
Abstract
The nuclear fertility restorer gene Rf5 in HA-R9, originating from the wild sunflower species Helianthus annuus, is able to restore the widely used PET1 cytoplasmic male sterility in sunflowers. Previous mapping placed Rf5 at an interval of 5.8 cM on sunflower chromosome 13, distal to a rust resistance gene R11 at a 1.6 cM genetic distance in an SSR map. In the present study, publicly available SNP markers were further mapped around Rf5 and R11 using 192 F2 individuals, reducing the Rf5 interval from 5.8 to 0.8 cM. Additional SNP markers were developed in the target region of the two genes from the whole-genome resequencing of HA-R9, a donor line carrying Rf5 and R11. Fine mapping using 3517 F3 individuals placed Rf5 at a 0.00071 cM interval and the gene co-segregated with SNP marker S13_216392091. Similarly, fine mapping performed using 8795 F3 individuals mapped R11 at an interval of 0.00210 cM, co-segregating with two SNP markers, S13_225290789 and C13_181790141. Sequence analysis identified Rf5 as a pentatricopeptide repeat-encoding gene. The high-density map and diagnostic SNP markers developed in this study will accelerate the use of Rf5 and R11 in sunflower breeding.
Similar content being viewed by others

Introduction
Cytoplasmic male sterility (CMS) is a phenomenon that destroys a plant’s ability to develop viable pollen, and this phenomenon is commonly seen in higher plants1. CMS is maternally transmitted, and its determinants are generated through rearrangements of the mitochondrial genome, which generally lack sequence homology across taxa, suggesting multiple origins2. Nuclear fertility restorer genes (Rf) can suppress the expression of mitochondrial CMS genes to restore the production of viable pollen. The CMS and nuclear Rf gene system is of considerable value for commercial hybrid seed production in crops, particularly in maize, rice, cotton, sunflower, and numerous vegetables3.
In sunflowers, more than 70 CMS cytoplasms have been described4. However, the global commercial hybrid sunflower seed production industry has been largely relying on a single CMS, PET1, identified from wild Helianthus petiolaris subsp. petiolaris Nutt. and its corresponding fertility restoration gene Rf1 from sunflower line T660006-2-1 for over 50 years since their first reports5,6,7. Talukder et al. (2019) investigated 159 male fertility restorer lines widely used in sunflower breeding programs and found that 130 lines (83%) retain the Rf1 gene8. The sole use of the CMS-PET1/Rf1 system in the global sunflower industry is potentially risky due to genetic vulnerability of sunflower hybrids. Further characterization of currently identified CMS/Rf gene systems would shed light on molecular mechanisms of interaction between cytoplasmic and nuclear genes, which could enable better use of currently identified, yet widely used, CMS/Rf gene systems and minimize the risk associated with genetic vulnerability9.
Seven Rf genes (Rf1, Rf3–Rf7, and Msc1) have been identified and mapped to different chromosomes equating to linkage groups (LGs) in the sunflower genome so far. RHA 266 derived Rf1 was first mapped to LG6 on a restriction fragment length polymorphism (RFLP) map10. RHA 271 derived Rf1 was separately mapped to LG13 and LG2 on RFLP-based maps by different research groups11,12. Later, Rf1 from RHA 325 was mapped on combined randomly amplified polymorphic DNA (RAPD) and amplified fragment length polymorphism (AFLP) maps without determined LGs13,14. There was discrepancy in naming of sunflower linkage groups before the sunflower public simple sequence repeat (SSR) map was published15. The most recent study placed Rf1 derived from RHA 439 to LG13 on an SSR and target region amplification polymorphism (TRAP)-based map16. Talukder et al. (2019) reported that 24 significant single nucleotide polymorphism (SNP) markers from LG13 were associated with Rf1 in a genome-wide association study8. Msc1 was mapped to LG12 on a RFLP map17. Rf3 from RHA 340 and RHA 280, respectively, were both mapped to LG7 on the SSR maps from two different investigations18,19. Both Rf4 and Rf6 from wild sunflower H. maximiliani and an amphiploid of H. angustifolius/P 21, respectively, were mapped to LG3 in two sunflower SSR maps20,21. Recently, Rf5 and Rf7, from wild H. annuus PI 613748 and RHA 428, were mapped to a location on LG13 close to Rf1 on SSR- and SSR/SNP-based maps, respectively8,22.
Because of its high value in commercial hybrid seed production, the genetic and molecular mechanisms underlying cytonuclear incompatibility have been extensively studied and well characterized in many crop species2. The first nuclear fertility restorer gene, Rf2, was cloned in maize and encodes a mitochondrial aldehyde dehydrogenase to restore fertility of CMS-T cytoplasm23,24. Since then, several Rf genes have been cloned in different plant species, including Rf-PPR592 in petunia25, Rfo (Rfk1) in radish26,27,28, Rf1a, Rf1b, Rf2, Rf4, Rf5, Rf17, and Rf98 in rice29,30,31,32,33,34,35,36,37, Rf1 in sorghum38, and Rf1 (bvORF20) in sugar beet39. Except for a few, all cloned Rf genes encode proteins containing pentatricopeptide repeat (PPR) motifs40,41,42. The PPR motifs are characterized by tandem arrays of 2–27 repeats each with degenerate 35-amino-acid sequences43,44. Most PPR proteins in plants target mitochondria or plastids for RNA processing41.
Rust incited by biotrophic fungus Puccinia helianthi Schw. is one of the most serious diseases of sunflower and is causing substantial yield and quality losses in many sunflower production countries of the world45. Resistance to rust in sunflower is often governed by single dominant genes. Currently, twelve rust resistance genes (R genes) have been reported and mapped on the sunflower genome, located on chromosomes 2 (R5), 8 (R1 and R15), 11 (R12 and R14), 13 (Radv, R4, R11, R13a, R13b, and R16), and 14 (R2)22,46,47,48,49,50,51,52,53,54,55. However, the rapid evolution of novel P. helianthi races has rendered many R genes ineffective, and only seven of 12 R genes (R11, R12, R13a, R13b, and R14–R16) remain effectively resistant to all P. helianthi races identified in North America so far51. Among six rust R genes mapped to chromosome 13, R11 is linked to a male fertility restorer gene, Rf5, both of which were transferred from wild H. annuus into cultivated sunflower22,56.
Previous research mapped Rf5 and R11 to a 7.1 cM interval, resulting in a large gap between the genes and markers on the SSR map22. The objective of this research was to fine-map Rf5 and R11 through the identification of additional genetic recombinants close to the genes from a large population and to identify genomic fragments carrying Rf5 using a sequencing-based chromosome walking guided by the two sunflower reference genome assemblies HA412-HO and XRQ57. The diagnostic markers developed in this study that are closely linked to or within R11 will facilitate sunflower rust-resistance breeding. In addition, fine mapping of Rf5 and R11 to a small genomic interval containing few candidate genes would lay the foundation for cloning the genes in the future.
