
Abstract
In the present work, culture-based and culture-independent investigations were performed to determine the microbiota structure of the coelomic fluid of Mediterranean sea urchin Paracentrotus lividus individuals collected from two distinct geographical sites neighboring a high-density population bay and a nature reserve, respectively. Next Generation Sequencing analysis of 16S rRNA gene (rDNA) showed that members of the Proteobacteria, Bacteroidetes and Fusobacteria phyla, which have been previously reported to be commonly retrieved from marine invertebrates, dominate the overall population of microorganisms colonizing this liquid tissue, with minority bacterial genera exhibiting remarkable differences among individuals. Our results showed that there is a correlation between microbiota structure and geographical location of the echinoderm collection site, highlighting over-representation of metagenomic functions related to amino acid and bioactive peptides metabolism in specimens inhabiting the nature reserve. Finally, we also described the developmental delay and aberrations exhibited by sea urchin embryos exposed to distinct bacterial isolates, and showed that these defects rely upon hydrophilic compound(s) synthesized by the bacterial strains assayed. Altogether, our findings lay the groundwork to decipher the relationships of bacteria with sea urchins in their aquatic environment, also providing an additional layer of information to understand the biological roles of the coelomic fluid.
Similar content being viewed by others
Introduction
Life on earth began in the oceans. Over millions of years, the organisms that inhabit the seas evolved strategies to handle a changing and sometimes invasive environment. Extending through a dynamic interaction from antagonistic to cooperative elements, marine eukaryotic organisms became hosts for large and complex community of microorganisms playing important roles in nutrition1, response to environmental stimuli2,3, disease-resistance and evolution4, as well as chemical defence against both predators and competitors5,6. In fact, the secondary metabolites produced by the symbionts include compounds showing anti-inflammatory, antitumor, antibiotic and antifungal activities, which are particularly interesting in drug discovery campaigns (reviewed in7).
Studies of bacterial communities associated with body tissues of marine animals have largely focused on sponges, corals, bryozoans and crustaceans8,9,10,11,12. Despite the scientific and economic importance of sea urchins, little attention has been given to their microbial community and the potential role of microorganisms in the host physiology. In particular, some reports focused on the microbial abundance and diversity of the digestive system of distinct species, such as Echinocardium cordatum13, Paracentrotus lividus14, and Lytechinus variegatus15,16, while few other works focused on the general role of microorganisms in nutrients digestion13,17 or in disease progression18,19.
Sea urchins are invertebrate animals distributed throughout the oceans of the world, where they represent central players in many benthic ecosystems. Besides their ecological role and economic importance as fishery products20, sea urchins have been used as model organisms for biological research for nearly two centuries, contributing significantly to our understanding of fertilization, molecular embryology, gene regulation, cell cycle regulation, evolution, population genetics, and toxicology21,22,23,24,25,26. The reason of their extensive use as experimental model likely relies in their phylogenetic position. Sea urchins are indeed basal deuterostomes belonging to Echinodermata, the sister phylum to the chordates27, and therefore they are closely related to humans28.
Similar to the blood of higher metazoans, the main circulatory medium of sea urchins is the coelomic fluid enclosed in the main body cavity. Such an internal fluid contains a heterogeneous mixture of organic molecules and circulating cells that ensures essential functions such as nutrient transport and immune activity29,30,31. The coelomic fluid is in equilibrium with the surrounding seawater environment and in direct contact with internal tissues, whereas the circulating immunocytes respond to pathogen challenge producing antimicrobial molecules32,33,34. Therefore, the coelomic fluid could be considered a rather complex tissue that mediates responses to wounding and microbial infections by undergoing reactions such as opsonization, coagulation, encapsulation and phagocytosis34. In light of this, the coelomic fluid of these echinoderms has been traditionally supposed to be a microorganism-free compartment35,36. Contrarily, recent reports described the identification of microbial communities associated with the coelomic fluid of holothurians, starfish and sea urchins37,38,39,40,41,42,43, suggesting that complex symbiotic associations could exist in such a perivisceral liquid of the echinoderms.
In the present study, we confirm this hypothesis describing the composition of the bacterial microbiota in the coelomic fluid from the Mediterranean sea urchin Paracentrotus lividus.
Results
Structure of the coelomic fluid bacterial microbiota of P. lividus
In order to collect sea urchin P. lividus specimens, two distinct geographical sites, arbitrarily named A and B, were chosen for their proximity to a high-density population bay and to a nature reserve, respectively. Based on the notion that the cultivability of marine microbial communities often ranges below 10% of total bacteria44,45, to obtain a comprehensive understanding of the microbiota structure we performed an analysis based on Next Generation Sequencing of V3-V4 region amplicons of 16S rDNA obtained from metagenomic DNA extracted from coelomic fluid of six P. lividus individuals from site A and four from site B (Supplementary Fig. S1, Table S1). Overall, this approach led to the identification of 227 genera of which 38 and 68 were found exclusively in samples derived from sites A and B, respectively (Supplementary Table S1, Fig. S2). The analysis of alpha-diversity (i.e. the ecological richness within each sample) revealed a variation (p < 0.05) in the number of observed Operational Taxonomic Units (OTUs) and Shannon entropy index (p < 0.01) among the samples collected in A and B sites, with B showing a diversity greater than A (Supplementary Fig. S3). The analysis of beta-diversity highlighted that the microbial community structure within the coelomic fluid of echinoderms collected from site A differs significantly with respect to that of individuals from site B (Supplementary Fig. S1), as revealed by weighed Unifrac and Bray–Curtis distances (p = 0.057 and p = 0.025, respectively).
