part in reaction, (4) V is associated with the hydrophobicity of a compound. The compound with the larger V is more prone to distribute to the organic phase. Figure 4 displayed the plot of the predicted versus observed log k values. As can be seen, the established model has good linear feature and high predictive power.

Plot of predicted versus observed values of log k.
Conclusions
This study demonstrated that DOM and its constituents such as catechol, gallic acid, syringic acid in waters could significantly suppress the measured amounts of H2O2 by using a peroxidase-mediated depletion of scopoletin fluorescence method, and the interfering extents were parallel for DOM and varied vastly for DOM constituents. The results will provide new insight into the accurate determination of H2O2 in natural waters, whereby implicating its environmental behavior. Based on the 14 physicochemical and quantum-chemical descriptors of 22 DOM constituents, a QSAR model with remarkable stability and strong predictability for evaluating the interfering ratio of DOM constituents to H2O2 measurement was established by multiple linear regression method, which could be used to predict the effect of DOM constituent analogues on H2O2 determination. Nevertheless, further works are needed to better understand the specific effect of DOM and its constituents on H2O2 measurement in situ in the future in light of the complexity of natural waters.
Methods
Chemicals
H2O2 (w/w 30%), phenol (99%), scopoletin (99%) and HRP (type II, EC 1.11. 1.7) were obtained from Sigma-Aldrich (St. Louis, MO). 24 kinds of natural DOM constituents including catechol (99%), resorcinol (99%), hydroquinone (99%), guaiacol (99%), p-methoxyphenol (99%), 3,4-dimethoxyphenol (99%), p-aminophenol (99%), vanillin (99%), syringaldehyde (99%), aniline (99%), o-anisidine (99%), p-anisidine (99%), benzoic acid (> 99%), syringic acid (99%), gallic acid (99%), p-hydroxybenzoic acid (99%), salicylic acid (99%), 3,5-dihydroxybenzoic acid (> 97%), caffeic acid (99%), p-aminobenzoic acid (99%), vanillic acid (99%), 2,5-dihydroxy-1,4-benzoquinone (99%), 2,6-dimethoxy-1,4-benzoquinone (99%) and veratryl alcohol (> 98%) were purchased from J & K Scientific Ltd. (Shanghai, China), and the corresponding molecular weight and structural formula were shown in Table S4. All the reagents were of high performance liquid chromatography (HPLC) grade or higher and used as received unless otherwise stated. Six forms of DOM containing Suwannee Rive Humic Acid (SRHA), Suwannee River Fulvic Acid (SRFA), Suwannee River Natural Organic Matter (SRNOM), Nordic Lake Fulvic Acid (NLFA), Nordic Lake Humic Acid (NLHA) and Pony Lake Fulvic Acid (PLFA) were purchased from International Humic Substance Society (IHSS), and the corresponding C, H, O content were displayed in Table S5.
A 50 mgC L−1 stock solution of each DOM measured by a Shimadzu TOC-L analyzer and the quantification was based on a standard calibration of potassium dihydrogen phthalate solution in the range of 1–20 mg L−1. 1 mM stock solution of each DOM constituent were prepared in ultrapure water obtained from Milli-Q purification system. The working standard solution of 20 μM H2O2 was prepared daily by dilution of 30% H2O2 stock solution calibrated via a titrimetric method using potassium permanganate titration solution just before use30.
Experimental procedures
All experiments were carried out in 10 mL colorimetric tubes. Each reaction solution was prepared in 0.01 M phosphate buffer solution (PBS) (pH 7.0), consisting of H2O2 with an initial concentration of 200 nM and a DOM or its constituent with a series of prescribed levels. It was noticed that the regulated concentration of H2O2 was a typical level in natural water2,31. A 8 mL sample was withdrawn from each tube in quadruplicate to a 5 mL Teflon tubes. Subsequently, an 80 μL of 5 × 10–6 M scopoletin was added to one of the tubes for 5 min, and then a 40 μL volume of the working solution containing 12.5 U mL−1 HRP and 1 mM phenol in 0.01 mM PBS (pH 7.0) was also added. After shaken for 5 min at room temperature, the mixture was analyzed in a 1 cm cuvette using a fluorometric method to determine the apparent concentration of H2O2. The remaining three samples were used to determine blanks. In detail, Blanks were analyzed to quantify the fluorescence response of catalase, the fluorometric reagent, and the sample solution. The catalase blank was determined by decomposing the aqueous H2O2 and organic peroxides in one sample: 50 μL of 2.0 U mL−1 catalase was added to each tube followed by a 5 min reaction time (CAT blank). Similarly, after addition of catalase for 5 min, 40 μL fluorometric reagent was added one of the remaining two tubes, and the reaction was allowed to proceed for an additional 5 min (FL blank). The last one sample acted as the natural blank of reaction solution (NAT blank). After fluorometric analysis of these blanks, the total blank was calculated from the following relationship:
Note, catalase was added to one of the samples prior to the fluorescence reagent to test whether the observed signal was arising from H2O2.
