
Abstract
The ATP-binding cassette transporter ABCG2 mediates the efflux of several chemotherapeutic drugs, contributing to the development of multidrug resistance (MDR) in many cancers. The most promising strategy to overcome ABCG2-mediated MDR is the use of specific inhibitors. Despite many efforts, the identification of new potent and specific ABCG2 inhibitors remains urgent. In this study, a structural optimization of indeno[1,2-b]indole was performed and a new generation of 18 compounds was synthesized and tested as ABCG2 inhibitors. Most compounds showed ABCG2 inhibition with IC50 values below 0.5 µM. The ratio between cytotoxicity (IG50) and ABCG2 inhibition potency (IC50) was used to identify the best inhibitors. In addition, it was observed that some indeno[1,2-b]indole derivatives produced complete inhibition, while others only partially inhibited the transport function of ABCG2. All indeno[1,2-b]indole derivatives are not transported by ABCG2, and even the partial inhibitors are able to fully chemosensitize cancer cells overexpressing ABCG2. The high affinity of these indeno[1,2-b]indole derivatives was confirmed by the strong stimulatory effect on ABCG2 ATPase activity. These compounds did not affect the binding of conformation-sensitive antibody 5D3 binding, but stabilized the protein structure, as revealed by the thermostabilization assay. Finally, a docking study showed the indeno[1,2-b]indole derivatives share the same binding site as the substrate estrone-3-sulfate.
Similar content being viewed by others


Introduction
Multidrug resistance (MDR) has been described as a major challenge in cancer therapy1. Although there are several known mechanisms of MDR, the overexpression of ATP-binding cassette (ABC) transporters is considered the leading cause for the development of drug resistance in many cancers2. These overexpressed ABC transporters increase the efflux of chemotherapeutic drugs, decreasing intracellular accumulation to subclinical concentrations3. ABC proteins are described as polyspecific due to their ability to export a wide range of drugs with unrelated chemical structures and cellular targets4. Actually the human genome codes for 48 ABC proteins5. However, the precise number of members contributing to clinical MDR is still under investigation. Three extensively studied ABC transporters have been implicated in the development of MDR: P-glycoprotein (P-gp/ABCB1), multidrug resistance associated protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2)2, 6.
A common strategy to overcome MDR is the use of inhibitors. Potent small molecule inhibitors have become a source of potential drug leads7. Despite the in vitro success of P-gp inhibitors, preclinical and clinical studies failed to improve the chemotherapeutic efficacy8. There are various reasons for failure in the clinic, including the fact that many drugs transported by P-gp are also transported by ABCG2 and other ABC transporters9. Since P-gp and ABCG2 are both overexpressed in several cancers, and despite recent advances, there are very few potent, non-toxic ABCG2 inhibitors available for clinical studies, demonstrating that the development of new ABCG2 inhibitors is an urgent necessity2, 10.
The first inhibitor of ABCG2 described was the fungal toxin fumitremorgin C (FTC)11. Although its selectivity toward ABCG2 is very good, FTC is neurotropic and triggers abnormal excitation of the central nervous system12, 13. Because such neurotoxicity precludes its clinical use, FTC analogues have been synthesized and tested as ABCG2 inhibitors, with the most promising candidate being Ko143. To date, this compound is considered as a reference inhibitor of ABCG2 for in vitro assays. However, the effect of Ko143 is not specific to ABCG2, because at higher concentrations, it also inhibits P-gp- and MRP1-mediated drug transport13, 14.
During the last few years we have identified certain classes of compounds as potent ABCG2 inhibitors, including chromones15, 16, stilbenes17, chalcones18 and indeno[1,2-b]indoles19, 20. Designed initially as casein kinase II (CK2) inhibitors, the indeno[1,2-b]indole derivatives seem to be the most promising. Previously, we showed that indeno[1,2-b]indole derivatives that inhibit CK2 can be successfully converted into potent ABCG2 inhibitors19, 20. In this study, further structural insights on rings A, B and D led us to synthesize new indeno[1,2-b]indole derivatives. In addition, the mechanism of ABCG2 inhibition and the differences between compounds that produce a complete versus partial inhibition of mitoxantrone transport were also studied.
Results
Chemistry
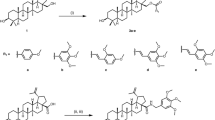
Tetrahydroindeno[1,2-b]indole-9,10-diones 5a–5n and 9-hydroxyindeno[1,2-b]indol-10-ones 6a, 6c–6e were synthesized on the basis of previous reported methods21, 22 as shown in Supplementary Fig. S1.
The synthesis began with the preparation of enaminones 2 by reacting primary amines on commercially available cyclohexane-1,3-diones 1a and 1b or cyclohexane-1,3-dione 1c23,24,25 (Supplementary Fig. S2). The latter was prepared by a consecutive Michael-Claisen cyclization26. Then, condensation of 2 with ninhydrins27 3a, 3b, 3c or 3d (Supplementary Fig. S2) led to the 4b,9b-dihydroxylated derivatives 4. It should be noted that the reaction with 4-hydroxyninhydrin 3b led to a mixture of 1-OH and 4-OH regioisomers not separable under classical conditions. The reaction with 3c led only to the 3-OH regioisomer, while the also expected 2-OH regioisomer was not obtained. Compounds 4 were then deoxygenated using tetraethylthionylamide (TETA) to obtain target compounds 5a–5n.
Generally, at this step, the regioisomers were separated by a chromatography column. Their structures were determined by NOESY experiments, as indicated in the supporting information and summarized in Supplementary Fig. S3.
For 5f, the assigned regiochemistry was confirmed by X-ray crystallography. ORTEP representation of 5f is shown in Supplementary Fig. S4. Structural resolution revealed that the crystal structure of 5f crystallizes in the monoclinic space group P 21/n with 4 formula units in the cell and confirmed results of RMN analysis. The structure was refined with a disorder in the cyclohex-2-enone moiety at a ratio of 35:65.
The O-prenylated derivatives 5j–5n (Supplementary Fig. S5) were obtained from their hydroxylated homologues by Williamson reaction with 3,3-dimethylallylbromide in the presence of NaOH at room temperature. The confirmation of their structures was also based on NOESY correlations and discussed in the supporting information.
In the last step, aromatization of the ketonic compounds 5 into the phenolic compounds 6, was carried out with 10% Pd–C at refluxing diphenyl ether28.
Altogether, 18 ketonic and phenolic indeno[1,2-b]indole derivatives were synthesized, and their various substituents are shown in Fig. 1.

Structure–activity relationship (SAR) study of indeno[1,2-b]indole derivatives: ABCG2 inhibition and cell cytotoxicity
Indeno[1,2-b]indole derivatives have been described as promising potent ABCG2 inhibitors19, 20. Structural optimization was done with compounds initially synthesized and tested on ABCG220. We introduced structural modifications favoring ABCG2 inhibition to obtain the eighteen novel derivatives (Fig. 1).
As shown in Supplementary Fig. S6, all compounds inhibited the transport activity of ABCG2 at 1 and 10 µM concentrations. In addition, many compounds showed a very high potency of inhibition, saturating at a concentration of 1 µM. To perform a precise SAR, a range of concentrations was assayed for each compound to determine and then compare their half maximal inhibition (IC50) values (Fig. 2C and S7). These results are summarized in Table 1.