At the phylum level, a predominance of taxa belonging to Proteobacteria, Bacteroidetes and Fusobacteria was observed in the microbiota of all specimens collected from either A or B sites, accounting for about 90% of abundance per each individual (Supplementary Fig. S1; Table S1). On the contrary, minority phyla (i.e. having a relative abundance lower than 1%) were overtly different among individuals (Supplementary Fig. S1).
Geographical variation of coelomic fluid bacterial microbiota composition
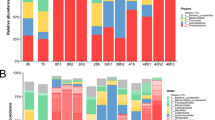
Looking at the average relative abundance, the most abundant genera (i.e. > 1.0%) in B site samples belonged to unclassified genera of Bacteroidetes phylum (31.6%) and of Desulfobulbaceae family (10.1%), and to Propionigenium (30.7%), Photobacterium (7.1%), Psychromonas (5,4%), Vibrio (4.3%) and Arcobacter (1.3%) genera. Using the same criterion, the most abundant bacteria in A site samples belonged to an unclassified phylum (3.3%) and to unclassified genera of Bacteroidetes phylum (12.5%), of Flavobacteriaceae (11.7%), of Desulfobulbaceae (2.2%) and of Thiotrichales incertae sedis families (1.4%), and to Propionigenium (46.3%), Arcobacter (11.1%), Photobacterium and Psychromonas (1.0%) (Fig. 1). It follows that a core in bacterial microbiota structure can be defined by the sum of the majority taxa (i.e. having an average relative abundance higher than 1%), as they collectively account for more than 75% in the relative abundance, irrespective of specimen collection site. In particular, such a core comprises the unclassified genera of Bacteroidetes phylum and Desulfobulbaceae family, and the Propionigenium, Photobacterium, Psychromonas and Arcobacter genera (Fig. 1).

Mean relative abundances of taxa, at genus level, of the coelomic fluid microbiota of P. lividus individuals collected from sites A and B. The corresponding phylum is highlighted by using shades of blue, green, and orange for Proteobacteria, Fusobacteria and Bacteroidetes, respectively. All bacterial genera with relative abundance < 1% are reported together and labeled as “Others”. The underlined taxa represent the microbiota core members.
The LEfSe analysis, revealing the majority bacterial genera that characterize A and B sites based on their relative abundance, highlighted 36 and 5 taxa in A and B sites, respectively (Fig. 2). Interestingly, besides the unclassified genera of Bacteroidetes and Desulfobulbaceae family (in samples from site B), and the unclassified genus of Thiotrichales incertae sedis family (in samples from site A), specific minority genera (i.e. having relative abundance lower than 1.0%) significantly enriched in A and B sites mostly belonged to the Proteobacteria phylum.

Comparative analysis of the most discriminant bacterial taxa in the coelomic microbiota of P. lividus individuals from site A vs site B. Log10 of LDA scores was calculated for the most discriminant bacterial taxa identified by LEfSe. Positive and negative LDA scores indicate the taxa enriched in the coelomic microbiota of P. lividus specimens from A and site B sites, respectively. Only taxa having a p < 0.05 (Wilcoxon rank-sum test) and LDA >|2.0| are shown.
Metabolic functions associated to the coelomic fluid microbiota
The Piphillin tool46 was used to examine the putative functional capabilities of the coelomic fluid microbiota of the P. lividus specimens collected from both the above-mentioned geographical sites, inferring the abundance of functional genes from 16S rDNA counts. Including only KEGG metabolic pathways of bacteria, 7 metagenomic functions were found to be differentially abundant (p < 0.01) between the microbiota of individuals collected from A and B sites. In particular, they were all over-represented in B site specimens, whereas 4 of them (arginine and proline metabolism, beta-alanine metabolism, taurine and hypotaurine metabolism, and cyanoamino acid metabolism) pertained the amino acid metabolism, and 1 (nonribosomal peptide structures) dealt with the synthesis of bioactive peptides (Fig. 3).

Putative functional capabilities differentially represented in the coelomic microbiota of specimens from A and B sites. A total of 7 differentially represented (p < 0.01) metabolic pathways were inferred by Piphillin using the KEGG database80.
Isolation and phylogenetic characterization of bacteria inhabiting the coelomic fluid of P. lividus
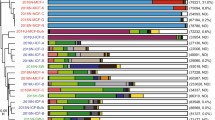
A culture-based investigation of the bacterial populations inhabiting the coelomic fluid of individuals collected from both the mentioned sites revealed an amount of 104–105 bacterial isolates/mL. Among these, a total of 70 colonies were picked and re-streaked on fresh marine agar plates. Bacterial isolates that shared similar colony morphology were grouped together and considered to be redundant. Then, a total of six isolates (namely A1-4 and B1-2 from A and B sites, respectively), each exhibiting representative colony morphology and/or pigmentation, were selected for phylogenetic analysis based on 16S rDNA sequence (Fig. 4). This analysis confirmed that the six isolates represented bacterial strains commonly found in marine organisms, with four isolates (A1, A2, B1 and B2) belonging to Vibrio genus and two isolates (A3 and A4) belonging to Bacillus and Shewanella genera, respectively.

Optimal phylogenetic tree of the six cultivable bacterial strains isolated from coelomic fluid of P. lividus specimens collected in A (A1–4) and B (B1–2) sites. The analysis involved 48 nucleotide sequences. The evolutionary history and distances were obtained using neighbor-joining and the maximum composite likelihood methods, respectively. Distances are in the units of the number of base substitutions per site.