Results
Saturation mapping of Rf5 and R 11 region
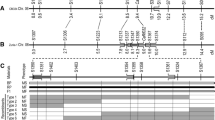
The previous SSR map placed Rf5 and R11 to a region of 7.1 cM, with R11 being 1.6 cM proximal to Rf522 (Fig. 1a). To saturate Rf5 and R11 regions, a total of 45 SFW-SNPs most likely to be around both gene loci on sunflower chromosome 13 were selected and converted into the PCR-based length polymorphism markers. The selected 45 SNP markers were screened between two parents, HA 89 and HA-R9, for polymorphism. Nine SFW-SNP markers, SFW01515, SFW01741, SFW02101, SFW03371, SFW04100, SFW04482, SFW04577, SFW05176, and SFW07542, were polymorphic with codominant nature. They were further genotyped in the F2 population of 192 individuals. Eight SFW-SNP markers were mapped to the Rf5 interval between ORS995 and ORS728, reducing the gene interval from 5.8 to 0.8 cM, while no SFW-SNP was mapped to the R11 interval between ORS728 and ORS45 (Fig. 1b). The genetic distance between Rf5 and R11 was comparable to that of the two genes in the previous SSR map, and three SFW-SNP markers, SFW01515, SFW04100 and SFW04577, were co-segregating with Rf5 and were 1.3 cM distal to R1122 (Fig. 1a,b).

Genetic maps of Rf5 and R11 on sunflower chromosome 13. (a) Rf5 and R11 basic map22; (b) Rf5 and R11 saturation map; and (c) Rf5 and R11 fine maps. *SSR markers.
Fine mapping of Rf5 and R 11 using SNP markers from whole-genome resequencing
Recombinant screens from a large population
To increase map resolution, a large population was screened to detect recombinants for both Rf5 and R11. Rf5 flanking markers, SNP SFW03371 and SSR ORS728, were used to screen 3517 F3 individuals selected from the previously characterized F2:3 families heterozygous for Rf5. A total of 87 recombinants were identified and were grown in the greenhouse for seeds. Among the 87 recombinants, 24 plants could not develop pollen and were considered sterile. Two fertile plants did not have enough seeds and were later excluded from fertility testing. The remaining 61 fertile recombinant families were grown in the field (35 seeds for each family) to evaluate their genotypes as homozygous or heterozygous, of which 37 were heterozygous fertile, and 24 were homozygous fertile.
Similarly, R11 flanking markers, SSR markers ORS728 and ORS45, were used to screen 8795 F3 individuals selected from the previously characterized F2:3 families heterozygous for R11 (Fig. 1b). A total of 112 recombinants were identified, and their advanced generation (20 seedlings for each family) was inoculated with P. helianthi race 336 for rust resistance testing. Among 112 recombinant families tested, 29 were homozygous susceptible, 18 homozygous resistant, and 65 segregating.
New SNP marker development and fine mapping
To further refine the positions of Rf5 and R11 in the target region, HA-R9 was sequenced at 40 × genome coverage to identify additional SNP markers within the region. The variants, including single nucleotide polymorphisms (SNPs) and insertion-deletions (InDels), were called in the target region of Rf5 from the two sunflower reference genome assemblies, spanning a 58.2 kb region (216,334,932–216,393,092 bp) on chromosome 13 in the HA412-HO genome and a 60.6 kp region (175,222,724–175,283,334 bp) in the XRQ genome, respectively. A total of 579 variants (536 SNPs and 43 InDels) from HA412-HO and 803 variants (752 SNPs and 51 InDels) from XRQ were identified, respectively. Eighty-four WGS-SNPs (31 from HA412-HO and 53 from XRQ) were selected from the two target regions and screened between the parents, HA 89 and HA-R9, with nine SNPs showing polymorphism. A total of 15 SNP markers (9 WGS-SNPs and 6 SFW-SNPs) were used to genotype 85 recombinants of Rf5 identified from 3517 F3 individuals. Linkage mapping placed Rf5 on a 0.00071 cM interval on chromosome 13, co-segregating with SNP marker S13_216392091 (Fig. 1c). Most of the WGS-SNP markers were physically positioned in accordance with their genetic positions in the XRQ genome assembly, but genetic and physical positions in the HA412-HO assemblies were reversed in order (Table 1). The flanking markers, S13_216392091 and C13_175253964, delineated Rf5 to within 35.6 and 30.6 kb regions in the HA412-HO and XRQ assemblies, respectively (Table 1).
In the saturation map, the flanking SSR markers, ORS728 and ORS45, delimited R11 to within a 3.4 Mb region (223,364,614–226,744,870 bp) in the HA412-HO assembly with no SNP marker mapped to this interval (Fig. 1b, Table 2). The SNPs/InDels were identified in the R11 target region by aligning HA-R9 sequence to the two reference genomes, spanning a 76.4 kb region (225,224,729–225,301,092 bp) in the HA412-HO assembly and a 1.5 Mb region (180,597,345–182,108,040 bp) in the XRQ assembly. A total of 559 SNPs/InDels were identified in the 76.4 kb region of HA412-HO, and 12,359 SNPs/InDels were found in the 1.5 Mb region of XRQ. A total of 34 SNPs (15 from HA412-HO and 19 from XRQ) were selected to screen between the parents, HA 89 and HA-R9. Ten polymorphic SNPs were used to genotype 112 recombinants of R11 identified from 8795 F3 individuals. Fine mapping placed R11 to a 0.00210 cM interval on chromosome 13, and the gene co-segregated with S13_225290789 and C13_181790141 (Fig. 1c). The flanking SNP markers, C13_181790141 and C13_181792157 delineated R11 to an interval of 2416 bp in the XRQ genome and 3197 bp in the HA412-HO genome (Table 2). R11 was approximately 8.9 and 6.5 Mb apart from Rf5 in the HA412-HO and XRQ genome assemblies, respectively (Tables 1 and 2).
Candidate genes for Rf5 and R 11 from the reference genomes
The gene annotation in the 58.2 kb (216,334,932–216,393,092 bp) and 58.8 kb (175,222,724–175,281,566 bp) genomic sequences of HA412-HO and XRQ, respectively, were analyzed on chromosome 13 encompassing the newly identified SNPs closely linked to Rf5 (https://www.heliagene.org/HA412.v1.1.bronze.20141015/; https://www.heliagene.org/HanXRQ-SUNRISE/). Three and two putative genes were found in the target regions of the HA412-HO and XRQ assemblies, respectively (Table 3). Two genes from the HA412-HO assembly, Ha412v1r1_13g048240 (7171 bp in length) and Ha412v1r1_13g048260 (1878 bp), featured pentatricopeptide repeats, a typical Rf gene motif identified from most other crops. One gene, HanXRQChr13g0420371, 40,234 bp in length on the XRQ assembly, also showed a typical tetratricopeptide-like helical domain (Table 3). Using Ha412v1r1_13g048260 sequence (1878 bp) as a query revealed that Ha412v1r1_13g048240 and HanXRQChr13g0420371 share 92 and 100% sequence identity with Ha412v1r1_13g048260, respectively. They were likely to be candidate genes for Rf5 based on functional domains and physical positions. Other genes in the target region included Ha412v1r1_13g048250, 1598 bp in length in HA412-HO, and HanXRQChr13g0420361, 2365 bp in length in XRQ, and both were predicted to encode AMP-dependent synthetase/ligase (Table 3).