Inhibition potency and cytotoxicity of compounds with the best therapeutic ratios. (A) Partial inhibitors (red) and (B) complete inhibitors (blue). (C) Representative IC50 curves of the partial and complete inhibitors. (D) Representative flow cytometry histograms of mitoxantrone accumulation in HEK293-ABCG2 cells. Overlay of histograms obtained by compounds at 10 µM mitoxantrone and Ko143. (E) Representative confocal microscopy images of Hoechst 33342 (1 µM) accumulation in HEK293-ABCG2 cells, using the program ImageJ2 (URL: https://imagej.net/Fiji). Effect produced by compounds at 10 µM compared to the reference inhibitor Ko143 at 0.5 µM.
The addition of a methoxy substituent in the ortho position of N5-phenethyl (5a) did not increase affinity (IC50 of 0.25 μM) by comparison with p4h (IC50 of 0.23 μM)19. The introduction of a polar substituent (hydroxyl) at position 1 (5i) increased by 3.5-fold the potency of ABCG2 inhibition comparing with 5a, as determined by IC50 values (5i 0.07 μM vs 5a 0.25 μM). In contrast with p4j (0.21 μM)19, we observed a 2-fold lower affinity for 5h (0.42 μM). Moving the hydroxyl substituent to position 4 was unfavorable, decreasing by 2.5-fold the compound’s affinity (5f 0.18 μM vs 5 g 0.44 μM). A relevant negative effect was observed when the hydroxyl substituent was located in position 3 (5d toward p4k), IC50 of 2.71 µM vs 0.31 µM19, respectively.
We found that the presence of O-prenylated group at position 4 led to a lower affinity, increasing 18-fold the IC50 values (p4h19 0.23 μM vs 5n 4.09 μM). However, the replacement of hydroxyl by O-prenyl at position 1 either decreased the affinity (5i 0.07 vs μM 5l 0.24 μM) or did not produce any effect (5h 0.42 μM vs 5j 0.48 μM). Shifting this substituent to position 4 either reduced the compound’s affinity (5j vs 5k, 0.48–0.92 μM) or did not produce any effect (5l vs 5m).
The presence of a methoxy group in ortho of N5-phenethyl increased the inhibition potency 22-fold (5m vs 5n) and 2.5-fold (5i vs 5f). With a methyl substituent at position 7 of the D-ring, the affinity was increased from 5- to 6-fold (5m vs 5k and 5i vs 5h). This favorable effect was also found in 5l by comparison to 5j (2-fold). In addition, phenolic derivatives with a methoxy substituent in meta of N5-phenethyl (6d) were 14-fold more potent than the ketonic derivative 5d (0.19 vs 2.71 μM).
Selection of the best inhibitors
Considering two important parameters, potency of ABCG2 inhibition and intrinsic cell cytotoxicity, we determined the therapeutic ratio (TR) values (Table 1). The highest TRs were observed with the less cytotoxic compounds 5e and 6a, with TRs higher than 400.
Surprisingly the indeno[1,2-b]indole derivatives showed different patterns related to the maximal inhibition (IMAX). Most derivatives produced a partial inhibition, with maximal inhibition of approximately 60%. To better understand this effect, the compounds were classified as either complete (IMAX ≥ 80% of ABCG2 inhibition) or partial (IMAX ≤ 80% of ABCG2 inhibition) inhibitors, by a threshold at 80% on IMAX values.
Considering that the best parameter to describe a promising inhibitor is the TR, the two best inhibitors of each class were selected. As shown in Fig. 2A, 5e and 6a were selected as the best partial inhibitors, showing TRs of > 470 and 453, respectively (Table 1). In addition, 6c and 6d were selected as the best complete inhibitors of the study, with TRs of 81 and 176, respectively (Fig. 2B and Table 1). These differences between partial and complete ABCG2 inhibitors identified by flow cytometry-based assays are illustrated by the IC50 curves of inhibition (Fig. 2C), and by flow cytometry histograms (Fig. 2D).
To confirm this pattern of inhibition, a different ABCG2 substrate, Hoechst 33342, was used. The intracellular fluorescence of this substrate was recorded by confocal microscopy (Fig. 2E). The Hoechst 33342-mediated efflux by ABCG2 confirmed the existence of two different patterns in the maximal inhibition produced by these derivatives. In addition, this results suggest that these indeno[1,2-b]indole derivatives are not substrate-specific inhibitors.
Indeno[1,2-b]indole derivatives are not transported by ABCG2
The selected indeno[1,2-b]indole derivatives showed a low intrinsic cytotoxicity, an advantageous characteristic when screening new inhibitors. However, the compounds produced a quantifiable cytotoxic effect at higher concentrations, leading us to check for possible cross-resistance. As shown in Fig. 3A, the intrinsic cytotoxicity produced by 6a and 5e was the same in ABCG2-transfected cells or control cells, indicating no apparent cross-resistance. In addition, a preferential cell death was observed in ABCG2-transfected cells treated with 6c and 6d, suggesting a mild collateral sensitivity (CS) effect. CS is characterized by compounds that trigger a preferential cell death in cells overexpressing ABC transporters and constitute a promising strategy to target resistant cancer cells; however, this suggestion needs further investigation. In summary, these results indicate that all the four indeno[1,2-b]indole derivatives are not transported by ABCG2.

Cytotoxicity of inhibitors and washout assay. Cell viability was determined by MTT assay. (A) Cell viability of HEK293-ABCG2 cells and HEK293 (wild-type) control cells upon 72 h treatment with inhibitors at increasing concentrations (0.1–100 µM), as indicated. (B) Washout assay performed on HEK293-ABCG2 cells with the compounds at 10 µM. The white bars represent the classical experiment (30 min of concomitant incubation with compounds and mitoxantrone before analysis), grey and black bars represent pre-treatment with compounds for 30 min followed by washing procedure at 0.5 and 3 h, as described in methods. The data are the mean ± SD of three independent experiments performed in duplicate.
To support the hypothesis of absence of transport, a washing assay was used to investigate if the compounds retain their inhibitory effect after removal from the culture medium. As shown in Fig. 3B, even after 3 h in the absence of compounds in the culture medium, the potency of inhibition was similar. Together, these data confirm that these compounds are not transported by ABCG2.
Selected indeno[1,2-b]indole derivatives sensitize ABCG2 expressing cells to anticancer drugs
To confirm that the indeno[1,2-b]indole derivatives sensitize the cells overexpressing ABCG2 to a chemotherapeutic, a cell viability assay was performed after 72 h of co-treatment using the active metabolite of Irinotecan, SN-38, with inhibitors in two conditions: a concentration that corresponds to the IC50 value and a saturation concentration. As shown in Fig. 4A, all compounds chemosensitized the ABCG2-transfected cells. The fold reversal values are summarized in Supplementary Table S1. In addition, these results also suggest that these compounds are not metabolized during a period of 72 h.