Developmental effects of coelomic fluid bacterial isolates on P. lividus
Since the bacterial strains isolated by culture-dependent analysis belong to genera whose members are capable to influence distinct health and physiological mechanisms of marine organisms43,47,48,49, we attempted to explore their effects on embryogenesis of P. lividus. Thus, sea urchin zygotes were cultured continuously after fertilization in the presence of intact bacteria resuspended in Millipore-filtered sea water (MFSW) at concentrations of 1 × 106/mL and 1 × 107/mL, or with equivalent amounts of bacterial lysate supernatants or methanolic extracts. In this analysis, we focused on Bacillus (A3) and Vibrio (B2) isolates that were randomly selected as representative of the two geographic sites described. Treated sea urchin embryos were observed at several time intervals during development, their overall morphology was examined and compared to that of control unperturbed embryos from the same batches. Development of treated embryos was apparently normal during early cleavage and until the mesenchyme blastula stage. From this stage onwards, significant fractions of the embryos treated with the bacteria suspensions showed overt developmental delay and defects (70%), as well as arrested development (20%). In particular, the morphology of most of these embryos remained roughly spherical during development, never evolving into the characteristic easel-like shape exhibited by control plutei (Fig. 5). Nonetheless, most of the assayed specimens eventually produced mesenchyme cells, archenteron and skeletal elements similar to control larvae (Fig. 5), indicating that both morphogenetic movements and specification of main tissues were not impaired by the exposure to the two bacterial strains examined. Intriguingly, in a small but reproducible fraction (~ 15%) of these embryos the body rod spicules extended parallel instead of converging toward the aboral apex, giving the embryos the characteristic shape of a chair (Fig. 5). All the observed anomalies arose in a dose-dependent manner, irrespectively of the bacterial strain. Worth to mention, exactly the same phenotypes were obtained following exposure of embryos with amounts of bacterial lysate supernatants corresponding to 1 × 107 cells/mL (Fig. 5), suggesting that the observed aberrations on embryogenesis rely upon hydrophilic compound(s) synthesized by the bacterial strains assayed. In support of this hypothesis, sibling embryos exposed to equivalent amounts of either methanolic extracts (Fig. 5) or methanolic hydrochloric acid, as well as PBS, developed normally compared to control unperturbed embryos. We also noticed that the swimming behaviour of larvae treated with either bacterial strains or the corresponding bacterial lysate supernatants changed markedly, ranging from abnormal locomotion to immobilization. In particular, while all of the control larvae did swim freely exhibiting rapid forward movements throughout the water column, roughly 50% and 80% of the embryos exposed to Bacillus (A3) and Vibrio (B2), respectively, were turning around their axis or circling slowly at the bottom of the culture plate.

Developmental effects of coelomic fluid bacterial strain exposure on P. lividus. Representative embryos cultured in the absence (control) or in the presence of the indicated bacterial strains, bacterial lysate supernatants or equivalent amounts of methanolic extracts, and observed at the pluteus stage.
Discussion
Individual asymmetries in the composition of the bacterial microbiota associated to distinct metazoans, spanning from Hydra to mammalians, have been reported by several authors50,51,52. For instance, Enomoto et al.37 reported a distinctive bacterial microbiota structure in the coelomic fluid of distinct holothurian individuals. These findings, coupled to the fact that the perivisceral liquid of echinoderms is in dynamic equilibrium with the surrounding seawater environment53, raise questions about the stability of the microbiota composition in the coelomic fluid of individual sea urchins across seasons and geographic sites. This point has been addressed in the present study by describing and comparatively analysing the bacterial microbiota residing in the coelomic fluid of sea urchins collected from two distinct geographical sites. Indeed, this analysis revealed that a core bacterial microbiota formed by members of the Proteobacteria, Bacterioidetes and Fusobacteria phyla is established in all the analyzed specimens, irrespective of the sampling site. On the other hand, minority taxa characteristically related to the geographical localization constituted the most variable part of the coelomic fluid bacterial microbiota structure. These findings could reflect environmental and physiological stimuli to each P. lividus individual, which can be highly variable in close subcostal areas of heavily anthropized regions. In strict accordance, the examination of differentially abundant metabolic pathways, inferred from 16S rDNA counts by a Piphillin-based analysis using the KEGG metabolic database, highlighted a possible over-representation of metabolic functions associated with amino acid metabolism and production of bioactive peptides in the P. lividus coelomic bacterial microbiota of site B specimens. Although this finding suggests that different metabolic functions are associated with the bacterial microbiota of specimens residing in site A and B, the exact role of these functional differences in the environmental contest is still elusive and, therefore, further dedicated studies are required to underline the crucial importance of bacteria-host association dynamics in the aquatic environment.
Worth to mention, all the taxa identified in the present study have been previously described as commonly associated with coastal marine ecosystems and especially with marine invertebrates, potentially having important metabolic and physiological roles in their host species54,55. For example, bacteria belonging to the Vibrio and Photobacterium genera are among the most recurring taxa in marine metazoan microbiotas, whereas their association may be (i) symbiotic56, (ii) neutral as commensal microorganisms57, or (iii) parasitic as agents of disease58. Bacteria belonging to Propionigenium genus were found associated with tyrian purple producing gland in the marine gastropod Dicathais orbita59, and the P. maris species is characterized for the capability of debrominating biogenic bromoaromatic compounds that are often distributed in intertidal marine sediments60. Desulphovibrio, usually found in marine sediments, was also revealed to be part of the sponge microbiota, having the characteristic property to conduct anaerobic reduction thus recycling marine inorganic elements61.