For the gene R11, the 60.0 kb (225,284,300–225,344,300 bp) and 70.981 kb (181,774,000–181,844,981) genomic sequences of HA412-HO and XRQ on chromosome 13 were extracted and analyzed, respectively. Two putative genes, Ha412v1r1_13g051750 (2166 bp) and Ha412v1r1_13g051760 (883 bp), were discovered from the HA412-HO assembly, and both were predicted to code for UDP-glucuronosyl/UDP-glucosyltransferase (Table 3). Three putative genes, HanXRQChr13g0422111, HanXRQChr13g0422121, and HanXRQChr13g0422131, were discovered from the XRQ assembly. Both HanXRQChr13g0422111 (19,151 bp) and HanXRQChr13g0422121 (1509 bp) were predicted to code for glucosyltransferase, while HanXRQChr13g0422131 (5201 bp) was predicted to code for a putative NB-ARC protein (Table 3).
Sequence comparison of HA-R9 with the candidate genes
HA-R9 whole-genome resequencing generated a total of 487,190,276 raw reads. After removal of 797,556 reads with adapters (0.16%) and 74,547 reads containing > 10% undetermined bases (0.02%), a total of 486,318,173 (99.82%) paired-end clean reads were used for assembly and gap repair. By using SOAPdenovo258, a total of 8,547,762 contigs were constructed, with most contigs (6,933,581 contigs; 81.12%) ranging between 100 and 500 bp in length. Only 20 (0.0002%) and 545,224 (6.38%) contigs were more than 10 and 1 kb in length, respectively. Additionally, a total of 7,010,438 scaffolds were identified, with most of them (6,032,509 scaffolds; 86.05%) ranging between 100 and 500 bp in length, while 11,749 (0.17%) and 565,585 (8.07%) scaffolds were more than 10 and 1 kb in length, respectively. The small size and similar total numbers of contigs and scaffolds were most likely due to the short reads (350 bp of 150 bp paired-end) resulting from the Illumina HiSeq/MiSeq sequencing platform and the wide distribution of repetitive sequences in the sunflower genome.
A stretch of 58.2 kb genomic sequence between 216,334,932 and 216,393,092 bp of chromosome 13 covering the Rf5 gene was extracted from the reference genome HA412-HO and used as query to search against the HA-R9 assembled contigs and scaffolds. A total of 106 contigs and 393 scaffolds were identified, and 2 contigs (C16577551 and C16613275) and 3 scaffolds (scaffold206293, scaffold545194, and scaffold550505) were selected based on their positions in the target region (Fig. 2). The selected contigs and scaffolds were aligned to the three candidate Rf5 genes, Ha412v1r1_13g048240, Ha412v1r1_13g048260, and HanXRQChr13g0420371, and showed high levels of sequence identity (Table 4). Contig C16613275 shared the highest level (99%) of identity with candidate gene Ha412v1r1_13g048240, followed by scaffold206293 with Ha412v1r1_13g048260 (97%) and HanXRQChr13g0420371 (97%). The aligned sequence between contig/scaffold and candidate gene was usually over 1 kb. Open reading frames (ORFs) were analyzed using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) among the selected contigs and scaffolds, and the longest ORF for each contig/scaffold was further analyzed by repeat and deduced amino acid numbers (Table 5). Not surprisingly, all of them belong to PPR superfamily with a series of degenerate 35-amino-acid repeats with different copy numbers, suggesting their candidacies for the Rf5 gene. The best ORFs from each contig/scaffold were aligned, and high similarity was found among them (Fig. 3).

Illustration of Rf5 gene along sunflower chromosome 13. (a) Extracted XRQ genomic region covering Rf5. Light and dark green bars highlight candidate Rf5 genes HanXRQChr13g0420361 and HanXRQChr13g0420371; (b) Fine map of Rf5 on sunflower chromosome 13 showing the relative positions of Rf5 and its linked markers; (c) Extracted HA412-HO genomic region covering Rf5. Red, orange and yellow bars highlight candidate Rf5 genes Ha412v1r1_13g048240, Ha412v1r1_13g048250 and Ha412v1r1_13g048260; (d) Solid purple and blue bars show the relative positions of HA-R9 contigs and scaffolds, respectively, aligned to the Rf5 targeted region of the HA412-HO reference genome, and the dotted line represents the gap.

Multiple alignment of amino acid sequences deduced from the best ORFs from two contigs and three scaffolds that are aligned to the Rf5 candidate gene Ha412v1r1_13g048260.
Comparative analysis of amino acid sequences of the candidate Rf5 gene, Ha412v1r1_13g048260, with other characterized Rf orthologues from petunia, radish, rice and sorghum was also performed to reveal the sequence similarity along the PPR motifs. Although multiple sequence alignment showed overall low sequence identity, relatively higher sequence similarity was found in the PPR domains (Supplementary Fig. S1). This comparative analysis indicated that the candidate Rf5 gene is phylogenetically distant to other characterized Rf orthologous from different plant species.
A stretch of 70.981 kb (181,774,000–181,844,981 bp) genomic sequence harboring the R11 gene was extracted from XRQ chromosome 13 and used as a query to search against HA-R9 assembled contigs and scaffolds to identify the R11 gene sequence. After analysis of numerous contigs and scaffolds aligned to the query sequence, one contig and seven scaffolds were selected based on their positions in the target sequence (Fig. 4, Table 6). The selected contig and scaffolds were aligned to the four candidate R11 genes, Ha412v1r1_13g051750, HanXRQChr13g0422111, HanXRQChr13g0422121, and HanXRQChr13g0422131. Scaffold607601 could be aligned to both Ha412v1r1_13g051750 and HanXRQChr13g0422111, respectively, while scaffold585166 and scaffold396498 showed high levels of sequence identity with HanXRQChr13g0422111 only (Table 6). Three scaffolds shared 92 to 98% sequence identity with HanXRQChr13g0422121, while one contig (C16653159) and one scaffold (scaffold433233) showed 95 to 96% sequence identity with HanXRQChr13g0422131.

Illustration of R11 gene along sunflower chromosome 13. (a) Extracted HA412-HO genomic region containing R11. Red and orange bars on HA412-HO assembly highlight candidate R11 genes Ha412v1r1_13g051750 and Ha412v1r1_13g051760; (b) Fine map of R11 on sunflower chromosome 13 showing the relative positions of R11 and its linked markers; (c) Extracted XRQ genomic region containing R11. Dark green, light green and brown bars on XRQ assembly highlight candidate R11 genes HanXRQChr13g0422111, HanXRQChr13g0422121, and HanXRQChr13g0422131; and (d) Solid purple and blue bars show the relative positions of HA-R9 contigs and scaffolds, respectively, aligned to the R11 targeted region of the XRQ reference genome, and the dotted line represents the gap.
SNP marker specificity for Rf5 and R 11
The eight SNP markers closely linked to Rf5 in the fine map were used to screen six sunflower lines: HA 89, HA 234, HA-R9, RHA 397, RHA 428, and RHA 464 (Supplementary Table S1). HA 89 was a recurrent parent for creating HA-R9 with Rf5, and HA 234 was a parental line used in mapping Rf7 from the RHA 428 line. RHA 397 carries an unknown Rf gene, while RHA 428 and RHA 464 harbor Rf7 and Rf1 mapped to chromosome 13 close to Rf5, respectively8. Out of eight SNP markers, two exhibited a unique PCR pattern in HA-R9, different from that of the other five lines, and were subsequently used to test a panel of 96 diversified sunflower lines. One dominant SNP marker C13_175260181 was a diagnostic marker for Rf5 (Fig. 5).

