
Abstract
Despite phages’ ubiquitous presence and great importance in shaping microbial communities, little is known about the diversity of specific phages in different ecological niches. Here, we isolated, sequenced, and characterized 38 Escherichia coli-infecting phages (coliphages) from poultry faeces to gain a better understanding of the coliphage diversity in the poultry intestine. All phages belonged to either the Siphoviridae or Myoviridae family and their genomes ranged between 44,324 and 173,384 bp, with a G+C content between 35.5 and 46.4%. Phylogenetic analysis was performed based on single “marker” genes; the terminase large subunit, portal protein, and exonucleases, as well as the full draft genomes. Single gene analysis resulted in six distinct clusters. Only minor differences were observed between the different phylogenetic analyses, including branch lengths and additional duplicate or triplicate subclustering. Cluster formation was according to genome size, G+C content and phage subfamily. Phylogenetic analysis based on the full genomes supported these clusters. Moreover, several of our Siphoviridae phages might represent a novel unclassified phage genus. This study allowed for identification of several novel coliphages and provides new insights to the coliphage diversity in the intestine of poultry. Great diversity was observed amongst the phages, while they were isolated from an otherwise similar ecosystem.
Similar content being viewed by others
Introduction
Bacteriophages (phages) are viruses that have the ability to specifically infect bacteria. They are estimated to be the most abundant form of life on Earth (~ 1031 organisms) and can be found in almost every ecosystem, including soil, wastewater, sewage water, seawater and in and on humans and animals1,2,3,4. Phages are thought to play essential roles in shaping the microbial ecology, including driving the diversity of the bacterial communities5. As no single gene is present in all phages, their taxonomic classification is based on host-range, physical characteristics, including size and morphology, genetic structure and composition, and overall genome similarity6,7. The phage classification scheme is regularly updated, refined and approved by the International Committee on the Taxonomy of Viruses (ICTV)8. Furthermore, in recent years several genome-based phage taxonomy schemes have been proposed7,9. According to the National Center for Biotechnology information (NCBI), as of February 2020, 9,238 complete phage genomes have been sequenced. However, despite a continuously rising number of sequenced phage genomes, most of them remain unclassified and poorly characterized. According to the ICTV, a phage genus can be defined as a group of viruses with > 50% nucleotide sequence similarity, which is distinct from viruses of other genera. Moreover, defining characteristics can be determined for each genus, including average genome length and number of coding sequences (CDSs), percentage of shared CDSs, and the presence of specific signature genes in genus members10.
Most phages that infect Escherichia coli, coliphages, belong the highly heterogeneous Caudovirales order, which constitute ~ 96% of all known isolated phages11. This order contains five families of tailed phages with dsDNA genomes: Myoviridae, Siphoviridae, Podoviridae, Ackermannviridae and Herelleviridae12. According to ICTV taxonomy (data of February 2020), these families comprise five, eleven, three, two, and five subfamilies, respectively, and 87, 210, 48, three, and 15 genera, respectively. The currently analysed Myoviridae coliphages belong to four subfamilies, including Ounavirinae, Peduovirinae, Tevenvirinae, and Vequintavirinae, and 17 genera. Siphoviridae coliphages are found in only two subfamilies; Guernseyvirinae and Tunavirinae, and in 13 genera. Podoviridae coliphages belong to two subfamilies, the Autographivirinae and the Sepvirinae, and to ten genera. Ackermannviridae coliphages belong only to the Cvivirinae subfamily and the Kuttervirus genus. To date, there have been no Herelleviridae coliphages isolated. To understand the diversity, relationships, and dynamics among any group of phages, nucleotide sequence information is needed1. For tailed phages, it has been reported that conserved genes such as the terminase large subunit, the portal protein and major capsid protein, can be used as phylogenetic phylomarkers for the diversity as well as their evolutionary relationship1,13.
Compared to their bacterial hosts, relatively few phages have been fully characterized14. Besides, despite the phages’ significant role and ubiquitous presence in various areas, little is known on the nature and extent of phage diversity in different ecosystems3. Recently, there has been an interest in the diversity of coliphages1,15,16. Here, we performed a detailed genome-based characterization and phylogenetic analysis of 38 fully sequenced coliphages, all isolated from a single, relatively unexplored environmental source: poultry faecal material.
Results
Phage isolation
In this study, 38 coliphages were isolated from poultry faecal samples collected from 27 Belgian poultry farms located in five different regions, including West Flanders, East Flanders, Antwerp, and Limburg. Between one and seven phages were isolated from each farm using E. coli C600 or K514 as host strain.
Phage morphological analysis
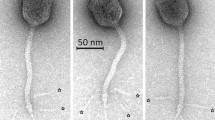
Based on a sequencing cut-off value of ≤ 95% nucleotide similarity, 18 coliphages were selected and subjected to TEM to determine phage morphology and confirm phage classification. Based on the morphological features, the phages were classified into the Caudovirales order and either the Siphoviridae family or the Myoviridae family. Analysis of the isolated Siphoviridae phages showed a long flexible non-contractile tail with a length varying between ~ 100 and ~ 200 nm and icosahedral heads with widths ranging from ~ 52 to ~ 77 nm (Fig. 1a–h). Among the isolated Myoviridae phages a long straight contractile tail was observed with a tailed length varied between ~ 100 and ~ 120 nm, head widths ranging from ~ 65 to ~ 84 nm, and head lengths from ~ 60 to ~ 110 nm (Fig. 1i–r). Taxonomic classification of each of the coliphages is shown in Table 1.