Irrespective of the enormous amount of microbial community data from the coelomic fluid, the mechanism of symbiotic establishment and its exact role within the sea urchin host remains mostly unclear. Establishment of symbiotic association in marine organisms has been recognized as a key feature for the correct differentiation of tissues and organs during embryogenesis56,62,63,64,65. The paradigmatic example is given by the functional maturation of the light organ of the Hawaiian bobtail squid Euprymna scolopes, which requires specific colonization by the luminous bacterium Vibrio fischeri during embryonic development56,62. Moreover, a pervasive example of microbial signalling in marine invertebrate development is the induction of metamorphosis and settlement of the benthonic larvae63. This transition is absolutely required for the completion of the life cycle of these animals, and in the case of sea urchins it roughly coincides with the beginning of coelomic fluid formation29. In light of this, an intriguing hypothesis is that specific bacterial colonization of the coelomic fluid could occur at the critical time of larval settlement. Thus, although a non-mutualistic partnership cannot be excluded, it could be reasonably supposed that the microbial–host interaction may provide sea urchins with benefits that include, among others, the interchange and assimilation of nutrients as well as defence barrier against pathogens. Another fascinating role in the host speciation and evolution processes could be potentially played by the bacteria of the Bacteroidetes phylum, which have been associated with alterations of the host reproduction64,65.
The six bacterial strains that we isolated by culture-dependent analysis belong to Vibrio, Bacillus and Shewanella genera, respectively, whose members are notoriously capable of: (i) influencing immune cell behaviour in marine invertebrates, as in the case of the Vibrio spp.66; (ii) producing biologically active metabolites and/or small molecules and enzymes, as in the case of Bacillus67; (iii) detoxifying environments due to their metabolic versatility, as in the case of Shewanella68. Therefore, these strains offer the unique possibility to be characterized for their possible roles in P. lividus development and physiology, as well as for potential use in biotechnological applications. Due to the importance of sea urchins in many scientific and economical fields like molecular embryology, marine ecology and fishery, detailed information on this issue is of outstanding interest.
In the present study, we show that a significant fraction of developing embryos exposed to either Bacillus (A3) or Vibrio (B2) bacterial strains isolated from coelomic fluid samples exhibited similar developmental delay and aberrations. The observed effects could depend on the bacterial doses used in our assay, which are presumably much higher than those encountered in the ocean by free-living echinoderm embryos. In support of this hypothesis, previous reports showed that many bacterial strains normally associate with healthy sea urchin larvae, where they behave as commensal or mutualistic microorganisms (reviewed in69). However, under dysbiosis conditions an excessive bacterial load can overwhelm larval defenses, thereby affecting larval welfare and physiology66,70. Moreover, though Bacillus (A3) and Vibrio (B2), as well as many other bacterial strains, inhabit the coelomic fluid of P. lividus, it should be emphasized that sea urchin larvae and juveniles in aquaculture facilities are apparently more susceptible to bacterial infections than adult echinoderms71.
In conclusion, our results not only provide an unexplored and unexploited source of microbial diversity, but also could be helpful for decoding the biological functions of the echinoderm coelomic fluid, for understanding the roles of bacteria in the physiology and ecology of echinoderms, and for the improvement of their aquaculture conditions.
Methods
Collection of sea urchins and DNA extraction from coelomic fluid
To counterbalance possible differences in the composition of the coelomic fluid due to different marine microenvironments, adult P. lividus specimens were collected during Spring 2015 from two distinct localities around the Eastern coast of Palermo (site A: 38.11° N 13.49° E, and site B: 38.21° N 13.24° E, respectively), chosen for their proximity to a high-density population urban area and to a nature reserve, respectively. Sampled individuals were transported in thermally insulated containers filled with cold sea water, and they arrived to the laboratory within one hour since collection. Immediately after, sampled individuals were rinsed thoroughly with sterile 0.45 µm-filtered sea water and kept in aseptic conditions. Six and four individuals (mean test diameter ranging between 55.0 ± 2.0 mm) from site A and B, respectively, were randomly selected and subjected to further analysis as follows. Approximately 7 mL of coelomic fluid were drawn, through the peristomial membrane surrounding the Aristotele’s lantern, using a sterile syringe with a 21-gauge needle, and 6.5 mM EDTA pH 8.0 was immediately added to prevent clot formation. The coelomic fluid was centrifuged at 6000 × g for 10 min at 4 °C in an Eppendorf 5804R centrifuge, and DNA extracted from the resulting cellular component by using the Genomic DNA purification kit (Thermo Scientific), following the manufacturer’s recommendations.
Generation of 16S rDNA amplicon library, next generation sequencing and bioinformatic analysis
The quality of metagenomic DNA samples extracted from coelomic fluid was preliminarily checked by end point PCR to amplify genes encoding 16S rDNA. PCR reactions were performed using 100 ng of DNA as template in 1X PCR RxN Reaction Buffer (Invitrogen), 3.5 mM MgCl2, 200 µM dNTPs (Invitrogen), 0.2 µM universal primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) and 1 U of Taq DNA Polymerase Recombinant (Invitrogen). Thermal cycling conditions were 94 °C for 3 min, followed by 30 cycles of 94 °C for 45 s, 50 °C for 1 min and 72 °C for 90 s, and finally 72 °C for 10 min. According to PCR-based quality test, the metagenomic DNA of 10 samples (6 from A site and 4 from B site) was considered suitable for NGS analysis that was performed by IGA Technology Services (Udine, Italy) using MiSeq (Illumina, San Diego, CA, USA) to generate 300 bp paired-end reads of 16S rDNA V3-V4 region (FWD: 5′-CCTACGGGNGGCWGCAG-3′; REV: 5′-GACTACHVGGGTATCTAATCC-3′). NGS data are available at the European Nucleotide Archive (https://www.ebi.ac.uk/ena) with the accession identifier PRJEB27034.