Sensitization of transfected and cancer cells overexpressing ABCG2 to SN-38. Cell viability was determined by MTT assay. (A) Cell viability of HEK293-ABCG2 and HEK293 (wild-type) control cells upon 72 h treatment with SN-38 at increasing concentrations, as indicated, and HEK293-ABCG2 cells upon co-treatment with SN-38 and inhibitors at either IC50 values or 5 µM. (B) Cell viability of H460MX20 and H460 control cells upon 72 h treatment with SN-38 at 10 nM, as indicated, and H460MX20 cells upon co-treatment with SN-38 and inhibitors at either IC50 values or 5 µM. The data are the mean ± SEM of three independent experiments performed in triplicate and compared using the Student's t test (2-sided) for independent samples. *p < 0.05 were considered significant for all tests.
Surprisingly, full chemosensitization was obtained with both partial inhibitors (Fig. 4A and Supplementary Table S1), suggesting that partial inhibition is enough to produce a full reversion of the chemoresistant phenotype. To confirm this hypothesis, a lung cancer cell line was used. The parental cancer cells, H460, and the cell line overexpressing ABCG2, H460MX20, were treated with SN-38 in the absence or presence of inhibitors (Fig. 4B). A similar behavior was observed, confirming that a partial inhibitor can chemosensitize cancer cells overexpressing ABCG2.
Specificity of indeno[1,2-b]indole derivatives toward ABCG2
The four selected indeno[1,2-b]indole derivatives were tested as inhibitors against two other important multidrug ABC transporters, ABCB1 (P-gp) and ABCC1 (MRP1). None of these four compounds inhibited P-gp-mediated transport (Fig. 5A). In contrast, three compounds produced a partial inhibition of MRP1-mediated transport at 10 μM, ranging from 30 to 60% of inhibition (Fig. 5B). These data suggest that indeno[1,2-b]indole derivatives inhibit preferentially ABCG2. Nevertheless, only 6a can be described as a specific ABCG2 inhibitor, while the selectivity toward ABCG2 for the other compounds is linked to the concentration used.

Selectivity of indeno[1,2-b]indole derivatives toward ABCG2. (A, B) Effect of compounds at 1 and 10 µM on ABCB1 (P-gp) for rhodamine 123 transport inhibition and on ABCC1 (MRP1) for calcein-AM transport inhibition. The data are the mean ± SD of three independent experiments performed in duplicate and compared using the Student's t test (2-sided) for independent samples. *p < 0.05 were considered significant for all tests.
Effect of indeno[1,2-b]indole derivatives on the ATPase activity and 5D3 binding to ABCG2
The transport activity of ABC transporters is dependent on ATP binding and hydrolysis. To further investigate the mechanism of inhibition, the ATPase activity was evaluated using High-Five insect cell total membranes overexpressing ABCG2 after incubation with indeno[1,2-b]indole derivatives at increasing concentrations. These compounds showed a strong stimulation effect on ABCG2 ATPase activity, with EC50 values of approximately 0.003 µM with phenolic compounds (6a, 6c and 6d) and 0.17 µM for ketonic compound (5e) (Fig. 6A and Supplementary Table S2). A second approach was used to evaluate possible conformational changes induced by these derivatives using the conformation-sensitive antibody 5D3, which recognizes extracellular loops of the ABCG2 transporter. As shown in Fig. 6B, the indeno[1,2-b]indole derivatives did not promote conformational changes that affect the 5D3 binding. Together, these results suggest that the binding of these compounds in the drug-binding pocket of ABCG2 promotes conformational changes affecting ATPase activity, but these conformational changes do not affect the extracellular loops recognized by the 5D3 antibody.

Studies on ABCG2 ATPase activity, binding of conformational antibody 5D3, biomodulation assay and thermostabilization assay. (A) Effect of compounds at increasing concentrations (0.001–5 µM) on basal ABCG2 ATPase activity. (B) Effect of compounds at 10 µM on the binding of conformational antibody 5D3. (C) Bimodulation assay using partial and complete inhibitors at 10 µM on ABCG2 transport activity using mitoxantrone as a substrate. (D) Thermostabilization assay with partial and complete inhibitors at saturating concentration of 10 µM. The data are the mean ± SD of three independent experiments performed in duplicate.
Bimodulation and thermostabilization assays
As previously described, the compounds were classified as either complete (IMAX ≥ 80%) or partial inhibitors (IMAX ≤ 80%). Partial inhibition at a saturating concentration allows the combination of these inhibitors (6a, 5e) with complete inhibitors (6c, 6d) to check for additive or antagonistic effects. As shown in Fig. 6C, all possible combinations decrease the maximal inhibition, indicating a possible antagonism effect between the indeno[1,2-b]indole derivatives. The decrease in the maximal inhibition was more evident when combining a partial and a complete inhibitor (purple bars). Since a partial inhibitor moderately suppresses the effect of a complete inhibitor, we hypothesize that compounds 6a and 5e may prefer to bind at sites that overlap with the binding sites of the complete inhibitors 6c and 6d.
The thermal stability assay showed that the indeno[1,2-b]indole derivatives trigger ABCG2 conformational changes stabilizing the protein structure (Fig. 6D). The partial inhibitors (5e and 6a) presented a moderate effect compared to complete inhibitors (6c and 6d). This effect can be observed by comparing the IT50 (temperature that reduces 50% of the ATPase activity) values from curves with or without ATP (Fig. 6D). For complete inhibitors, it was observed an overlap of both curves (-ATP and + ATP), suggesting a plausible explanation for the complete inhibition of substrate transport observed by flow cytometry.
Docking analysis of indeno[1,2-b]indole derivatives with ABCG2
Molecular docking was performed in order to gain insight into the structural determinants responsible for the behavior of partial and complete inhibitors with the ABCG2 transporter. The structure used for this study was the PDB 6HCO which shows a molecule of estrone-3-sulfate co-crystallized29. Docking results showed that the indeno[1,2-b]indole derivatives 5e, 6a, 6c and 6d share the same binding site as the estrone-3-sulfate (Fig. 7A), with similar binding energies, ranging from − 12.8 to − 12.3 kcal/mol and also share 6 common amino acids residues (Thr435, Asn436, Phe439, Thr542, Val546 and Met549) with estrone-3-sulfate (Supplementary Table S3). In agreement, recent studies showed that substrates (e.g. mitoxantrone, SN38, estrone-3-sulfate) and inhibitors (e.g. FTC, Ko143, MZ29) bind in the same drug-binding pocket in ABCG230,31,32. This small difference in energy is less than that of a weak hydrogen bond. Therefore, the energy profile of only four inhibitors did not provide a clear picture of their inhibitory profile. For this reason, the inhibitor-transporter interactions for all nine poses were examined carefully to distinguish their specific interactions. Hydrophobic interactions appear to be the most prevalent type of interactions between the central core of indeno[1,2-b]indole derivatives and hydrophobic amino acids residues, in particular the most frequent interactions with Phe439. Residues Phe432, Thr435, Asn436, Thr542, Val546 and Met549 are also involved (Fig. 7B). Although the hydrophobic interactions are clearly relevant, it is important to notice that some hydrogen bonds are present. An additional molecular docking analysis was performed with the PDB 6FFC30. The results confirmed that all four indeno[1,2-b]indole derivatives share the same binding site, showing the same interactions observed with the PDB 6HCO (Supplementary Fig. S8). Therefore, a molecular docking analysis was insufficient to highlight the residues responsible for discriminating between partial and complete inhibitors.