Negative staining electron microscopy images of Siphoviridae and Myoviridae coliphages. Siphoviridae phages: (a) Phage 17. (b) Phage 53. (c) Phage 54. (d) Phage 61. (e) Phage 70. (f) Phage 74. (g) Phage 76. (h) Phage 77. Myoviridae phages: (i) Phage 10. (j) Phage 11. (k) Phage 15. (l) Phage 18. (m) Phage 30. (n) Phage 55. (o) Phage 60. (p) Phage 62 (q) Phage 78. (r) Phage 79. The black bars represent 100 nm.
Phage genome sequence analysis and annotation
All 38 coliphages isolated in this study were characterized based on WGS data. An overview of the genomic characteristics and properties are listed in Table 1. According to FastQC parameters, good quality of the raw sequence data for all phages was confirmed. The phage genomes ranged in size between 44,324 and 173,384 bp, with a G+C content between 35.5 and 46.4%. Genomes smaller than 90,000 bp had a G+C content between 38.9 and 46.4%, whereas the larger genomes had a G+C content of 35.5–38%. For each coliphage, 72–275 putative CDSs were identified using both automatic and manual annotation. CDSs encoding the phage terminase small subunit, the phage terminase large subunit, the phage portal protein, and phage capsid and scaffold proteins were identified within all 38 coliphage genome sequences. They presented the same conserved genome structure with a general gene order: the terminase small subunit upstream from the terminase large subunit, the phage portal protein and two genes encoding phage capsid and scaffold proteins. In general, one phage terminase small subunit, one phage portal protein, and up to four phage capsid and scaffold proteins were found within each of the phage genomes. Besides, phage exonucleases were identified in all phage genomes. For each phage, one to three CDSs for exonucleases were found. No gene encoding for an integrase was found, indicating that these phages are strictly virulent/lytic phages. No known acquired resistance or virulence genes were detected in any of the 38 phage genomes.
Phage phylogeny and taxonomy
Taxonomic classification of the 38 isolated coliphages was performed through multiple WGS genome comparisons. These coliphages included 27 (71%) Siphoviridae coliphages and 11 (29%) Myoviridae coliphages. The Siphoviridae phages were compared with 146 published phages from this family. The Myoviridae phages were compared with 171 published Myoviridae phages. According to ICTV guidelines, phage family, subfamily and genus were predicted based on genome similarity. Results are shown in Table 1. All Siphoviridae phages belonged to the Tunavirinae subfamily, except for Phage 61. This phage was predicted to belong to the Tequintavirus genus, which do not have any ICTV subfamily. The ten phages, Phage 8, 53, 54, 63, 65, 68, 69, 71, 72, and 75 all belonged to the Hanrivervirus genus. Three phages belonged to the Rtpvirus genus, including Phage 17, 70, and 73. No existing ICTV genus could be assigned to the remaining 13 coliphages. Phage 28, 56_1 and 76 could be assigned to the same unknown genus. Phage 58 and Phage 74 were found to be in the same genus. Phage 47, 48, 59, 64, and 77 were predicted to belong to the same genus. Phage 52, 56_2, and 80 were predicted to belong to the same genus. The 11 Myoviridae belonged either to the Tevenvirinae or the Ounavirinae subfamily. Tevenvirinae phages included the six phages: Phage 10, 11, 15, 18, 30 and 55. Phage 10, 11 and 55 belonged to the Tequatrovirus genus, and Phage 18 and 30 belonged to the Mosigvirus genus. Ounavirinae phages included the remaining five phages; Phage 60, 62, 66, 78 and 79. All phages belonged to the Felixounavirus genus.
Phage diversity
To investigate the diversity of the coliphages, phage genomes were first clustered based on whole genome sequence. A total of 173 Siphoviridae and 182 Myoviridae coliphage genomes were included. Characteristics of selected reference genomes are listed in Supplementary Table S1. Siphoviridae phages isolated in this study were found in five different (sub)clusters, cluster A1–3, B and C, with a cut-off value of 0.82 (Fig. 2). Cluster A was divided into three subclusters. Fourteen of our phages, formed subcluster A1 together with the three pSf-1-like reference phages from the NCBI database. Phage 80, 28, 56_1, and 76 formed subcluster A2 with the three Swan01-like reference phages. Phages 69, 52, and 56_2 formed subcluster A3 with phage Jahat_MG145. Phage 73, 70, 17, 58 and 74 formed cluster B without any known reference phages. Phage 61 was placed in cluster C with 13 T5-like reference phages. For the Myoviridae phages, the resulting phylogeny placed phages isolated in this study in three different clusters with a cut-off height of 0.52 (Fig. 3). Phages 62, 78, 66, 60 and 79 formed cluster D with Felix01-like reference phage Alf5. Phage 30, 15 and 18 formed cluster E with 19 T4-like reference phages (cut-off height of 0.36). Phages 55, 11 and 10 were placed in cluster F with 57 reference phages. At the cut-off height of 0.39 Phage 55 was found in a different subcluster than Phage 10 and 11.

Phylogenetic analysis of Siphoviridae coliphages based on WGS sequence. Phages isolated in this study are highlighted. Each colour represents a cluster: Cluster A (blue), cluster B (green), and cluster C (red). Cluster A subclusters include A1 (light blue), A2 (blue), and A3 (dark blue). Distance matrices and clustering are based on kmer length = 10.