Reads were pre-processed using the MICCA pipeline v 1.5.072, and the overlapping paired-end reads were merged using micca mergepairs73. Forward and reverse primer trimming and quality filtering were performed using micca trim and micca filter, respectively. De novo greedy clustering and chimera filtering were performed by using micca otu: operational taxonomic units (OTUs) were assigned by clustering the sequences with a threshold of 97% pairwise identity, and their representative sequences were classified using micca classify with the RDP classifier v2.1174. Multiple sequence alignment was performed using the Nearest Alignment Space Termination (NAST)75 algorithm implemented in micca msa with the template alignment clustered at 97% similarity of the Greengenes database (release 13_05)76. The phylogenetic tree was inferred using micca tree77. Sampling heterogeneity was reduced rarefying samples at the depth of the less abundant sample (18,707 sequences).
Alpha- (number of observed OTUs and Shannon index) and beta- (weighed Unifrac and Bray–Curtis distances) diversity estimates were computed using the phyloseq R package78. Permutational MANOVA (PERMANOVA) was performed by using the adonis function of the vegan R package with 999 permutations. The identification of taxa differentially distributed in the groups of study was obtained by using the linear discriminant effect size analysis (LEfSe)79. Prediction of functional metabolic potential from metagenomic data was obtained by using Piphillin46 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) October 2018 database80 with a 99% identity cut-off. All statistical analyses were performed using R81.
Microbial isolation and phylogenetic characterization
Serial dilution method was used to count and isolate cultivable bacteria from coelomic fluid samples derived from individuals collected from both sites A and B. In particular, samples were serially diluted in sterile 0.45 µm-filtered sea water, 100 μl of each dilution was plated on marine agar medium (CONDA) and incubated for 24–48 h at 30 °C to allow bacterial growth. Then, a total of six isolates (namely A1-4 and B1-2 from A and B sites, respectively), each exhibiting representative colony morphology and/or pigmentation, were selected for phylogenetic analysis based on 16S rDNA sequence as previously described82,83. Briefly, 16S rDNA amplification was performed using the universal bacterial primers 27F and 1492R84. The PCR amplicons were purified according to the manufacturer’s protocol using PCR clean-up Gel extraction kit (Macherey–Nagel), and sequenced by Macrogen (http://www.macrogen.com/eng). The 16S rDNA sequences were reconstructed from raw forward and reverse sequence data by FinchTV software (Perkin Elmer) and analysed using the DECIPHER Find Chimeras web tool (http://decipher.cee.wisc.edu/FindChimeras.html). The six reconstructed bacterial 16S rDNA sequences were submitted to GenBank with the following accession numbers: MK492102 (A1); MK492110 (A2); MK492258 (A3); MK491620 (A4); MK492275 (B1); MK492272 (B2). These sequences were used as query for nucleotide BLAST interrogations against 16S ribosomal RNA sequences. The first ten hits for each query were selected, exported in FASTA format and then de-replicated. Then, a total of 48 16S rDNA sequences were used to perform a phylogenetic analysis by MEGA785, using the Neighbor-Joining and Maximum Composite Likelihood methods to infer and compute the evolutionary history and distances, respectively86. All positions containing gaps and missing data were eliminated. There was a total of 866 positions in the final dataset. The 16S rDNA sequence of Sulfolobus solfataricus strain IFO 15331 was used as outgroup. Confidence for tree topologies was estimated by bootstrap values based on 1000 replicates.
Preparation of bacterial samples and developmental toxicity assay
Overnight suspensions in Marine broth (CONDA) of individual bacterial strains isolated from coelomic fluid were split in two fractions. The first fraction was washed twice with MFSW by centrifugation at 6000 rpm for 10 min at 4 °C, and intact bacteria resuspended in MFSW. The bacterial pellet of the sister fraction was washed two times with phosphate buffered saline (PBS), resuspended in 500 μl of PBS and cells lysed by sonication using a Bandelin Sonopuls ultrasonic homogenizer. Following centrifugation at 12,000 rpm for 5 min, the supernatant containing hydrophilic substances was separated, while the pellet was incubated overnight with 0.5 M methanolic hydrochloric acid at room temperature, to extract lipophilic substances.
After gamete harvesting from fresh adult P. lividus echinoderms collected from the mentioned sites A and B, eggs were fertilized and cultured at 18 °C in MFSW, as previously described87,88,89. Individual bacterial strains from the above mentioned fraction were added to 24-well not-treated plates containing ~ 250 embryos/well at the concentration of 1 × 106 and 1 × 107/well. In parallel, sea urchin embryos from the same batches were exposed to bacterial lysate supernatants and methanolic extracts at the indicated amounts. Controls for this assay included sibling embryos exposed to equal amounts of PBS and methanolic hydrochloric acid solutions, respectively, as well as unperturbed embryos. Three replicates were reproduced for each experimental condition, and three independent experiments were performed using distinct embryo batches. Phenotypes of 100 embryos at the desired stage were examined under a Leica DM-4500B microscope, and digital images were processed using Adobe Photoshop CS10.
Data availability
The NGS datasets generated during and analysed during the current study are available in the European Nucleotide Archive repository (https://www.ebi.ac.uk/ena) with the accession identifier PRJEB27034. 16S rDNA sequences generated from the isolated bacterial strains are available in GenBank (https://www.ncbi.nlm.nih.gov/genbank) with the following accession numbers: MK492102 (A1); MK492110 (A2); MK492258 (A3); MK491620 (A4); MK492275 (B1); MK492272 (B2).
References
Webster, N. S. & Taylor, M. W. Marine sponges and their microbial symbionts: love and other relationships. Environ. Microbiol. 14, 335–346 (2012).
Ainsworth, T. & Hoegh-Guldberg, O. Bacterial communities closely associated with coral tissues vary under experimental and natural reef conditions and thermal stress. Aquat. Biol. 4, 289–296 (2009).