Cropped gel image from PCR amplification of SNP marker C13_175260181 diagnostic for Rf5 on a panel of 96 diversified sunflower lines including lines with Rf1 and Rf7 (Supplementary Table S1). Lane 37 is RHA428 with Rf7; lane 46 is HA-R9 carrying Rf5 and R11, and lane 49 is RHA 464 with Rf1. Diagnostic bands were 97 and 102 bp in length including 21/26 bp tail primer, respectively. Full-length gels are presented in Supplementary Figure S2.
The ten SNP markers closely linked to R11 in the fine map were used to screen six sunflower lines: HA 89, HA-R3, HA-R6, HA-R9, RHA 397, and RHA 464. HA-R3, HA-R6, and RHA 397 carry the rust resistance genes, R4, R13a, and R13b, respectively, all mapped to the lower end of chromosome 13, while RHA 464 harbors a rust R gene R12 mapped to chromosome 1148,51,52. Among 10 SNPs tested, three exhibited a unique PCR pattern in HA-R9, different from that of the other five lines, and were subsequently used to test a panel of 96 diversified sunflower lines. The two SNPs, C13_181790141 co-segregating with R11 and C13_181792157 proximal to R11 at 0.00011 cM genetic distance were diagnostic markers for R11 (Figs. 1c, 6).

Cropped gel image from PCR amplification of SNP marker C13_181792157 diagnostic for R11 on a panel of 96 diversified sunflower lines. Lane 23 is RHA 340 with Radv; and lane 46 is HA-R9 carrying Rf5 and R11 (Supplementary Table S1). Bottom diagnostic bands were 115 and 120 bp in length including 21/26 bp tail primer, respectively. Full-length gels are presented in Supplementary Figure S3.
Discussion
Sunflower chromosome 13, particularly its lower end, harbors a number of economically important genes locating within the cluster. This valuable gene cluster was further divided into two sub-clusters51, i.e. sub-cluster I including the rust R genes Radv and R11 and the male fertility restorer genes Rf1, Rf5 and Rf7, and sub-cluster II including the rust R genes R4, R13a, R13b, and R16, and the four downy mildew R genes Pl5, Pl8, Pl21, and Pl348,16,22,47,48,51,55. The two sub-clusters are approximately 23 Mb apart based on evidence from the two linked genes, Rf7 and Pl34, originally from the wild H. annuus species, accession PI 413157. Both genes were mapped to an interval of 5.8 cM genetic distance on chromosome 13 and located in the two sub-clusters, respectively8. The two SNP markers, NSA_001167 closely linked to Rf7 and SFW08875 closely linked to Pl34, located at the positions of 170,812,277 and 193,131,123 bp, respectively, in the XRQ genome, delimited the two genes to a physical interval of 22.3 Mb8. In the current study, sequencing-based chromosome walking combining with fine mapping delineated Rf5 and R11 to regions of 30.6 and 2.1 kb in the XRQ genome within the sub-cluster I, and diagnostic SNP markers for Rf5 and R11 were developed to facilitate marker-assisted breeding. Sequence alignment indicated that scaffold206293 from the HA-R9 sequence assembly shared 97% sequence identity with two candidate genes, Ha4121r1_13g048260 and HanXRQChr13g0420371, which codes a PPR protein, a motif of most cloned Rf genes, providing a starting point for Rf5 gene cloning in the future (Table 4). Its predicted ORF was 1188 bp in length and incomplete, suggesting the first step for future work is to retrieve the surrounding sequences for a complete ORF.
Clustering of Rf genes is common in plant species, having been reported in common bean, rice, and petunia25,42,59. The Rf gene cluster harboring five active genes (Rf1a, Rf1b, Rf4, Rf5 and Rf98) located on rice chromosome 10 shows extreme variation in structure and gene content37. In sunflower, among seven Rf genes reported, three of them, Rf1, Rf5, and Rf7, were mapped to sub-cluster I in the lower end of chromosome 138,16,22. Yue et al. (2010) localized Rf1 at a position 3.7 cM proximal to SSR marker ORS511 on LG 13, equivalent to chromosome 1316. Rf7 was mapped at a location 0.9 cM proximal to ORS511 in chromosome 138, while Rf5 shared a common SSR marker ORS316 with Rf7 in the target region22, suggesting the close genetic relationship of Rf1 and Rf7, as well as Rf5 (Fig. 7b,c,d).

Comparison of the male fertility restoration genes and rust resistance genes mapped to sub-cluster I of chromosome 13 in different studies. (a) the positions of the Radv gene47; (b) the positions of the Rf5 and R11 genes (current study); (c) the position of the Rf1 gene16; and (d) the position of Rf7 gene8.
Three Rf genes, Rf1, Rf5, and Rf7, originated from the different accessions of the sunflower wild species H. annuus collected from Texas (Rf1), Oklahoma (Rf5), and New Mexico (Rf7), respectively8,22,60. Recently, a genome-wide association study identified 24 significant SNP markers associated with Rf1, which are located in a region where Rf5 and Rf7 reside8. Among 24 SNPs associated with Rf1, only five and seven SNPs retained the Rf1 alleles in HA-R9 (Rf5) and RHA 428 (Rf7), respectively, suggesting that they are three different genes within a gene cluster. Owens et al. (2018) reported a candidate gene for Rf1, HanXRQChr13g0419821, which encodes an aldehyde dehydrogenase gene similar to a cloned Rf2 gene reported in maize23,24,61. HanXRQChr13g0419821 is located at the 174,022,089 bp position of the XRQ genome within the region where 24 SNPs associated with Rf1 reside. In the present study, fine mapping positioned Rf5 in an interval of 175,223,522–175,254,164 bp on chromosome 13 in the XRQ genome. Additionally, one of the candidate genes for Rf5, HanXRQChr13g0420371, was found at the 175,253,986 bp position of the XRQ assembly (Table 4), while Rf7 was reported in the interval 170,762,684–172,543,880 bp on chromosome 13 of the XRQ genome8 (Table 7). Taken together, we propose a hypothesis of three genes ordered within sub-cluster I: Rf7 near the 172 Mb position, Rf1 at 174 Mb, and Rf5 at 175 Mb.
Sub-cluster I with three the Rf genes also harbors two rust R genes, Radv and R11 linked to Rf5, which are positioned distal to a common SSR marker ORS316 at the genetic distances of 3.0 and 3.7 cM in the two maps, respectively22,47 (Fig. 7). Radv originated from a sunflower wild species H. argophyllus and encodes specific recognition to rust infection, different from that of R1151. Bachlava et al. (2011) reported that an NBS-LRR-encoding resistance gene candidate (RGC) marker RGC260 most closely linked to Radv was mapped to 0.2 cM distal to the Radv locus47 (Fig. 7a). Alignment of the RGC260 reverse primer sequence to the XRQ genome sequence indicated that RGC260 is located at the position of 178,056,184 bp in the XRQ genome assembly. In the present study, fine mapping delimited R11 to an interval between 181,789,941 and 181,792,357 bp in the XRQ genome, indicating that Radv and R11 are two closely linked, but different genes.