Phylogenetic analysis of Myoviridae coliphages based on WGS sequence. Phages isolated in this study are highlighted. Each colour represents a cluster: Cluster D (orange), cluster E (purple), and cluster F (brown). Distance matrices and clustering are based on kmer length = 10.
Coliphages were further assessed based on the presence/absence of families of orthologues genes in their pan genome. Similar clusters were observed with only minor changes. For the Siphoviridae phage analysis, 5,227 gene groups were included (Supplementary Fig. S1). The resulting phylogenetic analysis placed phages isolated in this study in the same five clusters, cluster A1–3, B and C, with a cut-off height of 0.81 (Supplementary Fig. S2). One additional reference phage was found in cluster B and C, including the T1-like reference phage CEB_EC3a and the T5-like reference phage EPS7, respectively. For the Myoviridae phage analysis, 9,420 gene groups were included (Supplementary Fig. S3). The resulting phylogeny placed phages isolated in this study in the same three clusters, cluster D, E, and F, with a cut-off height of 0.58 (Supplementary Fig. S4). For cluster D, additionally 13 Felix01-like reference phages were found. In contrast to the WGS-based analysis, at a cut-off height of 0.39, all cluster F phages isolated in this study, were found in one subcluster with 10 T4-like reference phages. The degree of topological and branch length agreement between the different phylogenetic methods were compared (Supplementary Table S2).
The coliphage diversity was further assessed based on three phage marker genes: the terminase large subunit and phage portal protein, and the phage exonuclease. Selected gene sequences from known phages were included for reference. Results are summarized in Table 1. For all three marker genes, cluster formation was in accordance with resulting clusters of the pan genome- and WGS-based phylogeny, cluster A–F, only with minor differences. Results based on the terminase large subunit analysis are shown below (Fig. 4).

Maximum likelihood tree based on the nucleotide sequences of the phage terminase large subunit. The analysis resulted in six clusters: A–F, according to phage family and subfamily. Cluster A and B: Siphoviridae, Tunavirinae, cluster C: Siphoviridae and Tequintavirus genus, cluster D: Myoviridae, Ounavirinae, and cluster E and F: Myoviridae, Tevenvirinae. Cluster A was divided into three subclusters: A1, A2 and A3. The tree was constructed using the MEGA X software54. The percent of data coverage for internal nodes is indicated. The scale bar indicates the number of nucleotide sequence substitutions per site. The analysis included 62 nucleotide sequences, including 24 reference phages listed in Supplementary Table S1 for comparison.
For cluster A, all coliphages isolated in this study were found within same subclusters as for the WGS-based phylogeny except for Phage 63, which was found in the A2 subcluster instead of A1. Analysis based on the phage portal protein resulted in the division of our A2 subcluster phages into two groups: Phage 56_1, 80 and 28 in one group and Phage 76 in the other group (Supplementary Fig. S5). Analysis based on the exonuclease resulted in multiple clusters of cluster C and F, as phages from these clusters encoded 2 or 2–3 exonuclease genes, respectively (Supplementary Fig. S6). Comparison of the cluster construction of the three single genes analysis showed only minor topological and branch length differences (Supplementary Table S2). Moreover, cluster construction was in accordance with phage subfamily defined based on the whole genome. Siphoviridae phages from cluster A and B belonged to the Tunavirinae subfamily, and Siphoviridae phages form cluster C had no defined ICTV subfamily. Myoviridae phages from cluster D belonged to the Ounavirinae subfamily, and Myoviridae phages from cluster E and F belonged to the Tevenvirinae subfamily.
Phage comparative genomics
Pan genome analysis of Siphoviridae and Myoviridae phages isolated in this study revealed that neither of the two groups had any core genes. Analysis of coliphage genomes from each of the six clusters, A–F, identified core genes (core and softcore) and accessory genes (shell and cloud). As cluster A phages had only five core genes (2% of the total genome), analysis of subclusters, A1, A2 and A3, were performed additionally. Results are summarized in Table 2. The pan genome included between 81 and 333 genes, and core genes constituted between 22 and 73% of the pan genome.
The level of synteny and genomic rearrangement within each cluster or subcluster of related phages was assessed by genome comparison. Results are summarized in Table 2. Eight comparisons were performed, corresponding to the eight (sub)clusters, A1, A2, A3, B, C, D, E, and F resulting from the phage diversity analysis above (Supplementary Fig. S7–S14). Genome comparison of the phages resulted in identification of local collinear blocks (LCBs), indicating homologues DNA regions shared by two or more genomes without sequence rearrangements. The LCBs comprised different modules of genes with different functions, including modules for DNA packaging, structural proteins, head and tail morphogenesis, and host cell lysis. Several modules comprised only hypothetical proteins with unknown function. The average level of conservation varied between the different type of genes.