Docking analysis in human ABCG2 (PDB 6HCO). (A) Representative docking poses of the inhibitors overlapping the binding site of estrone-3-sulfate (E3S – black, 6c—red, 6d—blue, 6a—cyan and 5e—yellow), in stick representation. The two monomers are colored differently, orange (chain A) and green (chain B). (B) Interacting residues on the best energy score pose of the inhibitors are represented as stick models. The images were generated using the program PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC (URL: https://pymol.org/2/).
Discussion
Currently, clinical trials using ABC drug transporter inhibitors were conducted exclusively targeting P-glycoprotein. The majority of these clinical trials failed to improve the chemotherapy efficacy, probably due several factors, including the myriad of multidrug resistance mechanisms found in tumors, and the absence of standardized protocols to identify a subset of patients based on tumor expression levels of ABC transporters. Considering the clinical relevance of P-glycoprotein and ABCG2, and the availability of many potent P-glycoprotein inhibitors, there is an urgent need for the identification of potent and specific ABCG2 inhibitors suitable for clinical use along with P-glycoprotein inhibitors.
As previously reported, indeno[1,2-b]indole derivatives that were initially designed as human casein kinase II (CK2) inhibitors can be converted into potent ABCG2 inhibitors19, 20. The novel screened compounds confirmed that derivatives containing N5-phenethyl on the C-ring and hydrophobic groups on the D-ring produce the most powerful ABCG2 inhibitors (Supplementary Fig. S9).
The potency of inhibition, normally reported as the IC50 of inhibition, gives a general idea about the affinity of compounds. Considering the IC50 values alone, the best indeno[1,2-b]indole derivative identified was the compound 5i (0.07 µM). However, this compound was one of the most cytotoxic (IG50 of 20 µM). Since the evidence that some potent and specific ABCG2 inhibitors, as FTC, cannot be used in pre-clinical models because of their neurotoxic effects13, an evaluation of intrinsic cell cytotoxicity has been included in studies describing new ABCG2 inhibitors. Considering that intrinsic cytotoxicity (IG50) and potency of inhibition (IC50) are the two most important parameters for screening new ABCG2 inhibitors, the concept of a therapeutic ratio (TR) was introduced16,17,18,19,20, 29, 33. Therefore, we selected the best inhibitors based exclusively on their TR values (Fig. 2A,B).
Another important consideration concerning ABCG2 inhibitors is their maximal inhibition. Many compounds produce a partial inhibition, such as 6-prenylchrysin34, methoxy stilbenes17, some chalcones18 and chromones15. These compounds reach approximately 70% inhibition. Some of the indeno[1,2-b]indole derivatives screened in this study also showed this effect when mitoxantrone was used as a substrate. The differences were explored in this study; we classified the inhibitors as either partial or complete (Table 1). To our knowledge, this is the first study investigating such characteristics of ABCG2 inhibitors. We selected the two best partial and complete inhibitors based on their TR values to continue characterization of the effects (Fig. 2A,B).
Surprisingly, the partial inhibitors 6a and 5e exhibited TR values (> 450) higher than the complete inhibitors 6c and 6d, with TRs of 176 and 81, respectively. In order to determine if a partial inhibitor would be useful in pre-clinical studies, the effects of both classes of compounds were evaluated in chemosensibilization assays. Despite their partial inhibition, 6a and 5e efficiently chemosensitize cells overexpressing ABCG2 (Fig. 4, Supplementary Table S1). This effect was confirmed in lung cancer cells overexpressing ABCG2, constituting a proof of concept that partial inhibitors can be useful clinically, since the chemoresistance phenotype was fully reversed. Further, the indeno[1,2-b]indole derivatives are not transported by ABCG2, and this is true for both partial and complete inhibitors (Fig. 3A,B). Additionally, the indeno[1,2-b]indole derivatives are not substrate-specific inhibitors, since they inhibit mitoxantrone (Fig. 2D and Supplementary Fig. S6 and Table 1), Hoechst 33342 (Fig. 2E) and SN-38 (Fig. 4A,B, Supplementary Table S1) efflux-mediated by ABCG2.
ABCG2 inhibitors are also classified as either selective toward ABCG2 or dual inhibitors, which include compounds that inhibit more than one ABC transporter. Despite the selectivity of the indeno[1,2-b]indole derivatives toward ABCG2 in comparison to P-gp, only compound 6a was completely selective to ABCG2, whereas 5e, 6c and 6d partially inhibited MRP1-mediated transport (Fig. 5B). These data are in agreement with those previously described for indeno[1,2-b]indole derivatives, which also showed a non-selective effect but a higher affinity for ABCG2 than MRP119,20. This dual effect of ABCG2/MRP1 inhibition is not common, because dual inhibition is normally observed for compounds that inhibit ABCG2 and P-gp35. Some of these non-selective inhibitors, such as GF120918, present higher affinity for P-gp than ABCG236. However, other compounds, such as the reference inhibitor of ABCG2, Ko143, show higher affinity for ABCG2 than P-gp14.
The transport of substrates through the cellular membrane mediated by ABC transporters is coupled to the binding and hydrolysis of ATP2. In contrast to the effect of Ko143, which inhibit the ATPase activity of ABCG2 transporter14, the indeno[1,2-b]indole derivatives were found to stimulate ATPase activity (Fig. 6A). Generally, compounds that inhibit the transport of substrates also inhibit the ATPase activity, however, several chemically unrelated ABCG2 inhibitors stimulate the ATPase activity, such as: sildenafil37, curcumin analogs38, alectinib39, stilbenes17, icotinib40, lapatinib41, imatinib42, AST130643, WHI-P15444, telatinib45 and selonsertib46. This effect is even more complex for some compounds, as nilotinib, which produces a dual effect: ATPase stimulation at low concentrations and ATPase inhibition at high concentrations47. In addition, some classical substrates, as mitoxantrone48, produce a mild stimulation of the ATPase activity of ABCG2 (40%), when compared to prazosin (100%). Although some of these compounds that stimulate the ATPase activity were suggested to be transported by ABCG2, further investigations are required to understand why certain ABCG2 modulators stimulate the ATPase activity. A plausible explanation about this complexity probably is related to the recent evidences about the overlap of substrate and inhibitor binding sites on ABCG232.
Considering only the effect on ATPase activity, the affinity of phenolic compounds was approximately tenfold higher than Ko143. The observed stimulation is similar to previous reports indicating that indeno[1,2-b]indole derivatives either stimulated or did not affect the ATPase activity20, and suggest allosteric interactions between the nucleotide-binding domain and the binding site of these new derivatives. ABCG2 inhibitors have also been found to trigger conformational changes that increase the 5D3 antibody binding49. However, the indeno[1,2-b]indole derivatives did not have any effect on 5D3 binding (Fig. 6B), suggesting an absence of allosteric interactions between the extracellular loop ECL3 and their respective binding sites.