H2O2 analyses
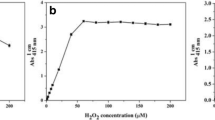
The concentration of H2O2 was measured by a HRP-mediated depletion of scopoletin fluorescence using a FluoroMax-4 fluorescence spectrometer (Horiba, France) with excitation centered at 354 nm and emission centered at 496 nm. The sampling intervals on excitation and emission modes were both set at 1 nm. The fluorometer was periodically calibrated with 100 nM quinine sulfate in 0.05 M sulfuric acid. Prior to calibration, the fluorometer was zeroed with ultrapure water. Decrease in the magnitude of the fluorescence signal was converted to concentrations of reacted H2O2 employing a calibration curve. The 8 mL of H2O2 standard solution was prepared in 0.01 mM PBS (pH 7.0) and in quadruplicate to 5 mL Teflon tubes, and the following analysis procedure is the same as above. Three blanks, i.e., CAT blank, FL blank and NAT blank, were parallelly performed. Linear ranges of standard operating curves for H2O2 concentration were from 10 to 500 nM. The detection limit, defined as 3 times the standard deviation of the blanks, was 10 nM.
Characterization of DOM and its constituents
The 5 mgC L−1 of each DOM and its constituent was prepared by dilution of the corresponding stock solution for characterization of UV–Vis spectra. The process was carried out with 1 cm quartz cuvette in Varian Cary 100 scan UV–Vis spectrophotometer, and the absorbance of each DOM at 254 nm and 365 nm was acquired, respectively.
Calculation of the molecular parameters
The structural and thermodynamic parameters of DOM constituents in water solution were computed by density functional theory (DFT) method and Onsager model in self-consistent field (SCRF) at the DFT/B3LYP/6-31G** level. The resulting structural and thermodynamic parameters including energy of the lowest unoccupied molecular orbital (Elumo), energy of the highest occupied molecular orbital (Ehomo), dipole moment (μ), the most positive net charge of hydrogen atoms (qH+), the most negative atomic net charge of molecule (q-), total energy (TE), molecular average polarizability (α), zero-point vibration energy (ZPE), Gibbs free energy (Gθ), enthalpy (Hθ), entropy (Sθ), corrected heat energy (Eth), molecular volume (V), and molar heat capacity at constant volume (Cvθ) were used to build QSAR models by the SPSS version 17.0 (Table S6). With the propose of obtaining optimum number of variables for the correlation model, stepwise multiple linear regression procedure was employed to establish the dependent equation by adopting structural and thermodynamic parameters calculated at the B3LYP/6-311G** level. The model stability was validated by variance analysis and standard regression coefficient.
Statistical analysis
All data were presented as means ± SD (standard deviation) from triplicated experiments. Intergroup differences were assessed using one-way analysis of variance (ANOVA) based on Tukey's multiple comparisons test by SPSS statistical package (ver. 16.0, SPSS Company, Chicago, USA). Differences between groups were considered as statistically significant if p < 0.05.
References
Clark, C. D. et al. Photochemical production of hydrogen peroxide in size-fractionated southern California coastal waters. Chemosphere 76, 141–146 (2009).
Cooper, W. J. et al. Photochemical formation of H2O2 in natural waters exposed to sunlight. Environ. Sci. Technol. 22, 1156–1160 (1988).
Scully, N. M., McQueen, D. J. & Lean, D. R. S. Hydrogen peroxide formation: The interaction of ultraviolet radiation and dissolved organic carbon in lake waters along a 43–75°N gradient. Limnol. Oceanogr. 41, 540–548 (1996).
Andrews, S. S., Carbon, S. & Zafirious, O. C. Photochemical oxygen consumption in marine waters: A major sink for colored dissolved organic matter. Limnol. Oceanogr. 45, 267–277 (2000).
Cooper, W. J. et al. Factors affecting the distribution of H2O2 in surface waters. In Environmental Chemistry of Lakes and Reservoirs; Baker, L. A., ed. 391–422 (American Chemical Society, Washington, DC, 1994).
Mullaugh, K. M. et al. Long-term temporal variability in hydrogen peroxide concentrations in Wilmington, North Carolina USA rainwater. Environ. Sci. Technol. 45, 9538–9542 (2011).
Scully, N. M. et al. Photochemical formation of hydrogen peroxide in lakes: Effects of dissolved organic carbon and ultraviolet radiation. Can. J Fish. Aquat. Sci. 52, 2675–2681 (1995).
Shaked, Y., Harris, R. & Klein-kedem, N. Hydrogen peroxide photocycling in the gulf of Aqaba. Red Sea. Environ. Sci. Technol. 44, 3238–3244 (2010).