Genes encoding the terminase large and small subunit, the major capsid protein, DNA primase, single-stranded protein, portal protein, recombinase, specific tail protein and holin were the most conserved genes between all phages, whereas genes with the lowest level of conservation included, specific tail fiber proteins, tail tape measure proteins and HNH homing endonucleases. Hypothetical proteins were found with large variation in level of conservation. Each phage genome comprised between four and 17 LCBs. Genome comparison of phages belonging to subcluster A1, A2 and A3 identified 16, seven and four LCBs, respectively. All phages in each cluster comprised all LCBs. All cluster B phages comprised all six LCBs. For the cluster C phages, between 6 and 10 LCBs were identified for each phage. Phage 61 isolated in this study comprised all nine regions. Variation in number of LCBs was due to a variable repeat region comprising multiple LCBs, which was found only in some of the cluster C phages. For the cluster D comparison, 14–17 different LCBs were identified for each phage. Variation in number of LCBs was due to four different small variable regions of which some of all were missing in the majority of the phages. Phages isolated in this study, including Phage 79, Phage 78, Phage 60, Phage 66, and Phage 62, comprised 17, 17, 16, 15, and 14 LCBs, respectively. Comparison of phages belonging to cluster E identified 18 LCBs. All phages lacked one or both of the same two LCBs. Phages isolated in this study, including Phage 30, Phage 15, and Phage 18, comprised 16, 17, and 17 LCBs, respectively. All 13 cluster F phages included in the comparison comprised all five LCBs. The comparison confirmed the presence of homologue regions between the phages within the clusters but also highlighted that re-arrangement and/or gain/loss of LCBs must have occurred at some point during the evolution of the phages. The region encoding the terminase large subunit and portal protein were present in a conserved region all genomes in all eight comparisons.
Discussion
In this study, 38 coliphages were isolated from poultry faecal material, sequenced and characterized. The high number of coliphage genomes included in the analysis allowed for a better understanding of both coliphage diversity in poultry and the global coliphage diversity within the Siphoviridae and Myoviridae families. However, one should be aware of the possible biases as the coliphages were isolated using two E. coli K12-derived host strains only and, as such, cannot be seen as the complete coliphage diversity. Both host strains are mutated for the FhuA (previously called TonA) gene, which is used as receptor for some phages, including Siphoviridae coliphages T1 and T517. However, phage adsorption is not always restricted to one receptor. Accordingly, we were able to isolate a T5-like phage using the selected host strains. Also, as none of the host strains has the F pili, phages utilising a F pili encoded receptor for absorption, such as Inoviridae phages, will most likely not be isolated17. The reasons why we could not isolate coliphages from the Podoviridae family remains obscure, however, this is in accordance with the finding of Korf et al.15, who also did not isolate any Podoviridae coliphages from poultry while they could isolate them from sewage and surface water. On the other hand, other studies have successfully isolated Podoviridae phages from poultry faeces18,19,20,21. Several factors might play a role in the type of phage being isolated, including culture and isolation method, host strain, and isolation source. In Podoviridae studies mentioned above, phages were isolated using a single or multiple E. coli host-strains and the DLA method was similar to this study. However, a notable difference is the type of host-strain used. In our study, laboratory strains were used whereas the Podoviridae study host-strains only included E. coli strains isolated from poultry. As our 11 Myoviridae phages were isolated from samples from 11 different farms, no correlation between phage isolated and geographical location (poultry farm) was found (data not shown). In accordance to the findings of Olsen16, the most prevalent genus of the Myoviridae and Siphoviridae phages isolated in our study was the Felixounavirus (45.5%) and the Hanrivervirus (33.3%), respectively. Both studies used E. coli K-12 derived laboratory strains as host strain for phage isolation. While in a collection of 50 coliphages isolated from surface water, manure, sewage, or animal faeces, 29 different E. coli host strains were used15. No Felixounavirus phages could be isolated and most Myoviridae phages belonged to the Tequatrovirus genus, followed by the Mosigvirus genus. Those two genera were also found in our study.
Currently, the polyphasic approach is the most commonly used for bacterial classification22,23, and a similar approach combining multiple methods is recommended when working with phages12. However, studying phage taxonomy has proven challenging since no universal conserved marker gene, as the 16S rRNA gene used for bacteria, exists throughout all phage families. Several semi-conserved family-specific marker genes have been proposed as candidates to support taxonomical classification of tailed phages, including DNA packaging and head assembly genes1,24,25. Accordingly, in this study we used the terminase large subunit, portal protein and exonucleases as markers, and were able to determine the family and subfamily to all coliphages isolated in this study in accordance with the TEM-based and whole genome-based classification, respectively. Furthermore, the analysis based on the terminase large subunit and portal protein resulted in a clear distinction between the two Tevenvirinae clusters, indicating different genera (Mosigvirus and Tequatrovirus). Thus, single gene-based analysis provides good initial indication in which taxonomic cluster the phages belong to. However, a single gene does not provide a global view of the structural organization of the phage nor accounts for genomic rearrangements, mutations, and mosaicism. Moreover, in accordance with finding from previous studies the selected gene was not always detected in the phage genomes24, thus excluding these phages form classification. When using single genes, one should be aware of the possibility of multiple distinct variants of the same gene within one genome. Several of our phages encoded up to three different exonuclease genes. Depending on which gene variant used for analysis, the risk of “false” cluster formation and distance, and hereby a faulty classification at subfamily or genus level was present. Thus, for more comprehensive phage taxonomy, including genus classification, the single gene analysis should be accompanied by whole genome-based analysis as well as functional gene studies. When investigating the evolutionary relationship between phages studies have shown the advantage of combining different proteomic and comparative genomic approaches, including WGS data and well-characterized reference dataset, which take into account the effect of horizontal gene transfer (HGT) and recombination events on the phage genome evolution7,26.