Interestingly, combining indeno[1,2-b]indole derivatives produced an antagonistic effect (Fig. 6C), suggesting a partial binding site overlap and competition. To verify this hypothesis, docking experiments were carried out using the cryo-EM structure of ABCG2 (PDB 6HCO co-crystallized with estrone-6-sulfate)29. The analysis showed that the inhibitors share the same binding site and three amino acid residues appear to be important for the inhibition behavior, Phe439 (the most interactive) followed by Val546 and Met549. Hydrophobic interactions are the most prevalent, with the most frequent interaction being π-π stacking between the phenyl ring of the Phe amino acid residue and the polycyclic core of the inhibitors. Asn436 in both ABCG2 monomers is involved in this interaction with the hydrophilic groups from the compounds, such as carbonyl, indolic nitrogen, methoxy and phenolic groups. This amino acid residue was already reported to be involved in interactions with an ABCG2 inhibitor (MZ29) in an ABCG2 cryo-EM structure30, and its mutation resulted in the loss of substrate transport (estrone-3 sulfate)29. The indeno[1,2-b]indole derivatives promoted conformal changes thus stabilizing the protein structure (Fig. 6D). The most pronounced effect was observed with the complete inhibitors 6c and 6d, suggesting a possible relationship with a more closed form of protein and the complete inhibition.
Considering the chemical structure of the indeno[1,2-b]indole derivatives, it is important to note that compound 6d (complete inhibitor) has a methoxy group in the meta position and compound 6a (partial inhibitor) has that group in the ortho position on the phenethyl moiety. Docking results for 6d showed interactions between 3-OMe-phenethyl group and Thr435-A, Phe432-A and Val546-B. In the case of 6a, interactions between 2-OMe-phenethyl group and Thr435-B, Phe432-B and Met549-B were noted. The different behavior of compounds 6a and 6d could also be explained by interactions induced by the 7-methyl group present in 6a but not in 6d or by the 3-hydroxyl group present in 6d but not in 6a. As 6d, multiple interactions are present between the indolic part of 6c (east part of this second complete inhibitor, Fig. 7B) and residues Thr435-A, Val546-B, Phe439-A, Phe432-A and Asn436-A. The result observed with 5e (partial inhibitor) showed that the presence of a ketonic ring D does not interact with any amino acid residues. The presence of aromatic rings A, B and C seemed to be as well important with several interactions (e.g. Phe439-A, Phe439-B, Met549-B, Phe432-A, Asn436-A, Thr435-A and Val546-B).
In summary, we describe in this report new indeno[1,2-b]indole derivatives that are potent ABCG2 inhibitors. These compounds chemosensitize cancer cells overexpressing ABCG2. Their very high TR values suggest that they are promising compounds that could be used in pre-clinical assays. Our findings are similar to those reported for chromones, which showed high TR values during in vitro screening16 and were successfully applied in vivo50. Further structural investigations are engaged to define more precisely the roles of the ketone function in position 10 (east part of partial inhibitors, Fig. 7B) and of the substituent in position 5 (west part of complete inhibitors, Fig. 7B).
Materials and methods
Chemistry
All intermediates and tested compounds used in this study are fully described in the Supporting Information. The chemical reactions were monitored by thin-layer chromatography (TLC) on GF254 plates that were visualized under an ultraviolet lamp (254 nm). Chromatographic separations of the compounds of a mixture were performed on silica gel columns by column chromatography (Kieselgel 300 − 400 mesh). Evaporation of solvent was performed in a vacuum with a rotary evaporator. Melting points were determined on an Electrothermal 9200 capillary apparatus19, 20.
The IR spectra were recorded on a PerkinElmer SPECTRUM TWO FT-IR spectrometer. The 1H and 13C NMR spectra were recorded at 400 MHz on a Bruker DRX 400 spectrometer. Chemical shifts are expressed in ppm (δ) downfield from internal tetramethylsilane (TMS), and coupling constants J are reported in hertz (Hz). The following abbreviations are used: bs, broad singlet; Cquat, quaternary carbons; d, doublet; dd, doubled doublet; dt, doubled triplet; q, quartet; m, multiplet; s, singlet; t, triplet. The mass spectra were performed by direct ionization (EI or CI) on a Thermo Finnigan MAT 95 XL apparatus19, 20.
The purity of the final compounds (greater than 95%) was determined by UHPLC-MS on an Agilent 1290 system using an Agilent 1290 Infinity ZORBAX Eclipse Plus C18 column (2.1 mm × 50 mm, 1.8 μm particle size) with a gradient mobile phase of H2O/CH3CN (90:10, v/v) with 0.1% of formic acid to H2O/CH3CN (10:90, v/v) with 0.1% of formic acid at a flow rate of 0.5 mL min−1, with UV monitoring at the wavelength of 254 nm with a run time of 10 min19, 20.
Cell lines and cultures
NIH3T3, BHK21 and HEK293 wild-type (WT) cells and transfected cells NIH3T3-ABCB1, BHK21-ABCC1 and HEK293-ABCG2 were kindly provided by Dr. Attilio Di Pietro (IBCP, Lyon, France). The human non-small lung cancer H460 wild-type cells (H460 WT) and mitoxantrone-induced H460 cells overexpressing ABCG2 (H460MX20) were provided by Dr. Attilio Di Pietro (IBCP, Lyon, France) and Dr. Susan Bates laboratory (HIH, Bethesda, MD). The human fibroblast HEK293 cell line was transfected with plasmid coding for human ABCG2 (HEK293-ABCG2) as previously described51. The NIH3T3/ABCB1 drug-resistant cell line was transfected with human MDR1/A-G18552 and BHK21 cells were transfected with plasmid coding for human ABCC1 (BHK21-ABCC1)53. All cells were maintained in high glucose Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin supplemented with either 0.75 mg mL−1 G418 (HEK293-ABCG2), 20 nM mitoxantrone (H460MX20), 60 ng mL−1 colchicine (NIH3T3-ABCB1) or 0.1 mg mL−1 methotrexate (BHK21-MRP1) at 37 °C and 5% CO2 under controlled humidity.
Cytotoxicity assays
Cell viability was evaluated with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay54. HEK293 WT and HEK293-ABCG2 cells were seeded into 96-well culture plates at a 1.5 × 104 cells/well density. After overnight incubation, the cells were treated with various concentrations of compounds (0–100 µM) for 72 h at 37 °C under 5% CO2. For sensitization tests (reversion), cells were concomitantly treated for 72 h with compounds (at a concentration that corresponds to IC50 or a saturation concentration) and an increasing concentration of SN-38 (1 nM–20 µM). Sensitization experiments were also carried out on H460 and H460MX20 cells. For these experiments, H460 and H460MX20 cells were seeded into 96-well culture plates at 1.0 × 104 cells/well density. After overnight incubation, the cells were concomitantly treated with compounds (at a concentration that corresponds to IC50 or a saturation concentration) and SN-38 at 10 nM for 72 h at 37 °C under 5% CO2. After the treatment, the culture medium was removed and 100 μL of a 0.5 mg.mL−1 MTT solution was added. Cells were then incubated for 4 h at 37 °C under 5% CO2. The formazan crystals were dissolved with a solution of DMSO/ethanol (1:1) and the absorbance was measured at 570 nm using a Multiscan FC microplate reader (Thermo Scientific). The results were expressed as percentage of viable cells versus control cells (0.1% DMSO, v/v) taken as 100%.