Although the success of Rf gene cloning has been reported in maize, peanut, radish, rice, sorghum, and sugar beet, its cloning from sunflower is precluded due to the large genome size and high proportion of repetitive sequences23,25,26,32,35,38,39,62. Sunflower is a diploid species with a genome size of approximately 3.6 Gb and more than 80% repetitive sequences. The availability of sunflower genome sequences of two inbred lines, HA412-HO and XRQ, has enabled the development of high density molecular markers and accelerated fine mapping and map or sequence-based gene cloning57. With reference-guided chromosome walking, we identified three and two candidate genes for Rf5 from the HA412-HO and XRQ assemblies, respectively. Among them, two from the HA412-HO assembly, Ha412v1r1_13g048240 and Ha412v1r1_13g048260, both had PPR motifs typical of Rf genes, and one from XRQ assembly, HanXRQChr13g0420371, showed a typical tetratricopeptide-like helical domain, which shares 100% sequence identity with Ha412v1r1_13g048260, indicating the sequence of Ha412v1r1_13g048260 is highly conserved in sunflower, at least among the sunflower lines we studied.
The majority of cloned Rf genes in plants encode a specific clade of the RNA-binding PPR protein family42,63. Duplicated PPR-containing genes residing within the Rf locus are habitual in plant species. A pair of duplicated PPR-containing genes, Rf-PPR591 and Rf-PPR592, was found to reside in the Rf locus in Petunia, share 93% sequence similarity and are identical in PPR organization, but only differ in the last 12 C-terminal amino acids25. Further functional characterization confirmed Rf-PPR592 was able to restore fertility to CMS plants, but not Rf-PPR591, suggesting Rf-PPR592 as the Rf gene in Petunia25. In the current study, the two candidate genes, Ha412v1r1_13g048240 and Ha412v1r1_13g048260, were located within a narrow 12.1 kb region at positions 216,342,878 and 216,354,988 bp in the HA412-HO assembly, respectively (Table 3). Both genes encode a PPR protein and share 92% sequence similarity, suggesting it is likely one of the two is the candidate Rf5 gene in sunflower.
As HA412-HO does not have the Rf5 gene, the candidate genes in HA412-HO could be rf or pseudo-alleles and fail to interact with cytoplasm for fertility restoration. Thus, it is important to determine the physical location of Rf5 in the HA-R9 genome. The contigs and scaffolds from HA-R9 whole-genome resequencing and assembly were searched with the use of the candidate gene sequences as queries, and contig C16613275 and scaffold206293 were determined to share very high sequence identity (97–99%) with queries. However, the predicted ORF of scaffold206293 is incomplete, and a large portion of it consists of undetermined nucleotides, which is not uncommon of the repetitive sequences in the sunflower genome and the sequencing system. All these targeted contigs and scaffolds feature a typical PPR motif with a series of degenerate 35-amino-acid repeats of different numbers (Table 5). Due to the unavailability of a stable transformation system in sunflower, we were unable to confirm their function to restore male sterility. Alternatively, we developed an EMS mutant population of HA-R9, and sterile male plants, resulting from mutations in the Rf5 locus in the M1 generation, were obtained in the M3 families. Target region sequencing of the Rf5 mutant plants and the Rf5 donor HA-R9 is underway using PacBio long-read sequencing for further functional analysis.
HA-R9 carrying Rf5 has been tested for its male fertility restoration to eight different CMS lines, including PET1, PET2, MAX1, GIG1, ANN2, ANN3, RIGX, and GIG256. The results indicated that Rf5 can only restore PET1 CMS, just as Rf1. Rf5 is approximately 6 Mb apart from R11 with a recombination ratio of 1.3% between the two genes (Fig. 1b). Therefore, Rf5 and R11 could be used as a linkage block in sunflower breeding programs. The introgression of these two genes into new hybrids is important, as Rf5 provides a new Rf gene to PET1 CMS, and R11 provides resistance to all P. helianthi races identified so far in North America. The high-density map and diagnostic SNP markers developed provide the information and tools required to accelerate the transfer of Rf5 and R11 into elite sunflower lines.
Methods
Plant materials
An F2 population with 192 individuals previously used to map Rf5 and R11 with SSR markers was used for saturation mapping in the current study22. In its original cross, the inbred line HA 89 was susceptible to rust, while the wild H. annuus accession PI 613748 was used as a donor of Rf5 and R11. A sunflower germplasm line, HA-R9, characterized as homozygous for both Rf5 and R11, released by USDA and North Dakota State University in 2013, was used as a gene donor for whole-genome resequencing56.
For fine mapping of Rf5 and R11, recombinants were screened from 3517 and 8795 F3 individuals selected from the previously characterized F2:3 families heterozygous for Rf5 or R11, respectively. Each selected heterozygous F3 family was considered a segregating F2 population for Rf5 or R11.
An evaluation panel was assembled consisting of 96 inbred sunflower lines with diverse origins, including 20 and 17 lines known to harbor the different male fertility restoration Rf genes and the rust R genes, respectively (Supplementary Table S1). This panel was used to validate the diagnostic DNA markers linked to Rf5 and R11.
Male fertility evaluation
F2:3 individuals and F2:4 families were visually scored as fertile or sterile based on the presence or absence of pollen. Plants that could develop anthers and shed pollen were considered fertile, while those that could not develop anthers or pollen were considered sterile. Recombinants selected from the fertility restoration segregating F2:3 population were grown in the greenhouse to evaluate male fertility. From 3517 F2:3 individuals, 86 were selected as recombinant between the markers SFW03371 and ORS728; of these recombinants, 23 were male-sterile, and 63 were fertile. The subsequent generation of 61 fertile F2:4 recombinant families (35 seeds each) were grown in a field at Glyndon, MN, in the summer of 2015 to evaluate their homozygosity and heterozygosity. The family was considered homozygous fertile if all the plants in the family were able to develop anthers and shed pollen. Conversely, the family was considered heterozygous fertile if the plants in the family were segregating for male fertility.
Rust resistance evaluation
The recombinants selected from the fine mapping population were evaluated for rust resistance in the greenhouse in 2015. Twenty seeds from each of the selected F2:4 recombinant families, together with the parents HA 89 and HA-R9, were grown in 4 × 9 cell plastic flats filled with Sunshine SB 100B potting mixture (SunGro Horticulture, Bellevue, WA, USA). Regular greenhouse maintenance was performed until seedlings reached the four-leaf stage. The P. helianthi isolate of race 336 was chosen for testing seedlings using an artificial inoculation procedure described by Qi et al. (2011)64. Leaves were inoculated with urediniospores of P. helianthi race 336. Resistance against rust was evaluated 12 to 14 days after inoculation for both infection types (ITs) based on the 0 to 4 scale described by Yang et al. (1986)65 and the percentage of leaf area covered with pustules (severity) described by Friskop et al. (2015)66. Infection types 0, 1, and 2 combined with pustule coverage of 0 to 0.5% were classified as resistant, and ITs 3 and 4 with pustule coverage > 0.5% were considered susceptible.