In this study, genome-based phylogenetic and taxonomic analysis were performed in combination with traditional morphological examination of the phage using TEM. Through the genome-based analysis we identified a potential new Siphoviridae genus. The three unclassified A3 subcluster from this study clustered together with the Jahat_MG145 reference phage, which was a singleton16. Thus, we propose that this group of phages represents a new unclassified genus with currently four phages, including Phage 52, Phage 56_2 Phage 69, and Jahat_MG145.
We aimed to expand our knowledge on the coliphage diversity, and observed great diversity among these phages, while they were isolated from a similar ecosystem. The diversity was characterised by a great span in genome size (44.3–173.1 kb) and G+C content range (35.5–46.4%), as well as cluster-specific characteristics of the six phage clusters, A–F. Cluster B phages had the smallest genomes and lowest number of CDSs followed by phages from cluster A, D, C, and E/F. Similar to findings from other studies16,27, lower genome size seemed to be correlated with an increase in G+C content. The largest variation in genome size and number of CDSs were observed for the group of Myoviridae phages, whereas the largest variation in G+C content (7.2%) was overserved for the group of Siphoviridae phages. Notably, the Tequintavirus Phage 61 showed to be more similar to the group of Myoviridae compared to the other Siphoviridae phages based on the above-mentioned characteristics. When omitting Phage 61, G+C content variation for the Siphoviridae phages was only 2.9%. Phage G+C content has been shown to be correlated with the G+C content of the phage host27,28. Accordingly, differences in G+C content observed for the coliphages might reflect phage-host interactions and co-evolution with past and current host(s). Gene content, including number of core genes appeared to be associated with the cluster. Moreover, number of exonucleases encoded by the phage appeared to be cluster associated as well, as only phages from cluster C and cluster F encoded multiple exonucleases. Encoding multiple exonucleases could be a result of adaptive evolution conferring fitness advantage over other phages. However, it could just as well reflect some of the challenges to accurate phage genome annotation, including false negatives (undetected genes) and incorrect functional annotation29,30. Gene content variation has been shown to be related to recombination events resulting in acquisition or loss of gene(s)31. Through our comparative genomics analysis of related phages, LCBs with modules with varying level of gene conservation were identified and highlighted the different levels of heterogeneity between different phage clusters. Repeat regions were observed in several of the phage genomes and resulted in variation of LCBs. The presence of these regions should be considered when assessing the gene content variation, as these regions are shown to be prone to genome assembly mistakes, and as such, might represent false level of variation32. LCBs modules are also called for mosaic section and the two terms have been used interchangeably throughout history. One definition is that mosaic sections refer to patches of low nucleotide similarity when two similar phage genomes are compared, and that modules refer to exchangeable sections between two or more phages in the population13. The genome comparison showed the mosaic nature of the phage genomes, with modules with high level of conservation interspersed with mosaic sections. The mosaic sections could be acquired though HGT from other phages, which is thought to happen when phages are found in the same host. Most often this happens through phage co-infection or single-phage infection of a host that carries one or more prophages33,34. Moreover, phages have been shown to acquire genes from their host35. The comparative genomics approach hereby underlines the continuous evolution of phage genomes as well as the great phage diversity.
In conclusion, this study has identified a potential new coliphage genus and several new species and provides insight not only to the coliphage diversity of the intestine of poultry but the global coliphage diversity as well. Moreover, classification of phages isolated in this study brings us one step closer to a more refined taxonomic understanding of coliphages. Our comparative genomic analysis showed different levels of heterogeneity between different phage clusters and highlighted the mosaic nature of the phage genomes as well as the continuous evolution of phages in a single environment source. However, to fully understand the complexity and underlying mechanisms of the phage diversity further studies are needed.
Methods
Phage isolation and propagation
The phages were isolated from poultry faecal material, collected randomly from 27 poultry houses in Belgium in 2013. Phages were propagated according to Adams and Bonilla et al. with minor modifications36,37. Briefly, the samples (5 g) were emulsified in Luria Bertani (LB) broth (Sigma-Aldrich, Saint Louis, MO, USA). The decanted supernatant obtained from each emulsion was enriched by the addition of two early-log phase host bacteria, E. coli K-12 derived laboratory strains C60038 or K51439, a non-restricting, modifying (rK−, mK+) derivative of strain C600. Suspensions were incubated overnight at 37 °C, with shaking (120 rpm) and were then centrifuged at 4,000 rpm for 30 min to pellet the cellular debris. The supernatant containing the phage(s) was re-centrifuged and filtered using a 0.45 µm membrane filter followed by a 0.2 µm Minisart Filter (Fisher Scientific, Waltham, MA, USA). The enriched phage suspensions were enumerated and tested for lytic activity on the host bacteria using the double-layer agar (DLA) technique36,40,41. Briefly, phage suspensions were serial diluted and spotted on an overlay of the respective host bacteria on solid LB medium supplemented with 0.8% agar and 0.5 mM CaCl2. A clear zone in the plate, a plaque, resulting from the lysis of host bacterial cells, indicated the presence of virulent coliphage(s). Samples with lytic activity against the indicator strain were further processed for single phage plaque isolation, including three rounds of plaque purification, and propagation. All phage lysates were stored at 4 °C until required.
Phage morphological analysis
The morphology of unique coliphages (≤ 95% nucleotide similarity) isolated in this study was investigated using transmission electron microscopy (TEM). Phage suspension was applied to the surface of Formvar carbon-coated grids, the phages were fixed using paraformaldehyde (PFA) (4% w/v), washed, and negatively stained with UrAC (1% w/v). After drying, grids were examined using a JEM-1400 Plus transmission electron microscope (JEOL, Benelux).