Ainsworth, T. D., Thurber, R. V. & Gates, R. D. The future of coral reefs: a microbial perspective. Trends Ecol. Evol. 25, 233–240 (2010).
Rosenberg, E., Koren, O., Reshef, L., Efrony, R. & Zilber-Rosenberg, I. The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362 (2007).
Paul, V. J. & Puglisi, M. P. Chemical mediation of interactions among marine organisms. Nat. Prod. Rep. 21, 189–209 (2004).
Paul, V. J., Ritson-Williams, R. & Sharp, K. Marine chemical ecology in benthic environments. Nat. Prod. Rep. 28, 345–387 (2011).
Leal, M. C. et al. Marine microorganism-invertebrate assemblages: perspectives to solve the “supply problem” in the initial steps of drug discovery. Mar. Drugs 12, 3929–3952 (2014).
Frias-Lopez, J., Klaus, J. S., Bonheyo, G. T. & Fouke, B. W. Bacterial community associated with black band disease in corals. Appl. Environ. Microbiol. 70, 5955–5962 (2004).
Wang, W. et al. A spiroplasma associated with tremor disease in the Chinese mitten crab (Eriocheir sinensis). Microbiology 150, 3035–3040 (2004).
Kittelmann, S. & Harder, T. Species- and site-specific bacterial communities associated with four encrusting bryozoans from the North Sea, Germany. J. Exp. Mar. Biol. Ecol. 327, 201–209 (2005).
Ritchie, K. B. Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar. Ecol. Prog. Ser. 322, 1–14 (2006).
Webster, N. S. et al. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082 (2010).
Thorsen, M. S., Wieland, A., Ploug, P., Kragelund, C. & Nielsen, P. H. Distribution, identity and activity of symbiotic bacteria in anoxic aggregates from the hindgut of the sea urchin Echinocardium cordatum. Mar. Biol. Res. 57, 1–12 (2003).
Meziti, A., Kormas, K. A., Pancucci-Papadopoulou, M. A. & Thessalou-Legaki, M. Bacterial phylotypes associated with the digestive tract of the sea urchin Paracentrotus lividus and the ascidian Microcosmus sp. Russ. J. Mar. Biol. 33, 84–91 (2007).
Hakim, J. A. et al. An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin. Lytechinus variegatus. Front. Microbiol. 6, 1047. https://doi.org/10.3389/fmicb.2015.01047 (2015).
Hakim, J. A. et al. The gut microbiome of the sea urchin, Lytechinus variegatus from their natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles. FEMS Microbiol. Ecol. https://doi.org/10.1093/femsec/fiw146 (2016).
Lawrence, J., Lawrence, A. & Watts, S. Feeding, digestion, and digestibility of sea urchins. In Sea Urchins: Biology and Ecology (ed. Lawrence, J. M.) 135–154 (Elsevier, Amsterdam, 2013).
Becker, P. T., Egea, E. & Eeckhaut, I. Characterization of the bacterial communities associated with the bald sea urchin disease of the echinoid Paracentrotus lividus. J. Invertebr. Pathol. 98, 136–147 (2008).
Becker, P. T., Gillan, D. C. & Eeckhaut, I. Characterization of the bacterial community associated with body wall lesions of Tripneustes gratilla (Echinoidea) using culture-independent methods. J. Invertebr. Pathol. 100, 127–130 (2009).
Lawrence, J. M. Edible Sea Urchins: Biology and Ecology (Elsevier, Amsterdam, 2007).
Davidson, E. H. A History of Embryology (Cambridge University Press, Cambridge, 1986).
Ernst, S. G. A century of sea urchin development. Am. Zool. 37, 250–259 (1997).
Cavalieri, V., Di Bernardo, M., Anello, L. & Spinelli, G. cis-Regulatory sequences driving the expression of the Hbox12 homeobox-containing gene in the presumptive aboral ectoderm territory of the Paracentrotus lividus sea urchin embryo. Dev. Biol. 321, 455–469 (2008).
Cavalieri, V., Melfi, R. & Spinelli, G. The Compass-like locus, exclusive to the ambulacrarians, encodes a chromatin insulator binding protein in the sea urchin embryo. PLoS Genet. 9, e1003847. https://doi.org/10.1371/journal.pgen.1003847 (2013).
Di Caro, V., Cavalieri, V., Melfi, R. & Spinelli, G. Constitutive promoter occupancy by the MBF-1 activator and chromatin modification of the developmental regulated sea urchin alpha-H2A histone gene. J. Mol. Biol. 365, 1285–1297 (2007).
Pellicanò, M. et al. The sea urchin embryo: a model to study Alzheimer’s beta amyloid induced toxicity. Arch. Biochem. Biophys. 483, 120–126 (2009).
Hyman, L. H. The Invertebrates: Echinodermata, The Coelomate Bilateria Vol. 4 (McGraw-Hill, London, 1955).
Sodergren, E. et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 314, 941–952 (2006).
Endean, R. The coelomocytes and coelomic fluids. In Physiology of Echinodermata (ed. Boolootian, R. A.) 301–328 (Interscience Publishers, New York, 1966).
Farmanfarmaian, A. The respiratory physiology of echinoderms in physiology of echinodermata. In Physiology of Echinodermata (ed. Boolootian, R. A.) 245–266 (Interscience Publishers, New York, 1966).
Smith, L. C. et al. The sea urchin immune system. Inv. Surv. J. 3, 25–39 (2006).
Holland, L. Z., Giese, A. C. & Phillips, J. H. Studies on the perivisceral coelomic fluid protein concentration during seasonal and nutritional changes in the purple sea urchin. Comp. Biochem. Physiol. 21, 361–371 (1967).