Saturation mapping, whole-genome resequencing, and SNP marker identification
For saturation mapping, a total of 45 SNP markers potentially mapped around Rf5 and R11 gene loci on chromosome 13 were selected after comparison with a published genetic map67 (hereafter referred to as SFW-SNPs, Supplementary Table S2).
HA-R9 was sequenced on the Illumina HiSeq/MiSeq sequencing platform at Novogene Corp. according to their protocols. Briefly, quality DNA samples were randomly fragmented using Covaris cracker to 350 bp in size for library construction, and later qualified libraries were pooled for sequencing according to effective concentrations and expected data volume. Raw reads resulting from next generation sequencing were trimmed and filtered to remove adapters, reads with > 10% undetermined bases, and reads with more than half of the bases of low quality (Qscore ≤ 5). After filtering, clean reads were separately mapped to the two publicly available sunflower reference genomes, HA412-HO (https://www.heliagene.org/HA412.v1.1.bronze.20141015/) and XRQ (https://www.heliagene.org/HanXRQ-SUNRISE/). All SNPs and InDels were identified using the genome-mapped reads. The SNP markers were named with prefix C13 or S13 followed by a number representing the physical position of each SNP along chromosome 13 of each reference genome assembly (hereafter referred to as WGS-SNPs, Supplementary Table S3a and S3b). The prefix C13 represents the SNP from the XRQ reference genome assembly, while S13 represents the SNP from the HA412-HO reference genome assembly.
Genotyping of PCR-based markers
SSR marker genotyping was performed as described by Qi et al. (2012)49. Genotyping of polymerase chain reaction (PCR)-based SNP markers was conducted as described by Qi et al. (2015)68 and Long et al. (2017)69. For each SNP, two-tailed forward allele-specific primers (AS-primers F1 and F2) and one common reverse primer were designed (Supplementary Table S4). A universal priming-element-adjustable primer (PEA-primer 5′-ATAGCTGG-Sp9-GCAACAGGAACCAGCTATGAC-3′) with an attached fluorescence tag at the 5′ terminus was used in each PCR. The PCR protocol for SNP genotyping was conducted as described by Ma et al. (2017)70. Upon amplification, PCR products were loaded on a 6.5% polyacrylamide gel for visualization using an IR2 4300/4200 DNA analyzer (LI-COR, Lincoln, NE, USA).
Sequence assembly, alignment and candidate gene identification
The clean paired-end reads from HA-R9 whole-genome resequencing were assembled using SOAPdenovo2 and gaps were repaired58. The two genomic regions, 216,334,932–216,393,092 bp and 225,224,729–225,301,092 bp on chromosome 13 from the HA412-HO genome sequence assembly were selected to identify contigs and scaffolds possibly having Rf5 and R11 genes, respectively. The sequences of the selected contigs and scaffolds are presented in Supplementary Table S5. A standalone BLASTN program downloaded from the NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) was used to conduct a BLAST search of the reference sequences of the two genomic regions mentioned above against the assembled HA-R9 contigs and scaffolds, at E-value e-20. Selected contigs and scaffolds showing sequence similarity were again aligned with candidate genes identified in the reference assemblies using the BLASTN suite (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Open reading frames (ORF) were identified among the selected contigs and scaffolds using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). Multiple sequence alignments of deduced amino acid sequences of the Rf5 candidate gene Ha412v1r1_13g048260 with the contigs and scaffolds from HA-R9 and the characterized Rf orthologues from different plant species were performed using MultAlin version 5.4.1 (http://multalin.toulouse.inra.fr/multalin/).
Ethical standards
The experiments were performed in compliance with the current laws of the USA.
References
Eckardt, N. A. Cytoplasmic male sterility and fertility restoration. Plant Cell 18, 515–517. https://doi.org/10.1105/tpc.106.041830 (2006).
Hanson, M. R. & Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16, S154–S169. https://doi.org/10.1105/tpc.015966 (2004).
Bohra, A., Jha, U. C., Adhimoolam, P., Bisht, D. & Singh, N. P. Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 35, 967–993. https://doi.org/10.1007/s00299-016-1949-3 (2016).
Serieys, H. Identification, study, utilization in breeding programs of new CMS sources in the FAO Subnetwork. in Proceedings of the Sunflower Subnetwork Progress Report Rome, 47–53 (2005).
Leclercq, P. Une stérilité mâle cytoplasmique chez le tournesol. Ann. Amélior Plantes 19, 99–106 (1969).
Kinman, M. L. New development in the USDA and state experiment station sunflower breeding programs. in Proceedings of the 4th International Sunflower Conference, Memphis, USA, June 23–25, 1970. (Paris: International Sunflower Association), 181–183 (1970).
Dimitrijevic, A. & Horn, R. Sunflower hybrid breeding: From markers to genomic selection. Front. Plant Sci. 8, 2238. https://doi.org/10.3389/fpls.2017.02238 (2018).
Talukder, Z., Ma, G., Hulke, B., Jan, C. C. & Qi, L. Linkage mapping and genome-wide association studies of the Rf gene cluster in sunflower (Helianthusannuus L.) and their distribution in world sunflower collections. Front. Genet. 10, 216. https://doi.org/10.3389/fgene.2019.00216 (2019).
Ma, G. et al. Molecular dissection of resistance gene cluster and candidate gene identification of Pl17 and Pl19 in sunflower by whole-genome resequencing. Sci. Rep. 9, 1–10. https://doi.org/10.1038/s41598-019-50394-8 (2019).
Gentzbittel, L., Vear, F., Zhang, Y. X. & Berville, A. Development of a consensus linkage RFLP map of cultivated sunflower (Helianthusannuus L.). Theor. Appl. Genet. 90, 1079–1086. https://doi.org/10.1007/BF00222925 (1995).
Berry, S. T., et al. Presentation of the Advanta sunflower RFLP linkage map for public research. in Proceedings of the 19th Sunflower Research Forum, Fargo, ND, USA, January 9–10, 1997 (Mandan: National Sunflower Association), 113–118. https://www.sunflowernsa.com/uploads/research/607/1997_berry_advanta.pdf (1997).
Jan, C. C., Vick, B. A., Miller, J. F., Kahler, A. L. & Butler, E. T. III. Construction of an RFLP linkage map for cultivated sunflower. Theor. Appl. Genet. 96, 15–22. https://doi.org/10.1007/s001220050703 (1998).
Horn, R., Kusterer, B., Lazarescu, E., Prufe, M. & Friedt, W. Molecular mapping of the Rf1 gene restoring pollen fertility in PET1-based F1 hybrids in sunflower (Helianthusannuus L.). Theor. Appl. Genet. 106, 599–606. https://doi.org/10.1105/tpc.015966 (2003).
Kusterer, B., Horn, R. & Friedt, W. Molecular mapping of the fertility restoration locus Rf1 in sunflower and development of diagnostic markers for the restorer gene. Euphytica 143, 35–43. https://doi.org/10.1007/s10681-005-1795-9 (2005).
Tang, S., Yu, J. K., Slabaugh, M. B., Shintani, D. K. & Knapp, S. J. Simple sequence repeat map of the sunflower genome. Theor. Appl. Genet. 105, 1124–1136. https://doi.org/10.1007/s00122-002-0989-y (2002).