ABCG2 inhibition assay
ABCG2 inhibition assays were carried out on HEK293 WT and HEK293-ABCG2 cells. Cells were seeded at the density of 1 × 105 cells/well in 24 well culture plates and incubated for 48 h at 37 °C under 5% CO2. Cells were treated with compounds and 10 μM of mitoxantrone for 30 min at 37 °C and 5% CO2. After incubation the cells were washed with PBS, trypsinized and resuspended in ice cold PBS. The intracellular drug fluorescence was monitored with a FACS Calibur cytometer (Becton Dickinson) using the FL4 channel, with at least 10,000 events collected. The maximal fluorescence (taken as 100%) was determined with the difference between the mean fluorescence of HEK-ABCG2 cells incubated with 0.5 μM Ko143 (ABCG2 reference inhibitor) or by using the HEK293 WT cell line as a control and cells without inhibitor (mitoxantrone alone). The percent of inhibition was determined as previously described18. For bimodulation assays the cells were concomitantly treated with 10 μM of one or two combined inhibitors under similar conditions as described previously. For washing assays, cells were treated with 10 μM concentrations of compounds for 30 min. Then, the medium with the inhibitor was removed and the cells were washed with PBS and incubated only with culture medium for different periods of time (0.5 and 3 h). After incubation, cells were exposed to 5 µM mitoxantrone for 30 min, and then analyzed by flow cytometry. The percentage of transport inhibition was calculated by the following equation:
where “C” corresponds to the intracellular fluorescence of cells in the presence of the tested compounds and substrate (mitoxantrone to ABCG2, Rhodamine 123 to ABCB1 and calcein to ABCC1), “S” corresponds to the intracellular fluorescence of cells in the presence of substrate alone, and “I” corresponds to the intracellular fluorescence of cells in the presence of the substrate and reference inhibitor (Ko143 to ABCG2, GF120918 to ABCB1 and verapamil to ABCC1).
ABCB1 and ABCC1 inhibition assay
ABCB1 (P-gp) and ABCC1 (MRP1) inhibition assays were carried out on NIH3T3 WT and NIH3T3-ABCB1 or BHK21 WT and BHK21-ABCC1 cell lines. Cells were treated with 5 μM rhodamine 123 or 0.2 μM calcein-AM as substrates, and with or without GF120918 (0.5 μM) or verapamil (30 μM) as reference inhibitors, for ABCB1 and ABCC1, respectively. Compounds were assayed at 1 and 10 µM. The same procedure and calculation to determine the percentage of inhibition were used for ABCG2 inhibition assays. The intracellular fluorescence was monitored with a FACS Calibur cytometer (Becton Dickinson) using the FL1 channel.
Confocal images
Microscopy analysis was carried out on HEK293 WT and HEK293-ABCG2 cells. Cells were seeded at a density of 1 × 105 cells/well in 24-well culture plates containing coverslips for microscopy and incubated for 48 h at 37 °C under 5% CO2. Cells that adhered to the coverslips were treated with compounds and 1 μM of Hoechst 33342 for 30 min at 37 °C and 5% CO2. After incubation, the coverslips were removed from the plate and placed on slides for microscopy. The slides were then read in a confocal microscopy Nikon A1R MP + (NIKON, Tokyo, Japan) using an oil-immersed 40X objective (with the numerical aperture of 1.15). A laser of 405 nm was used for excitation and the fluorescence emission was recorded using a bandpass filter of 425–475 nm. The software Nis Elements 4.20 (NIKON, Tokyo, Japan) was used for visualization of the images (https://www.microscope.healthcare.nikon.com/pt_AMS/products/software/nis-elements/viewer).
5D3 binding
The 5D3 binding assay was carried out on HEK293-ABCG2 cells. Cells were seeded at a density of 5 × 105 cells/well in 24 well culture plates and incubated for 48 h at 37 °C under 5% CO2. After incubation the cells were washed with PBS, trypsinized, resuspended in PBS and washed 1 × with PBS. The cell pellet was resuspended in 100 µL of PBS containing 4 µL of a BSA solution (1 mg/mL). Cells were treated with compounds at 10 μM for 10 min at 37 °C. The 5D3 primary antibody (Purified mouse anti-human CD338, BD Pharmigen—1:100) was added and incubated at 37 °C for 30 min. After incubation, centrifugation was performed at 1000 × g for 3 min. The cell pellet was resuspended in 100 µL of PBS and the secondary antibody conjugated with phycoerythrin (PE Goat anti-mouse IgG, Abcam—1:200) was added and incubated at 37 °C for 30 min. After incubation, centrifugation was performed at 1000 × g for 3 min and the pellet was resuspended in 300 µL of PBS. The fluorescence was monitored with a FACS Calibur cytometer (Becton Dickinson) using the FL2 channel, with at least 10,000 events collected.
ATPase activity
The ATPase assay was carried out as previously described55. High-Five insect cell total membranes overexpressing ABCG2 were used at a concentration of 5 µg protein/tube in a final volume of 100 µL. The membranes were incubated in assay buffer containing 50 mM Tris–HCl, pH 6.8, 150 mM N-methyl-D-glucamine (NMDG)-Cl, 5 mM sodium azide, 1 mM EGTA, 1 mM ouabain, 2 mM DTT, and 10 mM MgCl2, in the presence or absence of sodium orthovanadate (0.3 mM). The protein-buffer mix was treated with the compounds at increasing concentrations (0.001–5 µM) and incubated for 20 min at 37 °C in the presence of ATP (5 mM). The reaction was stopped with the addition of 100 µL of 5% SDS, for color development was added 400 µL of Pi solution (sulfuric acid 36.2 N, water, ammonium molybdate and antimony potassium tartrate) and 200 µL of 1% ascorbic acid. The absorbance was measured after 10 min at 880 nm using a spectrophotometer Ultrospec 3100 pro (Amersham Biosciences).
Thermostabilization assay
The thermal stability assay was performed according to the published56. Total membrane prepared from High Five insect cells overexpressing the ABCG2 at 3 µg protein/tube concentration were incubated with assay buffer composed by 50 mM Tris–HCl, pH 6.8, 150 mM N-methyl-d-glucamine (NMDG)-Cl, 5 mM sodium azide, 1 mM ouabain and 2 mM DTT in the presence or absence of 0.3 mM orthovanadate at final volume of 50 µL. To evaluate the effect of those inhibitors over thermal stability, each sample was prepared with 12.5 mM MgCl2 or 6.25 mM ATP and incubated with a temperature range from 37 to 71 °C for 10 min using a thermocycler C1000 Touch (Bio-Rad, Hercules, CA). After incubation, 10 µL of 25 mM ATP or 50 mM MgCl2 (5 and 10 mM final concentration, respectively) was added and incubated at 37 °C/20 min to allow ATP hydrolysis. The reaction was stopped with the addition of 50 µL of Pi solution containing 1% ammonium molibdate, 2.5 N H2SO4 and 0.014% potassium-antimony tartrate. To evaluate the absorbance, the samples were transferred to a 96 well plate (50 µL/well), added 150 µL of 0.33% sodium ascorbate solution. The absorbance was measured after 15 min incubation at room temperature using the microplate reader Spectramax iD3 (Molecular Devices, San Jose, CA). The sensitive activity to vanadate (Vi) was calculated as the difference between the activity in the absence of Vi minus the activity in the presence of Vi to each temperature.