Genomic DNA extraction and sequencing
DNA extraction from phage lysates was performed using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) as previously described42. The concentration and quality of the DNA was assessed using the NanoDrop (Thermo Scientific, Roskilde, Denmark) and Qubit fluorometer (Thermo Scientific, Roskilde, Denmark) according to the manufacturer’s instructions. Preparation of paired-end 2 × 250 bp sequencing libraries was done using the Nextera XT Kit (Illumina, San Diego, USA) with adaptations for phage genomes as shown elsewhere43 and sequenced on the Illumina MiSeq platform using MiSeq Reagent Kit v2 (500-cycles) and manufacturer’s instructions, yielding a total of 16,270–237,128 paired end reads for each phage lysate. Read-pair contigs were generated for each MiSeq cluster prior to assembly.
Phage genome sequence analysis and annotation
FastQC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), version 0.11.3, was used for quality control validation of the raw reads sequence data. Low-quality sequences were excluded from further analysis. The raw reads were trimmed for quality, adaptor sequences were removed using default parameters. The sequence reads were de novo assembled using QIAGEN Bioinformatics CLC Genomic Workbench, version 11.0.1, using default settings, with minimum contig length changed to 250 bp. An overview of assembly statistics is provided in Supplementary Table S3. The assembled phage sequences were compared with phage homologues from the National Center for Biotechnology Information (NCBI) nucleotide database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) using the Basic Local Alignment Search Tool (BLAST) software44, and from the custom PHAge Search Tool Enhanced Release (PHASTER) phage database45. Newly assembled phage sequences were compared using both BLAST and PHASTER to identify unique and identical (> 95% nucleotide sequence similarity) phages. Assembled contigs were submitted to the ResFinder database, version 3.246 and the VirulenceFinder database, version 2.047 to identify any acquired antimicrobial resistance and virulence associated genes, respectively. By default, selected threshold for %ID was 90% and 60% for minimum length. All 15 databases for antimicrobial resistance genes were selected. The taxonomic group of E. coli bacteria was selected for VirulenceFinder. PHASTER and The Rapid Annotation using Subsystem Technology (RAST) server and the SEED viewer, version 2.0, were used for identification of CDSs and initial annotation of the phage genomes, including identification of the phage terminase large subunit48. Gene function of genes defined as “hypothetical protein” was predicted by comparison to homologue genes with defined functions in other related phage genomes. The G+C content of the phages was calculated using the SEED viewer.
Phage phylogeny and taxonomy
Multiple genome alignment of the WGS sequences was performed using Applied Maths BioNumerics software, version 7.6. According to the ICTV taxonomy guidelines, the 38 coliphages were classified into phage family, subfamily and genera based on nucleotide similarity to known Siphoviridae and Myoviridae coliphages. Known reference coliphages included were limited to isolates with complete genomes found in the ICTV database and the NCBI database (data of November 2019).
Phage diversity
Phylogenetic trees based on the phage whole genome sequences were constructed using R, version 3.5.149, for comparison of the coliphages with published Siphoviridae or Myoviridae reference phage genomes (accessed from the NCBI database). The trees were constructed using unweighted pair group method with arithmetic mean (UPGMA) from a distance matrix of binary distances calculated from either, gene presence/absence within the full genomes of the phages determined using Roary version 3.12.050, Prokka, version 1.13.751 and prodigal, version 2.6.352, or Kmer presence/absence (using 10 and 21mer) based on de novo assembled contigs, as calculated using a python script (Supplementary Script kmer.py).
For phylogenetic analysis based on single marker gene, phage gene sequences were aligned using Clustal X, version 2.153. A maximum likelihood phylogenetic trees (unrooted) was constructed and supported by bootstrap analysis (inferred from 1,000 replicates) with default substitution model (Tamura-Nei model) to assess the diversity of the coliphages using the phylogenetic and molecular evolutionary analyses (MEGA) software, version X54. The phylogenetic trees were based on the nucleotide sequences of the CDSs of the following genes: phage terminase large subunit, phage portal protein, or phage exonucleases. The reference genomes included, represented the best matching published sequences to the phages in this study (selected based on the BLAST max score) and core reference genomes for comparison.
The degree of topological and branch length agreement between the different phylogenetic methods and between the three marker genes was investigated using the R packages Analysis of Phylogenetics and Evolution (ape)55 and phangorn56.
Phage comparative genomics
A more detailed analysis of the most closely related coliphage genomes was carried out. Genomes were re-annotated using Prokka and pan genome analysis was carried out with Roary using script “roary-e-n-s-p 20-i90*.gff”, including identification of core genome, including core and softcore genes, and accessory genome, including shell and cloud genes.
In order to investigate the level of synteny and genomic rearrangement, whole genome alignment and comparison of coliphages and related reference phages from each cluster or subcluster were performed with the Mauve software using progressiveMauve57 with default parameters. No more than 19 of the most related phages were included in each comparison for simplification. Relatedness of the phages were based on percentage of nucleotide similarity and number of shared core genes. Reference genomes were included for annotation references.