Smith, L. C. Host responses to bacteria: innate immunity in invertebrates. In The Influence of Cooperative Bacteria on Animal Host Biology. Advances in Molecular and Cellular Microbiology Vol. 10 (eds McFall-Ngai, M. et al.) 293–320 (Cambridge University Press, Cambridge, 2005).
Smith, L. C. et al. Echinoderm immunity. Adv. Exp. Med. Biol. 708, 260–301 (2010).
Unkles, S. E. Bacterial flora of the sea urchin Echinus esculentus. Appl. Environ. Microbiol. 34, 347–350 (1977).
Offret, C., Jégou, C., Mounier, J., Fleury, Y. & Le Chevalier, P. New insights into the haemo- and coelo-microbiota with antimicrobial activities from Echinodermata and Mollusca. J. Appl. Microbiol. 126, 1023–1031 (2019).
Enomoto, M., Nakagawa, S. & Sawabe, T. Microbial communities associated with holothurians: presence of unique bacteria in the coelomic fluid. Microbes Environ. 27, 300–305 (2012).
Pivkin, M. V. Filamentous fungi associated with holothurians from the sea of Japan, off the primorye coast of Russia. Biol. Bull. 198, 101–109 (2000).
Chen, J., Ren, Y., Li, Y. & Xia, B. Regulation of growth, intestinal microbiota, non-specific immune response and disease resistance of sea cucumber Apostichopus japonicus (Selenka) in biofloc systems. Fish Shellfish Immunol. 77, 175–186 (2018).
Nakagawa, S. et al. Microbiota in the coelomic fluid of two common coastal starfish species and characterization of an abundant Helicobacter-related taxon. Sci. Rep. 7, 8764. https://doi.org/10.1038/s41598-017-09355-2 (2017).
Brothers, C. J. et al. Ocean warming alters predicted microbiome functionality in a common sea urchin. Proc. Biol. Sci. 285, 1881. https://doi.org/10.1098/rspb.2018.0340 (2018).
Leon-Palmero, E. et al. Diversity and antimicrobial potential in sea anemone and holothurian microbiomes. PLoS ONE 13, e0196178. https://doi.org/10.1371/journal.pone.0196178 (2018).
Jackson, E. W., Pepe-Ranney, C., Debenport, S. J., Buckley, D. H. & Hewson, I. The microbial landscape of sea stars and the anatomical and interspecies variability of their microbiome. Front. Microbiol. 9, 1829. https://doi.org/10.3389/fmicb.2018.01829 (2018).
Amann, R. I., Ludwig, W. & Schleifer, K. H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169 (1995).
Köpke, B., Wilms, R., Engelen, B., Cypionka, H. & Sass, H. Microbial diversity in coastal subsurface sediments: a cultivation approach using various electron acceptors and substrate gradients. Appl. Environ. Microbiol. 71, 7819–7830 (2005).
Iwai, S. et al. Piphillin: improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 11, e0166104. https://doi.org/10.1371/journal.pone.0166104 (2016).
Luis-Villaseñora, I. E., Macías-Rodríguez, M. E., Gómez-Gil, B., Ascencio-Valle, F. & Campa-Córdova, A. I. Beneficial effects of four Bacillus strains on the larval cultivation of Litopenaeus vannamei. Aquaculture 321, 136–144 (2011).
Avella, M. A. et al. Application of multi-species of Bacillus in sea bream larviculture. Aquaculture 305, 12–19 (2010).
Hau, H. H. & Gralnick, J. A. Ecology and biotechnology of the genus Shewanella. Annu. Rev. Microbiol. 61, 237–258 (2007).
Fraune, S. & Bosch, T. C. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl. Acad. Sci. USA 104, 13146–13151 (2007).
Blasiak, L. C., Zinder, S. H., Buckley, D. H. & Hill, R. T. Bacterial diversity associated with the tunic of the model chordate Ciona intestinalis. ISME J. 8, 309–320 (2014).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Freire, C. A., Santos, I. A. & Vidolin, D. Osmolality and ions of the perivisceral coelomic fluid of the intertidal sea urchin Echinometra lucunter (Echinodermata: Echinoidea) upon salinity and ionic challenges. Zoologia 28, 479–487 (2011).
Kwon, S. W. et al. Idiomarina homiensis sp. nov., isolated from seashore sand in Korea. Int. J. Syst. Evol. Microbiol. 56, 2229–2233 (2006).
Zhou, Y. et al. High quality draft genome sequence of the slightly halophilic bacterium Halomonas zhanjiangensis type strain JSM 078169(T) (DSM 21076(T)) from a sea urchin in southern China. Stand. Genomic Sci. 9, 1020–1030 (2014).
McFall-Ngai, M. J. The importance of microbes in animal development: lessons from the squid-vibrio symbiosis. Annu. Rev. Microbiol. 68, 177–194 (2014).
Sun, Y. et al. Intraspecific competition impacts vibrio fischeri strain diversity during initial colonization of the squid light organ. Appl. Environ. Microbiol. 82, 3082–3091 (2016).
Osorio, C. R. et al. A transmissible plasmid-borne pathogenicity island confers piscibactin biosynthesis in the fish pathogen Photobacterium damselae subsp. piscicida. Appl. Environ. Microbiol. 81, 5867–5879 (2015).
Ngangbam, A. K., Baten, A., Waters, D. L. E., Whalan, S. & Benkendorff, K. Characterization of bacterial communities associated with the tyrian purple producing gland in a marine gastropod. PLoS ONE 10, e0140725. https://doi.org/10.1371/journal.pone.0140725 (2015).
Watson, J. et al. Reductively debrominating strains of Propionigenium maris from burrows of bromophenol-producing marine infauna. Int. J. Syst. Evol. Microbiol. 50, 1035–1042 (2000).