Computational methods
Molecular docking studies were performed using the ABCG2 structure deposited in the Protein Data Bank under the ID code 6HCO29 and ID code PDB 6FFC30. The inhibitors were structurally optimized by applying the Mopac 2016 program (MOPAC2016, James J. P. Stewart, Stewart Computational Chemistry, Colorado Springs, CO, USA, http://OpenMOPAC.net (2016) by using the PM6 method57. The structures for ABCG2 and inhibitors were prepared for use with AutoDock Vina58 using AutoDock Tools (v.1.5.6)59. First, molecular docking was performed in the entire protein considering several space sizes to search the region for the best fitting based on the empirical binding free energy scoring function calculated by Autodock Vina. After the identification of this region, we performed a search for a space size of 23 × 19 × 19 Å for 6HCO and 23 × 22 × 10 Å for 6FFC centered in the middle of the transporter. A maximum number of 9 binding modes was used. Selected side-chain residues were flexible to rotate, i.e. Asn436, Phe432, Met549, Phe439, Val546, Thr435 and Thr542, from both ABCG2 monomers, and the remaining residues were treated as rigid. Docked models of the inhibitors, binding sites and amino acid interactions were visualized with ABCG2 using PYMOL (Molecular Graphics System, Version 1.3, Schrödinger, LLC)60.
References
Gottesman, M. M., Lavi, O., Hall, M. D. & Gillet, J.-P. Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol. 56, 85–102. https://doi.org/10.1146/annurev-pharmtox-010715-103111 (2016).
Robey, R. W. et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 18, 452–464. https://doi.org/10.1038/s41568-018-0005-8 (2018).
Szakács, G., Váradi, A., Özvegy-Laczka, C. & Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 13, 379–393. https://doi.org/10.1016/j.drudis.2007.12.010 (2008).
Aller, S. G. et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722. https://doi.org/10.1126/science.1168750 (2009).
Fletcher, J. I., Haber, M., Henderson, M. J. & Norris, M. D. ABC transporters in cancer: more than just drug efflux pumps. Nat. Rev. Cancer 10, 147–156. https://doi.org/10.1038/nrc2789 (2010).
Gottesman, M. M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 53, 615–627. https://doi.org/10.1146/annurev.med.53.082901.103929 (2002).
Leonard, G. D., Polgar, O. & Bates, S. E. ABC transporters and inhibitors: new targets, new agents. Curr. Opin. Investig. Drugs 3, 1652–1659 (2002).
Tamaki, A., Ierano, C., Szakacs, G., Robey, R. W. & Bates, S. E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 50, 209–232. https://doi.org/10.1042/bse0500209 (2011).
Robey, R. W., Massey, P., Amiri-Kordestani, L., Bates, E.S. ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med. Chem. 10, 625–633. https://doi.org/10.2174/187152010794473957 (2011).
Szakács, G. et al. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219–234. https://doi.org/10.1038/nrd1984 (2006).
Rabindran, S. K. et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 58, 5850–5858 (1998).
Nishiyama, M. & Kuga, T. Central effects of the neurotropic mycotoxin fumitremorgin A in the rabbit. (I) Effects on the spinal cord. Jpn. J. Pharmacol. 50, 167–173. https://doi.org/10.1254/jjp.50.167 (1989).
Allen, J. D. et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 1, 417–425 (2002).
Weidner, L. D. et al. The inhibitor Ko143 is not specific for ABCG2. J. Pharmacol. Exp. Ther. 354, 384–393. https://doi.org/10.1124/jpet.115.225482 (2015).
Valdameri, G. et al. 6-Halogenochromones bearing tryptamine: one-step access to potent and highly selective inhibitors of breast cancer resistance protein. ChemMedChem 7, 1177–1180. https://doi.org/10.1002/cmdc.201200154 (2012).
Valdameri, G. et al. Substituted chromones as highly-potent nontoxic inhibitors, specific for the breast cancer resistance protein. J. Med. Chem. 55, 966–970. https://doi.org/10.1021/jm201404w (2012).
Valdameri, G. et al. Methoxy stilbenes as potent, specific, untransported, and noncytotoxic inhibitors of breast cancer resistance protein. ACS Chem. Biol. 7, 322–330. https://doi.org/10.1021/cb200435y (2012).
Valdameri, G. et al. Investigation of chalcones as selective inhibitors of the breast cancer resistance protein: critical role of methoxylation in both inhibition potency and cytotoxicity. J. Med. Chem. 55, 3193–3200. https://doi.org/10.1021/jm2016528 (2012).
Gozzi, G. J. et al. Converting potent indeno[1,2-b]indole inhibitors of protein kinase CK2 into selective inhibitors of the breast cancer resistance protein ABCG2. J. Med. Chem. 58, 265–277. https://doi.org/10.1021/jm500943z (2015).
Gozzi, G. J. et al. Phenolic indeno[1,2-b]indoles as ABCG2-selective potent and non-toxic inhibitors stimulating basal ATPase activity. Drug Des. Devel. Ther. 9, 3481–3495. https://doi.org/10.2147/DDDT.S84982 (2015).
Hemmerling, H. J. & Reiss, G. Partially saturated indeno[1,2-b]indole derivatives via deoxygenation of heterocyclic α-hydroxy-N, O-hemiaminals. Synthesis 2009, 985–999. https://doi.org/10.1055/s-0028-1087983 (2009).
Hundsdörfer, C. et al. Indeno[1,2-b]indole derivatives as a novel class of potent human protein kinase CK2 inhibitors. Bioorg. Med. Chem. 20, 2282–2289. https://doi.org/10.1016/j.bmc.2012.02.017 (2012).
Zymalkowski, F. & Kothari, M. Die Synthese einiger Benzylchinoline. Arch. Pharm. 8, 667–675 (1970).
Greenhill, J. V. & Moten, M. A. N-Alkylation of enaminones. Tetrahedron 39, 3405–3407 (1983).
Berry, N. M., Darey, M. C. P. & Harwood, L. M. Direct 4-alkylation of 1,3-cyclohexanediones via dianionic species. Synthesis 1986, 476–480 (1986).
Sharma, D., Shil, A. K., Singh, B. & Das, P. Consecutive Michael-Claisen process for cyclohexane-1,3-dione derivative (CDD) synthesis from unsubstituted and substituted acetone. Synlett 23, 1199–1204. https://doi.org/10.1055/s-0031-1290900(2012) (2012).
Marminon, C. et al. Microwave-assisted oxidation of indan-1-ones into ninhydrins. Tetrahedron Lett. 56, 1840–1842. https://doi.org/10.1016/j.tetlet.2015.02.086 (2015).
Poumaroux, A., Bouaziz, Z., Domard, M. & Fillion, H. Regiospecific hetero Diels–Alder synthesis of pyrido[2,3-b]- and pyrido[3,2-b]carbazole-5,11-diones. Heterocycles 45, 585–596 (1997).
Manolaridis, I. et al. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 563, 426–430. https://doi.org/10.1038/s41586-018-0680-3 (2018).
Jackson, S. M. et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 25, 333–340. https://doi.org/10.1038/s41594-018-0049-1 (2018).
Taylor, N. M. I. et al. Structure of the human multidrug transporter ABCG2. Nature 546, 504–509. https://doi.org/10.1038/nature22345 (2017).
Orlando, B. J. & Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 11, 1–11. https://doi.org/10.1038/s41467-020-16155-2 (2020).