Data availability
Raw reads data for phage genome sequences have been registered with the NCBI BioProject database and assigned BioProject ID: PRJNA631989. Project information are assessible with the following link https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA631989. Phage genomes sequences were assigned the following names and NCBI-Sequence Read Achieve (SRA) accession numbers: Phage 8: Escherichia phage vB_EcoS-P8 (SRX8360060), Phage 10: Escherichia phage vB_EcoM-P10 (SRX8360061), Phage 11: Escherichia phage vB_EcoM-P11 (SRX8360071), Phage 15: Escherichia phage vB_EcoM-P15 (SRX8360082), Phage 17: Escherichia phage vB_EcoS-P17 (SRX8360091), Phage 18: Escherichia phage vB_EcoM-P18 (SRX8360092), Phage 28: Escherichia phage vB_EcoS-P28 (SRX8360093), Phage 30: Escherichia phage vB_EcoM-P30 (SRX8360094), Phage 47: Escherichia phage vB_EcoS-P47 (SRX8360095), Phage 48: Escherichia phage vB_EcoS-P48 (SRX8360096), Phage 52: Escherichia phage vB_EcoS-P52 (SRX8360062), Phage 53: Escherichia phage vB_EcoS-P53 (SRX8360063), Phage 54: Escherichia phage vB_EcoS-P54 (SRX8360064), Phage 55: Escherichia phage vB_EcoM-P55 (SRX8360065), Phage 56_1: Escherichia phage vB_EcoS-P56_1 (SRX8360066), Phage 56_2: Escherichia phage vB_EcoS-P56_2 (SRX8360066), Phage 58: Escherichia phage vB_EcoS-P58 (SRX8360067), Phage 59: Escherichia phage vB_EcoS-P59 (SRX8360068), Phage 60: Escherichia phage vB_EcoM-P60 (SRX8360069), Phage 61: Escherichia phage vB_EcoS-P61 (SRX8360070), Phage 62: Escherichia phage vB_EcoM-P62 (SRX8360072), Phage 63: Escherichia phage vB_EcoS-P63 (SRX8360073), Phage 64: Escherichia phage vB_EcoS-P64 (SRX8360074), Phage 65: Escherichia phage vB_EcoS-P65 (SRX8360075), Phage 66: Escherichia phage vB_EcoM-P66 (SRX8360076), Phage 68: Escherichia phage vB_EcoS-P68 (SRX8360077), Phage 69: Escherichia phage vB_EcoS-P69 (SRX8360078), Phage 70: Escherichia phage vB_EcoS-P70 (SRX8360079), Phage 71: Escherichia phage vB_EcoS-P71 (SRX8360080), Phage 72: Escherichia phage vB_EcoS-P72 (SRX8360081), Phage 73: Escherichia phage vB_EcoS-P73(SRX8360083), Phage 74: Escherichia phage vB_EcoS-P74 (SRX8360084), Phage 75: Escherichia phage vB_EcoS-P75 (SRX8360085), Phage 76: Escherichia phage vB_EcoS-P76 (SRX8360086), Phage 77: Escherichia phage vB_EcoS-P77 (SRX8360087), Phage 78: Escherichia phage vB_EcoM-P78 (SRX8360088), Phage 79: Escherichia phage vB_EcoM-P79 (SRX8360089), and Phage 80: Escherichia phage vB_EcoS-P80 (SRX8360090).
References
Grose, J. H. & Casjens, S. R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 468, 421–443 (2014).
Güemes, A. G. C. et al. Viruses as winners in the game of life. Annu. Rev. Virol. 3, 197–214 (2016).
Hatfull, G. F. Dark matter of the biosphere: The amazing world of bacteriophage diversity. J. Virol. 89, 8107–8110 (2015).
Pope, W. H. et al. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife 4, e06416 (2015).
Clokie, M. R. J., Millard, A. D., Letarov, A. V. & Heaphy, S. Phages in nature. Bacteriophage 1(1), 31–45 (2011).
Ackermann, H. W. Phage classification and characterization. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions Vol. 501 (eds Clokie, M. R. J. & Kropinski, A. M.) 127–140 (Humana Press, New York, 2009).
Rohwer, F. & Edwards, R. The phage proteomic tree: A genome-based taxonomy for phage. J. Bacteriol. 184, 4529–4535 (2002).
Lefkowitz, E. J. et al. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 46, D708–D717 (2018).
Meier-Kolthoff, J. P. & Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404 (2017).
Adriaenssens, E. M. & Brister, J. R. How to name and classify your phage: An informal guide. Viruses 9, 1–9 (2017).
International Committee on Taxonomy of Viruses (ICTV), A. M. K. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses (Elsevier, New York, 2011).
Barylski, J. et al. Analysis of spounaviruses as a case study for the overdue reclassification of tailed phages. Syst. Biol. 69, 110–123 (2019).
Casjens, S. R. & Thuman-Commike, P. A. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411, 393–415 (2011).
Hatfull, G. F. & Hendrix, R. W. Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303 (2011).
Korf, I. H. E. et al. Still something to discover: Novel insights into Escherichia coli phage diversity and taxonomy. Viruses 11, 1–29 (2019).
Olsen, N. S., Kot, W., Junco, L. M. F. & Hansen, L. H. Exploring the remarkable diversity of Escherichia coli phages in the Danish Wastewater Environment, including 91 Novel Phage Species. https://www.biorxiv.org/content/10.1101/2020.01.19.911818v1 (2020).
Silva, J. B., Storms, Z. & Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 363, 1–11 (2016).
Mullally, J. et al. Complete genome sequence of Escherichia coli phage APC_JM32 isolated from a chicken cecum. Genome Announc. 6, e01292-17 (2018).