Vermilyea, A. W., Hansard, S. P. & Voelker, B. M. Dark production of hydrogen peroxide in the gulf of Alaska. Limnol. Oceanogr. 55, 580–588 (2010).
Zuo, Y. G. & Holgé, J. Formation of hydrogen peroxide and depletion of oxalic acid in atmospheric water by photolysis of iron (III)-oxalato complexes. Environ. Sci. Technol. 26, 1014–1022 (1992).
Moffett, J. W. & Zika, R. G. Reaction kinetics of hydrogen peroxide with cooper and iron in seawater. Environ. Sci. Technol. 21, 04–810 (1987).
Pettine, M. et al. The effect of organic compounds in the oxidation kinetics of Cr(III) by H2O2. Geochim. Cosmochim. Acta. 72, 5692–5707 (2008).
Sunda, W. G. & Huntsman, S. A. Photoreduction of manganese oxides in seawater. Mar. Chem. 46, 133–152 (1994).
Jones, M. R. & Lee, K. Precipitation of hydrogen peroxide during winter storms and summer typhoons. Sci. Total Environ. 733, 139377 (2020).
Abele, D. et al. Exposure to elevated temperatures and hydrogen peroxide elicits oxidative stress and antioxidant response in the Antarctic intertidal limpet Nacella concinna. Comp. Biochem. Physiol. Part B 120, 425–435 (1998).
da Rosa, C. E., Bianchini, A. & Monserrat, J. M. Antioxidant responses of Laeonereis acuta (Polychaeta) after exposure to hydrogen peroxide. Braz. J Med. Boil. Res. 41, 117–121 (2008).
Wong, G. T. F., Dunstan, W. M. & Kim, D. B. The decomposition of hydrogen peroxide by marine phytoplankton. Oceanol. Acta 26, 191–198 (2003).
Burns, J. M. et al. Methods for reactive oxygen species (ROS) detection in aqueous environments. Aquat. Sci. 74, 683–734 (2012).
Miller, G. W. et al. Hydrogen peroxide method intercomparision study in seawater. Mar. Chem. 97, 4–13 (2005).
Kieber, R. J. & Helz, G. R. Two-method verification of hydrogen peroxide determination in natural waters. Anal. Chem. 58, 2312–2315 (1986).
Miller, W. L. & Kester, D. R. Hydrogen peroxide measurement in seawater by (p-hydroxyphenyl)acetic acid dimerization. Anal. Chem. 60, 2711–2715 (1988).
Garcia, P. E., Queimalinos, C. & Dieguez, M. C. Natural levels and photo-production rates of hydrogen peroxide (H2O2) in Andean Patagonian aquatic systems: Influence of the dissolved organic matter pool. Chemosphere 217, 550–557 (2019).
Stevenson, F. J. Humus Chemistry: Genesis, Composition Reaction (Wiley, New York, 1994).
Park, J. W. et al. Effect of humic constituents on the transformation of chlorinated phenols and anilines in the presence of oxidoreductive enzymes or birnessite. Environ. Sci. Technol. 33, 2028–2034 (1999).
Mostofa, K. M. G. et al. Dissolved organic matter in natural waters. In Photobiogeochemistry of organic matter (eds Mostofa, K. M. G. et al.) 1–137 (Springer, New York, 2013).
Yang, Y. et al. Influence of natural organic matter on horseradish peroxidase-mediated removal of 17α-ethinylestradiol: Role of molecular weight. J Hazard. Mater. 356, 9–16 (2018).
Montero, L. et al. Determination of phenols in lake and ground water samples by stir bar sorptive extraction-thermal desorption-gas chromatography-mass spectrometry. J Chromatogr. A 1071, 163–169 (2005).
Davi, M. L. & Gnudi, F. Phenolic compounds in surface water. Water Res. 33, 3213–3219 (1999).
Dimou, A. D. et al. Determination of phenolic compounds in the marine environment of Thermaikos Gulf, Northern Greece. Int. J Environ. Anal. Chem. 86, 119–130 (2006).
Klassen, N. V., Marchington, D. & McGowan, H. C. E. H2O2 Determination by the I3− method and by KMnO4 titration. Anal. Chem. 66, 2921–2925 (1994).
Clark, C. D. et al. Hydrogen peroxide production in marine bathing waters: Implications for fecal indicator bacteria mortality. Mar. Pollut. Bull. 56, 397–401 (2008).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (21577059).
Author information
Authors and Affiliations
Contributions
S.G. conceived this project and support the research. Y.Z. designed, and accomplished the experiment. J.P., Y.Z., J.L., X.W., M.W., Z.G., and S.G. analyzed the data and finished the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, J., Zhang, Y., Li, J. et al. Interference of dissolved organic matter and its constituents on the accurate determination of hydrogen peroxide in water. Sci Rep 11, 22613 (2021). https://doi.org/10.1038/s41598-021-01016-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-01016-9
- Springer Nature Limited