Krapf, M. K., Gallus, J., Spindler, A. & Wiese, M. Synthesis and biological evaluation of quinazoline derivatives—A SAR study of novel inhibitors of ABCG2. Eur. J. Med. Chem. 161, 506–525. https://doi.org/10.1016/j.ejmech.2018.10.026 (2019).
Ahmed-Belkacem, A. et al. Flavonoid structure-activity studies identify 6-prenylchrysin and tectochrysin as potent and specific inhibitors of breast cancer resistance protein ABCG2. Cancer Res. 65, 4852–4860. https://doi.org/10.1158/0008-5472.CAN-04-1817 (2005).
Boumendjel, A. et al. Targeting the multidrug ABCG2 transporter with flavonoidic inhibitors: in vitro optimization and in vivo validation. Curr Med. Chem. 18, 3387–3401. https://doi.org/10.2174/092986711796504736 (2011).
De Bruin, M., Miyake, K., Litman, T., Robey, R. & Bates, S. E. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 146, 117–126. https://doi.org/10.1016/S0304-3835(99)00182-2 (1999).
Shi, Z. et al. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 71, 3029–3041. https://doi.org/10.1158/0008-5472.CAN-10-3820 (2011).
Murakami, M. et al. Synthetic analogs of curcumin modulate the function of multidrug resistance—linked ATP-binding cassette transporter ABCG2. Drug Metab. Dispos. 45, 1166–1177. https://doi.org/10.1124/dmd.117.076000 (2017).
Yang, K. et al. Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. Exp. Mol. Med. 49, e303–e313. https://doi.org/10.1038/emm.2016.168 (2017).
Wang, D. S. et al. Icotinib antagonizes ABCG2-mediated multidrug resistance, but not the pemetrexed resistance mediated by thymidylate synthase and ABCG2. Oncotarget 5, 4529–4542. https://doi.org/10.18632/oncotarget.2102 (2014).
Dai, C. L. et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 68, 7905–7914. https://doi.org/10.1158/0008-5472.CAN-08-0499 (2008).
Brendel, C. et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 21, 1267–1275. https://doi.org/10.1038/sj.leu.2404638 (2007).
Zhang, H. et al. AST1306, a potent EGFR inhibitor, antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Cancer Lett. 350, 61–68. https://doi.org/10.1016/j.canlet.2014.04.008 (2014).
Zhang, H. et al. WHI-P154 enhances the chemotherapeutic effect of anticancer agents in ABCG2-overexpressing cells. Cancer Sci. 105, 1071–1078. https://doi.org/10.1111/cas.12462 (2014).
Sodani, K. et al. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem. Pharmacol. 89, 52–61. https://doi.org/10.1016/j.bcp.2014.02.012 (2014).
Ji, N. et al. Selonsertib ( GS-4997), an ASK1 inhibitor, antagonizes multidrug resistance in ABCB1- and ABCG2-overexpressing cancer cells. Cancer Lett. 440–441, 82–93. https://doi.org/10.1016/j.canlet.2018.10.007 (2019).
Hegedüs, C. et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 158, 1153–1164. https://doi.org/10.1111/j.1476-5381.2009.00383.x (2009).
Özvegy, C. et al. Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem. Biophys. Res. Commun. 285, 111–117. https://doi.org/10.1006/bbrc.2001.5130 (2001).
Telbisz, Á. et al. Antibody binding shift assay for rapid screening of drug interactions with the human ABCG2 multidrug transporter. Eur. J. Pharm. Sci. 45, 101–109. https://doi.org/10.1016/j.ejps.2011.10.021 (2012).
Honorat, M. et al. MBL-II-141, a chromone derivative, enhances irinotecan (CPT-11) anticancer efficiency in ABCG2-positive xenografts. Oncotarget 5, 11957–11970. https://doi.org/10.18632/oncotarget.2566 (2014).
Robey, R. W. et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br. J. Cancer 89, 1971–1978. https://doi.org/10.1038/sj.bjc.6601370 (2003).
Cardarelli, C. O., Aksentijevich, I., Pastan, I. & Gottesman, M. M. Differential effects of P-glycoprotein inhibitors on NIH3T3 cells transfected with wild-type (G185) or mutant (V185) multidrug transporters. Cancer Res. 55, 1086–1091 (1995).
Chang, X. B., Hou, Y. X. & Riordan, J. R. ATPase activity of purified multidrug resistance-associated protein. J. Biol. Chem. 272, 30962–30968. https://doi.org/10.1074/jbc.272.49.30962 (1997).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983).
Ambudkar, S. V. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 292, 504–514. https://doi.org/10.1016/s0076-6879(98)92039-0 (1998).
Lusvarghi, S. & Ambudkar, S. V. ATP-dependent thermostabilization of human P-glycoprotein (ABCB1) is blocked by modulators. Biochem. J. 476, 3737–3750. https://doi.org/10.1042/BCJ20190736 (2019).
Stewart, J. J. P. Optimization of parameters for semi empirical methods V: modification of NDDO approximations and application to 70 elements. J. Mol. Model. 13, 1173–1213. https://doi.org/10.1007/s00894-007-0233-4 (2007).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. https://doi.org/10.1002/jcc.21334 (2009).
Morris, G. M. et al. AutoDock4 and autoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. https://doi.org/10.1002/jcc.21256 (2009).
Schrödinger, LLC. The {PyMOL} Molecular Graphics System, Version~1.8. (2015).
Acknowledgements
The authors acknowledge CENAPD at UNICAMP-SP, Brazil for computational resources. We thank George Leiman in the Laboratory of Cell Biology for editorial assistance. We also thank Attilio Di Pietro from IBCP for having provided the cell lines. This project was supported by: CNPq (grant number 400953/2016-1) and Fundação Araucária (Code 006 – 09/2016). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. DHK, SL and SVA were supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, Center for Cancer Research. B.B., C.M., Z.B., M.B. and M.L.B. thank the Algerian Ministry of Scientific Research and the Embassy of France in Algeria for their PROFAS B+ 2015-2016 programme for B.B. Ph.D.
Author information
Authors and Affiliations
Contributions
G.V. and M.L.B. designed the study. G.V. designed the biological experiments, wrote and edited the manuscript. D.H.K. and S.L. performed the biological experiments. G.P., F.G.M., S.V.A., and V.R.M. designed biochemical experiments and wrote parts of the text. M.B.G. and I.F.Z. designed and performed molecular modeling analysis. N.G., B.B., T.M., C.M. and B.Z. performed the synthesis and characterization of the chemical compounds. S.B. carried out the X-ray crystallography study. N.G., C.M. and B.Z. wrote chemical part of the text. M.B. and J.J. reviewed and edited the manuscript. All the authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kita, D.H., Guragossian, N., Zattoni, I.F. et al. Mechanistic basis of breast cancer resistance protein inhibition by new indeno[1,2-b]indoles. Sci Rep 11, 1788 (2021). https://doi.org/10.1038/s41598-020-79892-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79892-w
- Springer Nature Limited
This article is cited by
-
Advances in the structure, mechanism and targeting of chemoresistance-linked ABC transporters
Nature Reviews Cancer (2023)
-
A curated binary pattern multitarget dataset of focused ATP-binding cassette transporter inhibitors
Scientific Data (2022)