Li, Y. et al. Complete genome sequence of the novel lytic avian pathogenic coliphage NJ01. J. Virol. 86, 13874–13875 (2012).
Chen, M., Xu, J., Yao, H., Lu, C. & Zhang, W. Isolation, genome sequencing and functional analysis of two T7-like coliphages of avian pathogenic Escherichia coli. Gene 582, 47–58 (2016).
Lau, G. L., Sieo, C. C., Tan, W. S. & Ho, Y. W. Characteristics of a phage effective for colibacillosis control in poultry. J. Sci. Food Agric. 92, 2657–2663 (2012).
Vandamme, P. et al. Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol. Rev. 60, 407–438 (1996).
Prakash, O. et al. Polyphasic approach of bacterial classification—An overview of recent advances. Indian J. Microbiol. 47, 98–108 (2007).
Lopes, A., Tavares, P., Petit, M. A., Guérois, R. & Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 15, 1–17 (2014).
Sullivan, M. B. et al. Portal protein diversity and phage ecology. Environ. Microbiol. 10, 2810–2823 (2008).
Glazko, G., Makarenkov, V., Liu, J. & Mushegian, A. Evolutionary history of bacteriophages with double-stranded DNA genomes. Biol. Direct 2, 1–14 (2007).
Almpanis, A., Swain, M., Gatherer, D. & McEwan, N. Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb. Genom. 4, e000168 (2018).
Jacobs-Sera, D. et al. On the nature of mycobacteriophage diversity and host preference. Virology 434, 187–201 (2012).
McNair, K. et al. Phage genome annotation using the RAST pipeline. in Bacteriophages. Methods in Molecular Biology Vol. 1681 (eds Clokie, M., Kropinski, A. & Lavigne, R.) 231–238 (Humana Press, New York, 2018).
Salisbury, A. & Tsourkas, P. K. A method for improving the accuracy and efficiency of bacteriophage genome annotation. Int. J. Mol. Sci. 20, 3391 (2019).
Kupczok, A. et al. Rates of mutation and recombination in siphoviridae phage genome evolution over three decades. Mol. Biol. Evol. 35, 1147–1159 (2018).
Klumpp, J., Fouts, D. E. & Sozhamannan, S. Next generation sequencing technologies and the changing landscape of phage genomics. Bacteriophage 2, 190–199 (2012).
Hendrix, R. W. Bacteriophage genomics. Curr. Opin. Microbiol. 6, 506–511 (2003).
Hendrix, R. W., Smith, M. C. M., Burns, R. N., Ford, M. E. & Hatfull, G. F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. 96, 2192–2197 (1999).
Pedulla, M. L. et al. Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182 (2003).
Adams, M. H. Bacteriophages (Interscience Publishers, New York, 1959).
Bonilla, N. et al. Phage on tap—A quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ. https://doi.org/10.7717/peerj.2261 (2016).
Appleyard, R. K. Segregation of new lysogenic types during growth of a doubly lysogenic strain derived from Escherichia coli K12. Genetics 39, 440–452 (1954).
Colson, C., Glover, S. W., Symonds, N. & Stacey, K. A. The location of the genes for host-controlled modification and restriction in Escherichia coli K-12. Genetics 52, 1043–1050 (1965).
Kropinski, A. M., Mazzocco, A., Waddell, T. E., Lingohr, E. & Johnson, R. P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages. Methods in Molecular Biology Vol. 501 (eds Clokie, M. R. & Kropinski, A. M.) 69–76 (Humana Press, New York, 2009).
Oliveira, A. et al. Isolation and characterization of bacteriophages for avian pathogenic E. coli strains. J. Appl. Microbiol. 106, 1919–1927 (2009).
Jakočiūnė, D. & Moodley, A. A Rapid bacteriophage DNA extraction method. Methods Protoc. 1, 27 (2018).
Kot, W., Vogensen, F. K., Sørensen, S. J. & Hansen, L. H. DPS—A rapid method for genome sequencing of DNA-containing bacteriophages directly from a single plaque. J. Virol. Methods 196, 152–156 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Arndt, D. et al. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
Hasman, H. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644 (2012).
Joensen, K. G. et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510 (2014).
Overbeek, R. et al. The SEED and the rapid annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, 206–214 (2014).
R Core Team. R: A language and environment for statistical computing. http://www.R-project.org/ (R Foundation for Statistical Computing, 2018).
Page, A. J. et al. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Hyatt, D. et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Paradis, E. & Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Schliep, K. P. phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011).
Darling, A. E., Mau, B. & Perna, N. T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Acknowledgements
We thank Irene Mutuku for the isolation of the coliphages. We are grateful for the technical help of Liesbeth Couck with the transmission electron microscopy. We acknowledge funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant agreement no. 765147.
Author information
Authors and Affiliations
Contributions
P.S. and D.J. performed experimental work. P.S. and K.K. analysed the phage genome sequences. A.M. supervised the work at its early stages. W.V. contributed with analysis tools. A.G. and H.I. co-supervised the work. P.B. conceived supervised the research. P.S. wrote the article. All authors have reviewed and approved of the article before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sørensen, P.E., Van Den Broeck, W., Kiil, K. et al. New insights into the biodiversity of coliphages in the intestine of poultry. Sci Rep 10, 15220 (2020). https://doi.org/10.1038/s41598-020-72177-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72177-2
- Springer Nature Limited
This article is cited by
-
Metagenomic analysis of wastewater phageome from a University Hospital in Turkey
Archives of Microbiology (2022)