Yue, B., Vick, B. A., Cai, X. & Hu, J. Genetic mapping for the Rf1 (fertility restoration) gene in sunflower (Helianthusannuus L.) by SSR and TRAP markers. Plant Breed. 129, 24–28. https://doi.org/10.1111/j.1439-0523.2009.01661.x (2010).
Gentzbittel, L. et al. A composite map of expressed sequences and phenotypic traits of the sunflower (Helianthusannuus L.) genome. Theor. Appl. Genet. 99, 218–234. https://doi.org/10.1007/s001220051228 (1999).
Abratti, G., Bazzalo, M. E., & León, A. Mapping a novel fertility restoration gene in sunflower. in Proceedings of the 17th International Sunflower Conference: Córdoba, Spain, June 8–12, 2008, ed L. Velasco (Paris: International Sunflower Association), 617–621 (2008).
Liu, Z., Mulpuri, S., Feng, J., Vick, B. A. & Jan, C. C. Molecular mapping of the Rf3 fertility restoration gene to facilitate its utilization in breeding confection sunflower. Mol. Breed. 29, 275–284. https://doi.org/10.1007/s11032-011-9563-0 (2012).
Feng, J. & Jan, C. C. Introgression and molecular tagging of Rf4, a new male fertility restoration gene from wild sunflower Helianthus maximiliani L. Theor. Appl. Genet. 117, 241–249. https://doi.org/10.1007/s00122-008-0769-4 (2008).
Liu, Z. et al. Diversifying sunflower germplasm by integration and mapping of a novel male fertility restoration gene. Genetics 193, 727–737. https://doi.org/10.1534/genetics.112.146092/-/DC1 (2013).
Qi, L. L., Seiler, G. J., Hulke, B. S., Vick, B. A. & Gulya, T. J. Genetics and mapping of the R11 gene conferring resistance to recently emerged rust races, tightly linked to male fertility restoration, in sunflower (Helianthusannuus L.). Theor. Appl. Genet. 125, 921–932. https://doi.org/10.1007/s00122-012-1883-x (2012).
Cui, X., Wise, R. P. & Schnable, P. S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 272, 1334–1336. https://doi.org/10.1126/science.272.5266.1334 (1996).
Liu, F., Cui, X., Horner, H. T., Weiner, H. & Schnable, P. S. Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 13, 1063–1078. https://doi.org/10.1105/tpc.13.5.1063 (2001).
Bentolila, S., Alfonso, A. & Hanson, M. A. Pentatricopeptide repeat containing gene restores male sterility to male-sterile plants. Proc. Natl. Acad. Sci. U.S.A. 99, 10887–10892. https://doi.org/10.1073/pnas.102301599 (2002).
Brown, G. G. et al. The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. Plant J. 35, 262–272. https://doi.org/10.1046/j.1365-313X.2003.01799.x (2003).
Desloire, S. et al. Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep. 4, 588–594. https://doi.org/10.1038/sj.embor.embor848 (2003).
Koizuka, N. et al. Genetic characterization of a pentatricopeptide repeat protein gene, orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. Plant J. 34, 407–415. https://doi.org/10.1046/j.1365-313X.2003.01735.x (2003).
Akagi, H. et al. Positional cloning of the rice Rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeting PPR protein. Theor. Appl. Genet. 108, 1449–1457. https://doi.org/10.1007/s00122-004-1591-2 (2004).
Kazama, T. & Toriyama, K. A. pentatricopeptide repeat-containing gene that promotes the processing of aberrant apt6 RNA of cytoplasmic male-sterile rice. FEBS Lett. 544, 99–102. https://doi.org/10.1016/S0014-5793(03)00480-0 (2003).
Komori, T. et al. Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryzasativa L.). Plant J. 37, 315–325. https://doi.org/10.1046/j.1365-313X.2003.01961.x (2004).
Wang, Z. et al. Cytoplasmic male sterility of rice with Boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 18, 676–687. https://doi.org/10.1105/tpc.105.038240 (2006).
Fujii, S. & Toriyama, K. Suppressed expression of retrograde-regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. U.S.A. 106, 9513–9518. https://doi.org/10.1073/pnas.0901860106 (2009).
Itabashi, E., Iwata, N., Fujii, S., Kazama, T. & Toriyama, K. The fertility restorer gene, Rf2, for lead rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J. 65, 359–367. https://doi.org/10.1111/j.1365-313X.2010.04427.x (2011).
Hu, J. et al. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine rich protein GRP162. Plant Cell 24, 109–122. https://doi.org/10.1105/tpc.111.093211 (2012).
Tang, H. et al. The rice restorer Rf4 for wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts. Mol Plant 7, 1497–1500. https://doi.org/10.1093/mp/ssu047 (2014).
Igarashi, K., Kazama, T. & Toriyama, K. A gene encoding pentatricopeptide repeat protein partially restores fertility in RT98-type cytoplasmic male-sterile rice. Plant Cell Physiol. 57, 2187–2193. https://doi.org/10.1093/pcp/pcw135 (2016).
Klein, R. R. et al. Fertility restorer locus Rf1 of sorghum (Sorghumbicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor. Appl. Genet. 111, 994–1012. https://doi.org/10.1007/s00122-005-2011-y (2005).
Matsuhira, H. et al. Unusual and typical features of a novel restorer-of-fertility gene of sugar beet (Betavulgaris L.). Genetics 192, 1347–1358. https://doi.org/10.1534/genetics.112.145409 (2012).
Saha, D., Prasad, A. M. & Srinivasan, R. Pentatricopeptide repeat proteins and their emerging role in plants. Plant Physiol. Biochem. 45, 521–534. https://doi.org/10.1016/j.plaphy.2007.03.026 (2007).
Chen, L. & Liu, Y. G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 65, 579–606. https://doi.org/10.1146/annurev-arplant-050213-040119 (2014).
Melonek, J., Stone, J. D. & Small, I. Evolutionary plasticity of restorer of-fertility-like proteins in rice. Sci. Rep. 6, 35152. https://doi.org/10.1038/srep35152 (2016).
Small, I. D. & Peeters, N. The PPR motif—A TPR-related motif prevalent in plant organellar proteins. Trends Biochem. Sci. 25, 46–47. https://doi.org/10.1016/S0968-0004(99)01520-0 (2000).
Lurin, C. et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16, 2089–2103. https://doi.org/10.1105/tpc.104.022236 (2004).
Ma, G. J., Seiler, G. J., Markell, S. G., Gulya, T. J. & Qi, L. L. Registration of two double rust resistant germplasms, HA-R12 and HA-R13 for confection sunflower. J. Plant Regist. 10, 69–74. https://doi.org/10.3198/jpr2015.05.0033crg (2016).
Yu, J. K. et al. Towards a saturated molecular genetic linkage map for cultivated sunflower. Crop Sci. 43, 367–387. https://doi.org/10.2135/cropsci2003.3670 (2003).
Bachlava, E. et al. Downy mildew (Pl8 and Pl14) and rust (RAdv) resistance genes reside in close proximity to tandemly duplicated clusters of non-TIR-like NBS-LRR-encoding genes on sunflower chromosomes 1 and 13. Theor. Appl. Genet. 122, 1211–1221. https://doi.org/10.1007/s00122-010-1525-0 (2011).