Zhang, D. et al. Phylogenetic diversity of sulphate-reducing Desulfovibrio associated with three South China Sea sponges. Lett. Appl. Microbiol. 60, 504–512 (2015).
McFall-Ngai, M. J. & Ruby, E. G. Symbiont recognition and subsequent morphogenesis as early events in an animal–bacterial mutualism. Science 254, 1491–1494 (1991).
Hadfield, M. G. Biofilms and marine invertebrate larvae: What bacteria produce that larvae use to choose settlement sites. Annu. Rev. Mar. Sci. 3, 453–470 (2011).
Bordenstein, S. R. Symbiosis and the origin of species. In Insect Symbiosis (eds Bourtzis, K. & Miller, T. A.) 283–304 (CRC Press, Boca Raton, 2003).
Hunter, M. S., Perlman, S. J. & Kelly, S. E. A bacterial symbiont in the Bacteroidetes induces cytoplasmic incompatibility in the parasitoid wasp Encarsia pergandiella. Proc. R. Soc. Lond. B 270, 2185–2190 (2003).
Schuh, N. W. et al. Bacterial exposure mediates developmental plasticity and resistance to lethal Vibrio lentus infection in purple sea urchin (Strongylocentrotus purpuratus) Larvae. Front. Immunol. 10, 3014. https://doi.org/10.3389/fimmu.2019.03014 (2020).
Mondol, M. A. M., Shin, H. J. & Islam, M. T. Diversity of secondary metabolites from marine Bacillus species: chemistry and biological activity. Mar. Drugs 11, 2846–2872 (2013).
Dash, H. R., Mangwani, N., Chakraborty, J., Kumari, S. & Das, S. Marine bacteria: potential candidates for enhanced bioremediation. Appl. Microbiol. Biotechnol. 97, 561–571 (2013).
Carrier, T. J. & Reitzel, A. M. Symbiotic Life of Echinoderm Larvae. Front. Ecol. Evol. 7, 509. https://doi.org/10.3389/fevo.2019.00509 (2020).
Brown, S. P., Cornforth, D. M. & Mideo, N. Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol. 20, 336–342 (2012).
Böttger, S. A., Walker, C. W. & Unuma, T. Care and maintenance of adult echinoderms. Methods Cell Biol. 74, 17–38 (2004).
Albanese, D., Fontana, P., De Filippo, C., Cavalieri, D. & Donati, C. MICCA: a complete and accurate software for taxonomic profiling of metagenomic data. Sci. Rep. 5, 9743. https://doi.org/10.1038/srep09743 (2015).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. https://doi.org/10.7717/peerj.2584 (2016).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Env. Microbiol. 73, 5261–5267 (2007).
DeSantis, T. Z. et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34, W394-399. https://doi.org/10.1093/nar/gkl244 (2006).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Team, R. C. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, 2012).
Gallo, G. et al. Adaptative biochemical pathways and regulatory networks in Klebsiella oxytoca BAS-10 producing a biotechnologically relevant exopolysaccharide during Fe(III)-citrate fermentation. Microb. Cell. Fact. 11, 152. https://doi.org/10.1186/1475-2859-11-152 (2012).
Milanesi, C. et al. Spoilage of oat bran by sporogenic microorganisms revived from soil buried 4000 years ago in Iranian archaeological site. Int. Biodeter. Biodegr. 104, 83–91 (2015).
Frank, J. A. et al. Critical Evaluation of Two Primers Commonly Used for Amplification of Bacterial 16S rRNA Genes. Appl. Environ. Microbiol. 74, 2461–2470 (2008).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Tamura, K., Nei, M. & Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 101, 11030–11035 (2004).
Cavalieri, V. & Spinelli, G. Ectopic hbox12 expression evoked by histone deacetylase inhibition disrupts axial specification of the sea urchin embryo. PLoS ONE 10, e0143860. https://doi.org/10.1371/journal.pone.0143860 (2015).
Cavalieri, V., Melfi, R. & Spinelli, G. Promoter activity of the sea urchin (Paracentrotus lividus) nucleosomal H3 and H2A and linker H1 α-histone genes is modulated by enhancer and chromatin insulator. Nucleic Acids Res. 37, 7407–7415 (2009).
Cavalieri, V., Bernardo, M. D. & Spinelli, G. Regulatory sequences driving expression of the sea urchin Otp homeobox gene in oral ectoderm cells. Gene Expr. Patterns. 7, 124–130 (2007).
Acknowledgments
We warmly thank Concetta Catania, Veronica La Fiora and Giuseppina Turturici for technical assistance in the initial stages of this project.
Author information
Authors and Affiliations
Contributions
A.M.P. and V.C. conceived and designed the experiments. T.F., F.A., F.F., C.R., E.P., F.S. and V.C. performed the experiments, while all the authors analyzed the data. T.F., E.P., F.S., G.G., and V.C. participated in drafting the manuscript. V.C. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Faddetta, T., Ardizzone, F., Faillaci, F. et al. Composition and geographic variation of the bacterial microbiota associated with the coelomic fluid of the sea urchin Paracentrotus lividus. Sci Rep 10, 21443 (2020). https://doi.org/10.1038/s41598-020-78534-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78534-5
- Springer Nature Limited
This article is cited by
-
Spotting disease disrupts the microbiome of infected purple sea urchins, Strongylocentrotus purpuratus
BMC Microbiology (2024)
-
Alterations in sea urchin (Mesocentrotus nudus) microbiota and their potential contributions to host according to barren severity
npj Biofilms and Microbiomes (2023)
-
The digestive tract sections of the sea cucumber Isostichopus badionotus reveal differences in composition, diversity, and functionality of the gut microbiota
Archives of Microbiology (2022)