Qi, L. L., Hulke, B. S., Vick, B. A. & Gulya, T. J. Molecular mapping of the rust resistance gene R4 to a large NBS-LRR cluster on linkage group 13 of sunflower. Theor. Appl. Genet. 123, 351–358. https://doi.org/10.1007/s00122-011-1588-6 (2011).
Qi, L. L., Gulya, T. J., Hulke, B. S. & Vick, B. A. Chromosome location, DNA markers and rust resistance of the sunflower gene R5. Mol. Breed. 30, 745–756. https://doi.org/10.1007/s11032-011-9659-6 (2012).
Qi, L. L., Ma, G. J., Long, Y. M., Hulke, B. S. & Markell, S. G. Relocation of a rust resistance gene R2 and its marker-assisted gene pyramiding in confection sunflower (Helianthusannuus L.). Theor. Appl. Genet. 128, 477–488. https://doi.org/10.1007/s00122-014-2446-0 (2015).
Gong, L., Gulya, T. J., Markell, S. G., Hulke, B. S. & Qi, L. L. Genetic mapping of rust resistance genes in confection sunflower line HA-R6 and oilseed line RHA 397. Theor. Appl. Genet. 126, 2039–2049. https://doi.org/10.1007/s00122-013-2116-7 (2013).
Gong, L., Hulke, B. S., Gulya, T. J., Markell, S. G. & Qi, L. L. Molecular tagging of a novel rust resistance gene R12 in sunflower (Helianthusannuus L.). Theor. Appl. Genet. 126, 93–99. https://doi.org/10.1007/s00122-012-1962-z (2013).
Zhang, M., Liu, Z. & Jan, C. C. Molecular mapping of a rust resistance gene R14 in cultivated sunflower line PH3. Mol. Breed. 36, 32. https://doi.org/10.1007/s11032-016-0456-0 (2016).
Ma, G. J., Song, Q. J., Markell, S. G. & Qi, L. L. High-throughput genotyping-by-sequencing facilitates molecular tagging of a novel rust resistance gene, R15, in sunflower (Helianthusannuus L.). Theor. Appl. Genet. 131, 1423–1432. https://doi.org/10.1007/s00122-018-3087-5 (2018).
Liu, Z. et al. Molecular mapping of the downy mildew and rust resistance genes in a sunflower germplasm line TX16R. Mol. Breed. 39, 19. https://doi.org/10.1007/s11032-018-0921-z (2019).
Qi, L. L. & Seiler, G. J. Registration of a male fertility restorer oilseed sunflower germplasm, HA-R9, resistant to sunflower rust. J. Plant Regist. 7, 353–357. https://doi.org/10.3198/jpr2013.04.0016crg (2013).
Badouin, H. et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546, 148–152. https://doi.org/10.1038/nature22380 (2017).
Luo, R. et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18. https://doi.org/10.1186/2047-217X-1-18 (2012).
Jia, M. H., He, S., Vanhouten, W. & Mackenzie, S. Nuclear fertility restorer genes map to the same linkage group in cytoplasmic male-sterile bean. Theor. Appl. Genet. 95, 205–210. https://doi.org/10.1007/s001220050549 (1997).
Korell, M., Mösges, G. & Friedt, W. Construction of a sunflower pedigree map. Helia 15, 7–16 (1992).
Owens, G. L., Baute, G. J., Hubner, A. & Rieseberg, L. H. Genomic sequences and copy number evolution during hybrid crop development in sunflowers. Evol. Appl. 11, 1–12. https://doi.org/10.1111/eva.12603 (2018).
Kazama, T. & Toriyama, K. A fertility restorer gene, Rf4, widely used for hybrid rice breeding encodes a pentatricopeptide repeat protein. Rice 7, 28. https://doi.org/10.1186/s12284-014-0028-z (2014).
Dahan, J. & Mireau, H. The Rf and Rf-like PPR in higher plants, a fast-evolving subclass of PPR genes. RNA Biol. 10, 1469–1476. https://doi.org/10.4161/rna.25568 (2013).
Qi, L. L., Gulya, T. J., Seiler, G. J., Hulke, B. S. & Vick, B. A. Identification of resistance to new virulent races of rust in sunflowers and validation of DNA markers in the gene pool. Phytopathology 101, 241–249. https://doi.org/10.1094/PHYTO-06-10-0162 (2011).
Yang, S. M., Antonelli, E. F., Luciano, A. & Luciani, N. D. Reactions of Argentine and Australian sunflower rust differentials to four North American cultures of Puccinia helianthi from North Dakota. Plant Dis. 70, 883–886. https://doi.org/10.1094/PD-70-883 (1986).
Friskop, A. J. et al. Effect of fungicide and timing of application on management of sunflower rust. Plant Dis. 99, 1210–1215. https://doi.org/10.1094/PDIS-10-14-1036-RE (2015).
Bowers, J. E. et al. Development of a 10,000 locus genetic map of the sunflower genome based on multiple crosses. G3 Genes Genomes Genet. 2, 721–729. https://doi.org/10.1534/g3.112.002659 (2012).
Qi, L. L., Long, Y. M., Jan, C. C., Ma, G. J. & Gulya, T. J. Pl17 is a novel gene independent of known downy mildew resistance genes in the cultivated sunflower (Helianthusannuus L.). Theor. Appl. Genet. 128, 757–767. https://doi.org/10.1007/s00122-015-2470-8 (2015).
Long, Y. M., Chao, W. S., Ma, G. J., Xu, S. S. & Qi, L. L. An innovative SNP genotyping method adapting to multiple platforms and throughputs. Theor. Appl. Genet. 130, 597–607. https://doi.org/10.1007/s00122-016-2838-4 (2017).
Ma, G. J., Markell, S. G., Song, Q. J. & Qi, L. L. Genotyping-by-sequencing targeting of a novel downy mildew resistance gene Pl20 from wild Helianthusargophyllus for sunflower (Helianthusannuus L.). Theor. Appl. Genet. 130, 1519–1529. https://doi.org/10.1007/s00122-017-2906-4 (2017).
Acknowledgements
The authors would like to thank Angelia Hogness for technical assistance. This project was supported by the USDA-ARS CRIS Project No. 3060-2100-043-00D. The mention of trade names or commercial products in this report is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: L.L.Q., G.J.M., Y.M.L. Performed the experiments: G.J.M., Y.M.L., Z.I.T., L.L.Q. Analyzed data: L.L.Q., Q.J.S., G.J.M., Y.M.L., M.S. Wrote the paper: G.J.M., L.L.Q. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, G., Long, Y., Song, Q. et al. Map and sequence-based chromosome walking towards cloning of the male fertility restoration gene Rf5 linked to R11 in sunflower. Sci Rep 11, 777 (2021). https://doi.org/10.1038/s41598-020-80659-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80659-6
- Springer Nature Limited
This article is cited by
-
Whole-genome sequencing enables molecular dissection and candidate gene identification of the rust resistance gene R12 in sunflower (Helianthus annuus L.)
Theoretical and Applied Genetics (2023)
-
Molecular mapping of the Rf9 gene from RCMG 1 for CMS ANN3 derived from wild sunflower (Helianthus annuus L.)
Euphytica (2023)
-
Discovery and mapping of two new rust resistance genes, R17 and R18, in sunflower using genotyping by sequencing
Theoretical and Applied Genetics (2021)